
Abstract
Araucaria (Araucaria angustifolia (Bert.) O. Ktze) is a primarily dioecious species threatened with extinction that plays an important social and economic role especially in the southern region of Brazil. The aim of this work is to investigate the diversity and likely determinants of genetic lineages in this species for conservation management. For this, a collection of 30-year-old Araucaria was used. Accessions collected from 12 sites across the species range were analyzed, with ten individuals per site. The SSR genotyping was conducted with 15 loci and the data were analyzed using several complementary approaches. Descriptive statistics among sampling sites were used and diversity was partitioned non-hierarchically to estimate the size and composition of genetic clusters using a Bayesian assignment method. To explore possible biological implications of differences between Niche Models and habitat suitability, a series of statistical procedures were used, and tests were carried out using the software ENM Tools and Maxent. Populations from the southernmost zone showed higher genetic variation and a lower inbreeding coefficient compared to the northernmost zone, which may correlate with their isolation. A positive relation between genetic differentiation and geographic distance was observed. Two genetic groups (southernmost and northernmost zones) were evident. The Niche modelling showed separate ranges for each genetic lineage suggesting that differences in selection pressure may be playing a role in the apparent differentiation and may be adaptive. Finally, an evident correlation was observed between genetic data and habitat suitability. The two distinct groups observed must be considered as independent units for conservation and hybridization in breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Many forest species from diverse biomes are currently threatened with extinction. The establishment of criteria for genetic conservation is a difficult task. The use of niche model and population genetic studies are tools currently applied to study the distribution and diversity of natural populations indicating populations for genetic conservation. This article aims to identify araucaria populations groups aiming at genetic conservation of this species.
Conservation of genetic diversity is a challenge for strongly fragmented species as result of anthropic activities, since the biological aspects, as diversity, phylogeny and reproductive pattern, among others are still unknown. Although Brazil is mega-biodiverse detainer, the strong fragmentation of forests, as a consequence of exploitation and change in land use, has been responsible for threats and extinction of thousands of animal and plant species (Pimm et al. 2014). The knowledge on the quantity and distribution of genetic diversity is crucial not only to support both in situ and ex situ conservation programs, but also to sustain breeding enterprises for the potential reforestation of forest tree species.
Araucaria angustifolia is a primarily dioecious gymnosperm tree native to the Atlantic Forest domain (Ombrophilous Mixed Forest) that plays an important social, economic, and ecological role especially in Southern Brazil. araucaria occurs more continuously in Southern Brazil (southernmost zone), with scattered populations in highlands of the Southeastern Brazil (northernmost zone) and small populations in Argentina and Paraguay (Carvalho 2003). This species used to be more widespread with an original area estimated in almost 250,000 Km2 (Lamprecht 1989). Due to overexploitation during the 20th Century, araucaria experienced a reduction of 97% in its original area in Brazil (Enright et al. 1995) and only small forest remnants persist. Consequently, araucaria is listed as critically endangered (Farjon 2006) in the International Union for Conservation of Nature and Natural Resources (IUCN) Red List and as vulnerable in the MMA Official list of flora threatened by extinction (MMA 2014).
As A. angustifolia occurs across a geographic region, variation between populations is expected and has been observed, since the first studies with quantitative traits (Gurgel and Gurgel Filho 1965; Gurgel Filho 1980; Shimizu and Higa 1980; Kageyama and Jacob 1980; Pires et al. 1983; Sebbenn et al. 2004), and later using biochemical (Mazza 1997; Shimizu et al. 2000; Sousa 2001; Auler 2002; Sousa et al. 2004; Mantovani et al. 2006) and molecular markers (Hampp et al. 2000; Stefenon et al. 2003, 2007, 2008a, b; Bittencourt and Sebbenn 2007; Souza et al. 2009; Patreze and Tsai 2010; Duarte 2011; Medina-Macedo et al. 2015, 2016). Studies on population genetics have shown that variation among populations is mostly attributed to geographic distances, and consequently, mating isolation by distance pattern (Gurgel Filho 1980; Gurgel and Gurgel Filho 1965; Shimizu and Higa 1980; Kageyama and Jacob 1980; Pires et al. 1983). Variation in genetic structure patterns is today better understood from the historical process and evolutionary forces acting on this species.
Palynological studies of lakebed pollen depositions (De Oliveira and Colinvaeux 1992; Ledru 1993; Behling 1997, 2002, 2003; Behling and Lichte 1997; Stefenon et al. 2008a; Jeske-Pieruschka et al. 2010) have been used to reconstruct historical dynamics in range expansion and contraction of this species. Historically there is evidence of the species occurring in the northernmost region and then expansion to the southernmost region, especially as a result of the climatic oscillations (temperature and humidity). Recent studies based on plastid DNA sequence information revealed a structuring of araucaria populations with strong biogeographic patterns both at a regional sampling scale (Lauterjung et al. 2018) and across the whole distribution range of the species in South America (Stefenon et al. 2019).
Nowadays prioritizing populations for conservation is hard given the strong forest fragmentation. Furthermore, the history and diversity of the remaining fragments is not known. However, the use of niche modeling together with genetic analysis to guide genetic conservation programs for threatened species facing extinction is challenging since establishing rational conservation is hampered by lack of relevant data.
Ecological Niche Modeling (ENM) is a tool successfully used to delimit the potential range of a species. According to Wiens (2011), Holt (2009) based on the general concept of Hutchinson (1957), a niche is a set of biotic and abiotic conditions where a species can persist. Species range limits are essentially the expression of a species ecological niche in geographic space (Sexton et al. 2009), once “range limits create biogeographic patterns and ecological niches create range limits” (Wiens 2011). Niche models have been used successfully to guide collecting of new populations of rare and endangered species (Menon et al. 2010), and in understanding contributors to phylogenetic diversity (Alvarado-Serrano and Knowles 2014; Stefenon et al. 2019). Niche modeling together with population genetic studies have also been helpful to estimate the environmental and geographic contributors to the current distribution and diversity of natural populations (Diniz-Filho et al. 2009; Reeves and Richards 2010; Temunović et al. 2012). A direct relation between ecological niche suitability of biotic and abiotic conditions has been shown, as well as the distribution of certain species, so that the occurrence limit coincides with the niche limit (Lee-Yaw et al. 2016).
In this study we focused on using nuclear SSR markers and niche modeling in order to (i) estimate the degree of genetic diversity and differentiation among sampled populations, (ii) correlate these patterns with spatially explicit models of habitat suitability and (iii) evaluate the level of genetic structure retained by the Active Germplasm Collection (AGC) of EMBRAPA Florestas.
Materials and methods
Populations and sampling
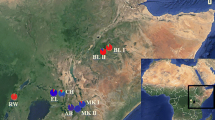
Analysis was performed on 120 individuals sampled at the Active Germplasm Collection (AGC) of EMBRAPA Florestas, Colombo, Paraná State, Brazil (25° 17′ 30″ latitude S, 49°, 13′ 27″ W, and 1027 m above sea level). These 120 individuals comprised plants propagated from seed at the AGC. Seeds were randomly collected from 12 natural populations located across Brazil (Fig. 1) in 1980, with 10 individuals sampled per population.

Blue circles represent the sampled populations and the red circle represents the location where the AGC was implanted in 1980. Populations collected within the northernmost zone of the species occurrence (1 = Barbacena-MG, 2 = Ipuiúna de Caldas-MG, 3 = Congonhal-MG, 4 = Campos do Jordão-SP, 5 = Itapeva-SP, 6 = Itararé-SP), and populations collected within the southernmost zone (7 = Telêmaco Borba-PR, 8 = Quatro Barras-PR, 9 = Irati-PR, 10 = Três Barras-SC, 11 = Caçador-SC, 12 = Chapecó-SC); AGB = Active Germplasm Collection in Colombo-PR (AGC). The smaller blue circles are incidence records used for environmental niche models
Populations were collected in a geographic region ranging from 43° 50′ 00″ W to 52° 36′ 00″ W and 21° 00′ 00″ S to 27° 07′ 00″ S at altitudes varying between 560 and 1800 m above sea level. Because the distribution of populations naturally reaches two Brazilian geographic regions, they were categorized here northernmost, as and southernmost zone of the species occurrence (Fig. 1 and Table 1). Populations from the northernmost zone are scattered and therefore smaller and more isolated than those from the southernmost zone.
Genotyping procedure
Samples of cambium tissue were stored dry in tubes with silica gel at room temperature during transportation and processing. Samples were disrupted using a Mixer Mill MM 300 (Qiagen). Genomic DNA was extracted using DNasy 96 Plant Kit (Qiagen) and stored at − 18 °C. Genotypes of all samples were scored at 15 loci: Aang01, Aang03, Aang04, Ang14, Aang17, Aang22, Ang23, Aang24, Ang28, Ang30, Ang35, Aang35, Aang37, Aang43, Aang45 and Aang47 (Schmidt et al. 2007).
Microsatellite sizing data was collected using M13 tailing and fluorescent capillary electrophoresis on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The 5′ tailed primers were produced by adding the M13 sequence 5′-CACGACGTTGTAAAACGAC-3′ to the forward primers during primer synthesis (GBT oligos, Brazil). The M13 primers with 5′ fluorescent dye labels (6-FAM, VIC, NED, or PET, Applied Biosystems) were used to label and detect PCR products (Steffens et al. 1993; Schuelke 2000). The PCR reaction mix consisted of 10 ng template DNA, 1X GoTaq® Flexi Buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 2.5 U GoTaq® Flexi DNA Polymerase (Promega), 2.0 pmol reverse primer, 0.4 pmol 5′ tailed forward primer, and 2.0 pmol of labelled M13 primer (CACGACGTTGTAAAACGAC), in 10 μL total volume. Thermal cycling regime was carried out in Peltier Thermal Cycler PTC 200 (MJ Research) and consisted of a denaturing step at 94 °C, 3 min; followed by 10 touchdown cycles of 94 °C for 30 s; 65–1 °C/cycle for 30 s; 72 °C for 45 s with subsequent amplification of 30 cycles at 94 °C for 30 s; 55 °C for 30 s; 72 °C for 45 s, and final extension of 72 °C for 7 min. The PCR products were multiplexed and diluted for analysis in Hi-Di Formamide (Applied Biosystems). The internal molecular weight standard for the ABI3130xl was Genescan 600-LIZ (Applied Biosystems). Fragment sizes were evaluated and scored using GeneMapper Version 4.0 Software (Applied Biosystems).
Intrapopulation genetic diversity analysis
Hardy–Weinberg equilibrium tests were performed, as well as linkage disequilibrium test for individual locus and also for pairs of loci using GDA Software through Exact test of Fisher after 3200 permutations of allele among individuals. After, we characterized the genetic diversity within each collection site. Several parameters of genetic diversity were estimated. Mean number of alleles per locus (A), allelic richness per locus and sample (Rs), expected heterozygosity (He), observed heterozygosity (Ho), and the Weir and Cockerham’s (1984) estimator of inbreeding (f) were estimated using GDA (Lewis and Zaykin 2001) and FSTAT version 2.9.3.2. (Goudet 2002).
Population genetic structure analysis
We assessed the genetic structure of the collection, using two complimentary approaches, based on (i) a priori population designations, and (ii) on model-based clustering (Dyer 2009). Genetic studio a suite of programs for analysis of genetic marker, the edges in the graph represent genetic variance among sites and the node diameter represents genetic variance within sites. Evidence of an isolation-by-distance pattern among populations of araucaria was inferred by plotting estimates of pairwise genetic differentiation (estimated as FST/(1 − FST); Rousset 1997) against pairwise geographic distance. Pairwise FST estimations were performed using the software FSTAT version 2.9.3.2 (Goudet 2002).
Spatial analysis of molecular variance (SAMOVA) was used to determine the most likely number of genetic clusters based on the multilocus allelic frequency and the geographic location of each population, using the software Samova v. 1.2 (Dupanloup et al. 2002), with the number of geographic groups K set from 1 to 10. The molecular distance was determined as the sum of squared size difference (RST) and 500 computations were performed for each K. This program finds the best number of geographic groups (K-value) by maximizing FCT value between K groups of geographically adjacent populations. In addition, a population graph of genotypes among the 12 sampling populations was performed using the analysis of molecular variance (AMOVA) framework using the software Genetic Studio (Dyer 2009).
A Bayesian assignment analysis, as performed in the software Structure 2.2.3 (Pritchard et al. 2000) was used to identify distinct genetic clusters and estimate admixture analysis of multilocus genotypes without using a priori population of origin information. Bayesian analysis of population structure was performed using the admixture and allele frequency correlated allele models with 50,000 burn-in periods and 500,000 Markov chain Monte Carlo Repts. Number of K was set from 2 to 10 and 10 replicates were run for each K. The most probable number of clusters (K) was selected by computing the second-order rate of change of the likelihood function with respect to K (Evanno et al. 2005) using the software Structure Harvester (Earl 2012). Analysis was also performed by a priori defining two groups: northernmost [municipalities of Barbacena, Ipuiúna de Caldas, Congonhal in Minas Gerais state (MG), and Campos do Jordão, Itapeva and Itararé in São Paulo state (SP)] and southernmost [Municipalities of Telêmaco Borba, Quatro Barras, Irati in Paraná state (PR), and Três Barras-SC, Caçador-SC and Chapecó in Santa Catarina state (SC)].
Environmental Niche modeling
Environmental niche modeling was performed using Maxent 3.3.3k (Phillips et al. 2006). Geographical coordinates were obtained from the active gene collection sites as well as from herbarium records of the species. A total of 126 incidence records (progenies) were used to build the model, 91 of which corresponded to a southernmost geographical group and 35 to a northernmost geographical group. These groups were proposed based on previous evidence from independent research on araucaria (Sousa 2001; Sousa et al. 2009; Stefenon et al. 2019) species. Climatic layers included 19 biologically-relevant variables (P1: Annual Mean Temperature, P2: Mean Diurnal Range (Mean period max–min), P3: Isothermality (P2/P7); P4: Temperature Seasonality (Coefficient of Variation), P5: Max Temperature of Warmest Period, P6: Min Temperature of Coldest Period, P7: Temperature Annual Range (P5–P6), P8: Mean Temperature of Wettest Quarter, P9: Mean Temperature of Driest Quarter, P10: Mean Temperature of Warmest Quarter, P11: Mean Temperature of Coldest Quarter, P12: Annual Precipitation, P13: Precipitation of Wettest Period, P14: Precipitation of Driest Period, P15: Precipitation Seasonality (Coefficient of Variation), P16: Precipitation of Wettest Quarter, P17: Precipitation of Driest Quarter, P18: Precipitation of Warmest Quarter, and P19: Precipitation of Coldest Quarter) derived from temperature and moisture data (the “BioClim” layers), plus altitude. Thirty arc-second grids were obtained from WorldClim (http://www.worldclim.org) and cropped to the study region. Habitat suitability models were calculated using all sites, and for the two geographical groups separately, using Maxent default settings. Twenty five percent of sites were set aside as test localities for model validation, which included an examination of omission rates on test localities and the area under the receiver operating characteristic curve (AUC), as well as application of a binomial test meant to determine whether the model computed by Maxent predicts the presence at test sites better than a random model (Anderson et al. 2002; Phillips et al. 2006).
To explore possible biological implications of differences between niche models for the northernmost and southernmost groups, a series of statistical procedures were employed, described in detail by Warren et al. (2008) and Glor and Warren (2011). Tests were carried out using the software ENMTools 1.3 (Warren et al. 2010). Measures of niche overlap (I, D) were calculated for all tests and compared to approximate null distributions (generated by resampling) using one-sample Z-test. The “niche identity test” considers the null hypothesis that two niche models of interest are no more different than models derived by random bipartition of the sampled sites. The “background similarity test” considers whether the models are no more different than random given the habitat available in the regions sampled, thus providing a correction for spatial autocorrelation of environmental variables not offered by the niche identity test. Background regions were defined using rectangles to enclose each geographical group. Null distributions for niche identity and background similarity tests were established using 500 replicates.
“Range break tests” consider whether the geographic dividing line between two groups is associated with a significant change in environment (Glor and Warren 2011). Both “linear” and “blob” models were used to partition data and develop a null distribution. The former splits the sampled sites using a line of random slope, while the latter uses a nucleating approach to partition data. Because of geographic clustering within groups, the number of pseudo replicate data sets that can be created for the null distribution is limited; bipartitions selected at random are often non-independent. We attempted to include all possible non-identical bipartitions by first generating 10,000 replicate samples at random using linear and blob models for data bisection, then filtering those samples to remove duplicated instances of the same bipartition. For the linear range break test, 144 unique data sets were found and used to construct the null distribution (blob test, 69).
Results
Population genetic structure
All, from 15 loci, were in Hardy–Weinberg equilibrium (at 5% level) and the analysis of pairs of loci showed 19.0% in linkage disequilibrium. However, Medina-Macedo et al. (2014) studying Araucaria angustifolia (10 loci) after applying Bonferroni correction to observed data, obtained 51% of pairwise loci in genotypic disequilibrium for adult trees. They attributed this result to the strong spatial genetic structure of populations. Despite of this these loci were recommended for genetic studies on genetic diversity and structure, mating system and gene flow of the species. Therefore, the proportion of 19.0% including 15 loci is much lower than the results observed in Medina-Macedo et al. (2014), and there is no reason to remake our analysis in a different way.
A total of 167 alleles were observed across the 15 loci, with an average of 11.13 alleles/locus. The mean number of alleles/locus at population level ranged from 4.33 to 6.07 (Table 2). And the mean alleles/locus values for the southernmost and northernmost groupings were 10 and 8, respectively. A higher number of private alleles were detected for southernmost populations (22 alleles) while for northernmost 11 alleles private allele was observed (Table 2). Chapéco, Barbacena and Itapeva populations presented more number of private alleles.
Most populations showed heterozygous deficit except for Quatro Barras-PR and Chapecó-SC, both from the southernmost zone (Table 2). In average, observed heterozygosity (Ho = 0.61) was slightly lower than expected heterozygosity (He= 0.67) (Table 2). Southernmost populations showed higher genetic variation and lower inbreeding compared to northernmost populations considering other parameter, such as average number of alleles per locus (A), Allelic Richness per locus and sample (Rs,), and gene diversity (H), Nei (1987).
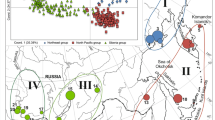
In the SAMOVA analysis, the FCT estimations were all significant (p < 0.001) and continuously decreased from K = 2 (FCT = 0.187) up to K = 10 (FCT = 0.152), suggesting the existence of two main genetic groups (Fig. 2a). One group is formed by populations Barbacena, Ipuiúna and Campos do Jordão, while the other group is composed by the all the other populations. Although population Congonhal is geographically located near populations Barbacena, Ipuiúna and Campos do Jordão (Fig. 1), the presence of two individuals with assignment to the southernmost populations (Fig. 2c) grouped it in the second group in the SAMOVA analysis.

Analyses of population structure (Bayesian based). a Spatial Analysis of Molecular Variance (SAMOVA) for K ranging from 2 to 10. b Population Graph of genotypes among sampling sites. Edges in the graph represent genetic variance among sites, and the node diameters represent genetic variance within site, the blue circles represent populations from northern and red circles represent populations from southern region Bar Barbacena, Ipiu Ipuiúna de Caldas, Con Congonhal, CJ Campos do Jordão, Itap Itapeva, Itara Itararé, Tele Telêmaco Borba, Irata Irati, QB Quatro Barras, Tres Três Barras, Caca Caçador, Cha Chapecó. c Bayesian-bases analysis of population structure for K = 2 (L’(K) = 462.95 and ΔK = 1516.41)
The highest likelihood values for the different replicates (10) used in Bayesian assignment analysis was observed for K = 2 that contributed for a high ΔK (1516.41). The Ln (K) and ΔK values ranged from − 163.18 (k = 8) to 462.95 (K = 2) and 0.0063 (K = 9) to 1516.41 (K = 2), respectively. The populations were structured in northernmost and southernmost groups, with one population (Itapeva) positioned as a transition population between groups, presenting a clear mixture of genotypes from both groups (Fig. 2c). The same pattern of population structuring was revealed by the population graph of genotypes based on the population graph framework (Fig. 2b), with two main groups and population Itapeva connecting both clusters. This population shows a higher genetic similarity to the northernmost region although it is geographically closer to the southernmost region. A positive relationship was observed between genetic differentiation and geographic distance (Fig. 3), suggesting an isolation-by-distance pattern.

Differentiation among populations plotting Rousset’s distance (FST/(1 − FST)) and geographic distance (Km). Regression R2 = 0.53, P < 0.0001. FST estimated according to Weir and Cockerham (1984)
Environmental Niche modeling
Niche models were able to capture the fundamental environmental factors driving the distribution of sampled araucaria populations (Fig. 4a). Omission rates on test sites across cumulative threshold values were comparable to predicted values for the northernmost data set and the data set containing all sites.

Niche model showing two different groups of suitability araucaria for northern and southernmost populations. Warmer color show areas with better predicted conditions. a Northern and southern populations correlated with an. b Environmental discontinuity between regions
Rainfall and temperature are the two main climatic differences between both region (northern and southern). Soil also differs greatly between them. Rainfalls amount is higher in the southern, as well as its distribution. It’s rain more in winter than in summer, while in northern rainfalls are more concentrated in the summer. In addition, the minimum temperatures are lower in the Southern, in comparing with northern boundaries. Occurrence of frost is also higher in the southern. The summer water deficit in the southern is higher than the rest of the region. The soils of the southern half are sandy and shallow.
The northernmost group omission rates were much lower than expected across a broad range of threshold values, most likely because of the small sample size and few geographically clustered sites, leading to non-independence between test and training sites. The AUC values for test data were high across data sets (overall = 0.866, northernmost = 0.971, southernmost = 0.869) and the binomial test of omission indicated that models performed significantly better than random, regardless of threshold applied (p < 1.7 × 10−5).
The niche identity test indicated that niche models generated by northernmost and southernmost groups were significantly less similar to one another than paired models generated by random sampling of occupied sites (p < 1 × 10−9). The test is trivial, however, when the ranges under consideration are allopatric. The background similarity test compensates for the spatial autocorrelation of environmental variables. Using this test, northernmost and southernmost niche predictions were found to be no more different than expected given random sampling of the underlying geographic ranges (p > 0.14 for all comparisons). Given that the groups originate from different geographic regions with associated differences in environmental conditions, this result was expected.
Range break tests indicate that the geographic line separating the northernmost and southernmost groups marks a region of significant environmental change (Fig. 4b). Null distributions for range break tests were computed using all possible unique bipartitions of the data. However, all distributions, except that calculated for the blob range break test using D, were non-normal (Shapiro–Wilk test, p < 0.009). Nevertheless, the overlap value for the two geographic groups was always the lowest value observed for a given distribution.
Finally, an evident relation was observed between genetic data and habitat suitability (Fig. 5). The overall pattern is likely the result of the interplay between a known biogeographic process (the recent suitability of the southernmost highlands, 3000 years BP) and barriers to gene flow.

Genetic data pattern correlated with habitat suitability (Niche model). The grey line represent the transition area between groups (Itapeva-SP population)
Discussion
Genetic diversity and differentiation among sampled populations
Our estimates of genetic diversity from these ex situ provenance trial collections is similar to estimates of diversity from samples taken directly from the field (Inza et al. 2018; Patreze and Tsai 2010; Medina-Macedo et al. 2015; Bittencourt and Sebbenn 2007). The levels of genetic diversity measured as expected heterozygosity (mean multilocus He = 0.67; Table 2) is quite similar to values estimated for natural populations (mean multilocus estimations ranging from He = 0.59 to He = 0.74). Stefenon et al. (2008b) suggested that planted forests may have abundant genetic diversity for species conservation when comparing natural populations and commercial plantations of A. angustifolia with SSR and AFLP markers. Analogously, the estimations obtained in this study suggest that the EMBRAPA’s AGC can be considered an important repository of the genetic resources of araucaria from the populations where samples were collected to establish this active germplasm collection.
Estimates of regional differentiation derived from three complementary analytical approaches confirmed that the species is split into two genetic groups. This discontinuity in genetic structure was estimated from SAMOVA analysis where K = 2 was the most likely, Structure analysis where K = 2 had the highest change in posterior probability and in a visual rendering of genetic structure though a reticulate network (a population graph). In the latter case, the pivotal population that had the most admixture among two genetic groups was the population identified as the link uniting two separate subnetworks (Fig. 2b). This overall pattern of genetic structure is congruent with studies reported for natural populations evaluated with isozymes (Sousa et al. 2004), nuclear SSR and AFLP markers (Stefenon et al. 2007; Souza et al. 2009). In addition, the structure estimated in this study using polymorphic nuclear SSR markers correlated broadly to genetic structure inferred for cpDNA haplotype (Stefenon et al. 2019).
The properties of this latitudinal split in genetic linages are also broadly supported by the partition of environmental parameters by niche modeling. The region where most genetic divergence occurs is also a region where the environmental gradients are diverging. It’s especially, fraught to support a set of statistical metrics supporting this environmental split since at this spatial scale geography and climate are confounded. However, the overlap value for the two geographic groups was always the lowest value observed for a given distribution. Our results indicate that the pattern is likely the result of the interplay between a known biogeographic process (the recent suitability of the Southern highlands, 3000 years BP) and barriers to gene flow. If the northernmost regions are considered the refugia during interglacial periods, the southernmost highlands may be the location of an expanding species range. Contemporary genetic structure may reflect processes that involve biogeographic range expansion and contraction including fluctuations in effective population size leading and differences in inbreeding and diversity between the two lineages. Our data suggest that the two genetic groups do differ in genetic diversity and in average inbreeding. However, it is clear that the current levels of genetic diversity are greatly affected by ongoing anthropogenic impacts on the species range and demographic dynamics.
Since A. angustifolia occurs with many related species in the Atlantic Forest Biome, such as broad-leaved tree species and Podocarpus, the only other coniferous genera native to Brazil, it might be possible to apply these results more generally at the community scale. For instance, the Mixed Rain Forest or Ombrophilous Mixed Forest, is composed of a number of key forest species whose genetic differentiation has been shaped by paleo climatic dynamics. Using palynological surveys, which serve as markers of past species distributions, it is clear that a pattern of expansion and contraction have occurred in the interglacial periods of the last 50,000 years BP. Such patterns have been described for Podocarpus spp. (Ledru et al. 2007). In Ledru et al. (2007) study, evidence gleaned from palynological data and molecular markers defined three main region groups composed of mixtures of diversifying species. In this particular case two of the main group (the southernmost 2) correspond closely with the genetic and environmental data in this study, lending support to the application of these groups across species and possibly ecologically defined community types.
Although Podocarpus spp and Araucaria angustifolia occurs in the Forest Atlantic Domain (which comprises the Ombrophilous Mixed Forest, Atlantic Rainforest, and Semi Seasonal Forest), Podocarpus has a wider natural area, also occurring in the northernmost region of Brazil, especially in the Atlantic Rainforest. Ledru et al. (2007), using botany, palynology, and genetics to characterize the current and past distribution of these genera, observed three groups: one in the southeastern geographic region, a Central group (which matches the northernmost group proposed in this study for A. angustifolia); and a southern group (which matches the southernmost group of A. angustifolia). Therefore, the two large groups observed for the A. angustifolia clearly match Podocarpus. In addition, Ledru et al. (2007) have found a small subgroup within the Central group (northernmost in our work), compatible with the Campos do Jordão region, which is already reported here as a controversial and different population
The levels of inbreeding of the EMBRAPA’s AGC (mean multilocus f (c)= 0.09; Table 2) are within the values estimated for natural populations (mean multilocus estimations ranging from f = − 0.09 to f = 0.17). Positive inbreeding coefficient has been detected by many researchers for northernmost and southernmost populations as well (Bittencourt and Sebbenn 2007).
In this work, the southernmost group had clearly higher values of genetic variation. On the other hand, higher inbreeding coefficients and higher fixation index (Fis) were present for northernmost populations. It reflects the current conditions of fragmented and isolated populations with a supposed smaller effective population size, thus characterized as peripheral population. According to Eckert et al. (2008), although several studies have shown that the difference in genetic diversity between central and peripheral populations may not be large, on average; there is a diversity decline and increased differentiation, besides the association of these trends.
Patterns of genetic structure and spatially explicit models of habitat suitability
Because araucaria occurs in a wide geographic range, variation between populations was expected, considering different environmental conditions as soil, climate, and other variables, continuously acting on populations in its natural occurrence range.
Since the first observations on the genetic variation of araucaria (Gurgel and Gurgel Filho 1965; Gurgel Filho 1980; Shimizu and Higa 1980; Kageyama and Jacob 1980), isolation-by-distance has been identified even before the use of molecular markers. Later, the use of this new tool allowed inferring the relation between genetic data and geographic distance (Stefenon et al. 2008b). Beyond the great distances between the populations of A. angustifolia, this species has a low potential gene flow, flotation ratio was demonstrated to be the principal reason for it (Sousa and Hattemer 2003), which hinders its dispersion and contribute with a spatial genetic structure, intensifying the differentiation between them. This is an unusual situation for this conifer species since pollen is normally more easily dispersed. Moreover, spatial genetic structure has been described using isoenzymes (Mantovani et al. 2006), as well as for microsatellite markers (Bittencourt and Sebbenn 2007; Stefenon et al. 2008a), contributing with an unequal variability distribution, as well as related individuals in a short distance.
Given that all possible random bipartitions were included in the niche modeling analysis, we can conclude that niche similarity between geographic groups is less than the random expectation. In other words, niche divergence is maximized when sampled sites are split into the northernmost and southernmost groups. This suggests that selection pressures may likely be different in the distinct genetic lineages, leading to the potential for adaptive differentiation.
Analyzing Itapeva population (Fig. 5) by several genetic parameters, it seems a transition between the two large detected groups. Floristic studies from this region corroborate this transition assumption. In fact, studies show that various floristic types are found, including species from the Cerrado (Brazilian Savannah), Pantanal (Wetlands), and Mixed Rain Forest or Ombrophilous Mixed Forest. According to Garcia et al. (2015), floristic surveys of Itapeva Station (Forestry Institute of São Paulo) have shown predominance of the Fabaceae family, typical of the Brazilian Savannah regions. Both populations are no contiguous they are located almost 25 km away. They appear in scattered populations. Itararé correspond better to populations from colder altitude areas of the Southeast and may come from the edge of the Devonian Escarpment. Itapeva population seems to belong to the araucaria transition where appear more frequently, but sparingly, under a slightly higher thermal regime and more severe winter drought conditions. Furthermore, Garcia et al. (2015) mention the species Clethra scabra Pers. reported by other researchers (Torres et al. 1994; Toniato et al. 1998) in Pantanal (associated vegetation formation or a mix of vegetation) or species from typical formations of Mixed Rain Forest (Ilex paraguariensis, Sanquetta et al. 2002).
Based on cpDNA and a wide sample covering the whole distribution range of the species, Stefenon et al. (2019) demonstrated the existence of three main genetic groups of araucaria: a Northern group (matching the northernmost group observed in this study), a central group and a southernmost group. Besides to southward forest expansion, the presence of glacial refugia in the northernmost and southernmost zones of araucaria distribution was the argument for explaining the species genetic structuring, although the anthropogenic influence in araucaria populations cannot be discarded (Stefenon et al. 2008b; Lauterjung et al. 2018).
Environmental characteristics directly influence the habitats of species and the distribution of genetic variation. Gene flow and consequently spatial structure of genetic variation could be affected. In this work, two different niche models could historically explain the observed genetic differences among groups since a strong fragmentation is recent compared to the long history and longevity of A. angustifolia. Thus, the genetic structure will be more affected in future generations than the old generation analyzed in this work. Based on these models, it is clear that, even before the settling, a gene flow between of the two large regions was very difficult. Therefore, the main barrier between population groups was niche suitability. Methods combining GIS (geographic information system) and genetic data are critical in linking patterns with process and support conservation efforts for other important species of the Ombrophilous Mixed Forest in Brazil.
Araucaria angustifolia is associated with other tree species of economic interest (Ocotea porosa, Ocotea catharinensis, Ocotea odorifera, and Ilex paraguariensis, among others) whose conservation is critical for most of them. The historic past for these populations is related in some degree to the history of the A. angustifolia. However, broad-leaved species have a very recent origin compared to A. angustifolia, which are more competitive and efficient in general. The understanding of two different niches for the Araucaria Forest can be helpful in an effort of genetic conservation for those associated species in Ombrophilous Mixed Forest. Currently, global warming is a great threat to the remnants of the species, besides the anthropogenic effect that still persists. Global climate change could change the natural occurrence, strongly limiting and reducing it to small areas where conditions remain favorable (Wrege et al. 2009, 2017). The temperature and humidity will be the variable climatic more limiting.
From the assessment of genetic data and Niche model analysis (suitability of habitat), the importance of environmental effects is evident, as well as genetic forces (gene flow, migration, drift, mutation, and selection), in the shaping of these populations along their history. The two distinct groups observed must be considered as independent units for conservation and intraspecific hybridization in breeding programs.
References
Alvarado-Serrano DF, Knowles LL (2014) Ecological niche models in phylogeographic studies: applications, advances and precautions. Mol Ecol Resour 14:233–248
Anderson RP, Gómez-Laverde M, Peterson AT (2002) Geographical distributions of spiny pocket mice in South America: insights from predictive models. Glob Ecol Biogeogr 11:131–141
Auler NMF (2002) The genetics and conservation of Araucaria angustifolia: genetic structure and diversity of natural populations by means of non-adaptative variation in the state of Santa Catarina, Brazil. Genet Mol Biol 25:239–338
Behling H (1997) Late Quaternary vegetation, climate and fire history from the tropical mountain region of Morro de Itapeva, SE Brazil. Palaeogeogr Palaeoclimatol Palaeoecol 129:407–422
Behling H (2002) South and Southeast Brazilian grasslands during Late Quaternary times: a synthesis. Palaeogeogr Palaeoclimatol Palaeoecol 177:19–27
Behling H (2003) Late glacial and Holocene vegetation, climate and fire history inferred from Lagoa Nova in southeastern Brazilian lowland. Veg Hist Archeobot 12:263–270
Behling H, Lichte M (1997) Evidence of dry and cold climatic conditions at glacial times sin tropical southeastern Brazil. Qua Res 48:348–358
Bittencourt JVM, Sebbenn AM (2007) Patterns of pollen and seed dispersal in a small, fragmented population of the wind-pollinated tree Araucaria angustifolia in southern Brazil. Heredity 99:580–591
Carvalho PER (2003) Espécies Arbóreas Brasileiras. Embrapa Informação Tecnológica, EMBRAPA-CNPF, Brasília, DF
De Oliveira PE, Colinvaeux PA (1992) Vegetational and climatic changes of the late Quaternary in southeastern Brazil. In: 8th International Palynological Congress Programmer and Abstracts, Aixen-Provence, p 34
Diniz-Filho JAF, Nabout JC, Bini LM, Soares TN, Telles MPC, JrP Marco, Collevatti RG (2009) Niche modelling and landscape genetics of Caryocar brasiliense (“Pequi” tree: Caryocaraceae) in Brazilian Cerrado: an integrative approach for evaluating central–peripheral population patterns. Tree Genet Genomes 5:617–627
Duarte RI (2011) Caracterização da diversidade genética de uma população de Araucaria angustifolia (BERT.) O. Kuntze. Dissertação, Universidade Federal de Santa Catarina
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Dyer RJ (2009) Genetic Studio a suite of programs for the analysis of genetic marker data. http://dyerlab.bio.vcu.edu
Earl DA (2012) Structure harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Enright NJ, Hill RS, Veblen TT (1995) The Southern Conifers. An introduction. In: Enright NJ, Hill RS (eds) Ecology of the Southern Conifers. Melbourne University Press, Melbourne, pp 1–9
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Farjon A (2006) Araucaria angustifolia. In: IUCN 2011. IUCN red list of threatened species. V. 2011.2. http://www.iucnredlist.org. Accessed 17 Feb 2012
Garcia CA, Santos AG, Barreira S (2015) Estrutura fitossociológica de uma área de cerrado na estação ecológica de Itapeva, São Paulo, Brasil. Rev Agric Neotrop 2:77–85
Glor RE, Warren D (2011) Testing ecological explanations for biogeographic boundaries. Evol Int J Org Evol 65(3):73–683
Goudet J (2002) FSTAT (Version 1.2): a computer program to calculate F statistics. J Hered 86:485–486
Gurgel Filho OA (1980) Silvicultura da Araucaria angustifolia (Bert.) O. Ktze. IUFRO Meeting on forestry problems of the genus Araucaria. Anais, Curitiba, pp 29–68
Gurgel JTA, Gurgel Filho OA (1965) Evidências de raças geográficas no pinheiro brasileiro, Araucaria angustifolia (Bert.) O. Ktze. Ciênc Cult 17(1):33–39
Hampp R, Mertz A, Schaible R, Schwaigerer M, Nehls U (2000) Distinction of Araucaria angustifolia seeds from different locations in Brazil by a specific DNA sequence. Trees 14:429–434
Holt RD (2009) Bringing the Hutchinsonian niche into the 21st century: ecological and evolutionary perspectives. Proc Nat Acad Sci 106:19659–19665
Hutchinson GE (1957) Concluding remarks. Cold Spring Harbor Symp 22:415–427
IBAMA (Instituto Nacional do Meio Ambiente e dos Recursos Naturais Renováveis) (1992) Lista Oficial das Espécies da Flora Brasileira Ameaçadas de Extinção. http://www.ibama.gov.br/flora/extincao.htm. Accessed 22 Feb 2012
Inza MV, Aguirre NC, Torales SL, Pahr NM, Fassola HE, Fornes LF, Zelener N (2018) Genetic variability of Araucaria angustifolia in the Argentinean Parana Forest and implications for management and conservation. Trees 32(4):1135–1146
Jeske-Pieruschka V, Fidelis A, Bergamin RS, Vélez E, Behling H (2010) Araucaria forest dynamics in relation to fire frequency in southern Brazil based on fossil and modern pollen data. Rev Paleobot Palynol 160:53–65
Kageyama PY, Jacob WS (1980) Variação genética entre e dentro de populações de Araucaria angustifolia (Bert.) O. Ktze. IUFRO Meeting on Forestry Problems of the Genus Araucaria. Anais, Curitiba, pp 83–86
Lamprecht H (1989) Silviculture in the tropics: tropical forest ecosystems and their tree species; possibilities and methods for their long-term utilization. Deutsche Gesellschaft für Technische Zusammenarbeit (GTZ), Bonn, p 296
Lauterjung MB, Bernardi AP, Montagna T, Candido-Ribeiro R, da Costa NCF, Mantovani A, dos Reis MS (2018) Phylogeography of Brazilian pine (Araucaria angustifolia): integrative evidence for pre-Columbian anthropogenic dispersal. Tree Genet Genomes 14(3):36
Ledru MP (1993) Late quaternary environmental and climatic changes in central Brazil. Quat Res 39:90–98
Ledru MP, Salatino MLF, Ceccantini G, Salatino A, Pinheiro F, Pintaud JC (2007) Regional assessment of the impact of climatic change on the distribution of a tropical conifer in the lowlands of South America. Divers Distrib 13:761–771
Lee-Yaw JA, Kharouba HM, Bontrager M, Mahony C, Csergörgo AM, Noreen AME, Li Q, Shuster R, Angert ALA (2016) Synthesis of transplant experiments and ecological niche models suggests that range limits are often Nicht limits. Ecol Lett 9:710–722
Lewis P, Zaykin D (2001) Genetic data analysis: computer program for the analysis of allelic data (software), v. 1.1. http://lewis.eeb.uconn.edulewishome/software.html
Mantovani A, Morellato PC, Reis MS (2006) Internal genetic structure and outcrossing rate in a natural population of Araucaria angustifolia (Bert.) O. Kuntze. J Hered 97:466–472
Mazza MCM (1997) Use of RAPD markers in the study of genetic diversity of Araucaria angustifolia (Bert.) populations in Brazil. In: Bruns S, Mantell S, Tragardh Viana AM (eds) Recent advances in biotechnology for tree conservation and management. International Foundation for Science, Stockholm, pp 103–111
Medina-Macedo L, Lacerda AEB, Ribeiro JZ, Bittencourt JVM, Sebbenn AM (2014) Investigating the Mendelian inheritance, genetic linkage, and genotypic disequilibrium for ten microsatellite loci of Araucaria angustifolia. Silvae Genet 63(1–6):234–239
Medina-Macedo L, Sebbenn AM, Lacerda AE, Ribeiro JZ, Soccol CR, Bittencourt JV (2015) High levels of genetic diversity through pollen flow of the coniferous Araucaria angustifolia: a landscape level study in Southern Brazil. Tree Genet Genomes 11:1–14
Medina-Macedo L, de Lacerda AEB, Sebbenn AM, Ribeiro JZ, Soccol CR, Bittencourt JVM (2016) Using genetic diversity and mating system parameters estimated from genetic markers to determine strategies for the conservation of Araucaria angustifolia (Bert.) O. Kuntze (Araucariaceae). Conserv Genet 17:413–423
Menon S, Choudhury BI, Khan ML, Peterson AT (2010) Ecological niche modeling and local knowledge predict new populations of Gymnocladus assamicus a critically endangered tree species. Endanger Species Res 11(2):175–181
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Patreze CM, Tsai SM (2010) Intrapopulational genetic diversity of Araucaria angustifolia (Bertol.) Kuntze is different when assessed on the basis of chloroplast of nuclear markers. Plant Syst Evol 284:111–122
Phillips SJ, Anderson RP, Schapire RE (2006) Maximum entropy modeling of species geographic distributions. Ecol Model 190:231–259
Pimm SL, Jenkins CN, Abell R, Brooks TM, Gittleman JL, Joppa LN, Raven PH, Roberts CM, Sexton JO (2014) The biodiversity of species and their rates of extinction, distribution, and protection. Science 344(6187):1246752
Pires CLS, Barbin D, Gurfinkel J, Marcondes MAP (1983) Teste de progênies de Araucaria angustifolia (Bert.) O. Ktze em Campos do Jordão. Silvicultura, São Paulo, pp 437–439
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Reeves PA, Richards CM (2010) Species delimitation under the general lineage concept: an empirical example using Wild North American Hops (Cannabaceae: Humulus lupulus). Syst Biol 60:45–49
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Sanquetta CR, Pizatto W, Netto SP, Figueiredo Filho A, Eisfeld RDL (2002) Estrutura vertical de um fragmento de Floresta Ombrófila Mista no centro-sul do Paraná. Floresta 32:267–276
Schmidt AB, Ciampi AY, Guerra MP, Nodari RO (2007) Isolation of microsatellite markers for Araucaria angustifolia (Araucariaceae). Mol Ecol Notes 7:340–342
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Sebbenn AM, Pontinha AAS, Freitas SA, Freitas JA (2004) Variação genética em cinco procedências de Araucaria angustifolia (Bert.) O. Ktze. no sul do estado de São Paulo. Rev Inst Florest 16(2):91–99
Sexton JP, McIntyre PJ, Angert AL, Rice KJ (2009) Evolution and ecology of species range limits. Annu Rev Evol Syst 40:415–436
Shimizu JY, Higa AR (1980) Variação genética entre procedências de Araucaria angustifolia (Bert.) O. Ktze, na região de Itapeva, estimada até o 6o ano de idade. IUFRO Meeting on Forestry Problems of the Genus Araucaria. Anais, Curitiba, pp 78–82
Shimizu JY, Jaeger P, Sopchaki SA (2000) Variabilidade genética em uma população remanescente de araucária no Parque Nacional do Iguaçu. Embrapa Florestas-Artigo em periódico indexado (ALICE)
Sousa VA (2001) Population genetic studies in Araucaria angustifolia (Bert.) O. Ktze, 1st edn. Cuvillier Verlag Goettingen, Goettingen
Sousa VA, Hattemer HH (2003) Pollen dispersal and gene flow by pollen in Araucaria angustifolia. Aust J Bot 51:309–317
Sousa VA, Robinson IP, Hattemer HH (2004) Variation and population structure at enzyme gene loci in Araucaria angustifolia (Bert.) O. Ktze. Silvae Genet 53:12–19
Sousa VA, Valgas RA, Lavoranti OJ, Chaves Neto A, Shimizu JY (2009) Genetic differentiation among Araucaria populations in Brazil. In: XIII Congreso Forestal Mundial, Desarrollo forestal: equilibrio vital, FAO, 2009, Buenos Aires
Souza MIF, Salgueiro F, Carnavalle-Bottino M, Félix D, Alves-Ferreira M, Bittencourt JVM, Margis R (2009) Patterns of genetic diversity in southern and southeastern Araucaria angustifolia (Bert.) O. Kuntze relict populations. Genet Mol Biol 32(3):546–556
Stefenon VM, Nodari RO, Reis MS (2003) Padronização de protocolo AFLP e sua capacidade informativa para análise da diversidade genética em Araucaria angustifolia. Sci For 64:163–171
Stefenon VM, Gailing O, Finkeldey R (2007) Genetic structure of Araucaria angustifolia (Araucariaceae) populations in Brazil: implications for the ex situ conservation of genetic resources. Plant Biol 9:516–525
Stefenon VM, Behling H, Gailing O, Finkeldey R (2008a) Evidences of delayed size recovery in Araucaia angustifolia populations after post-glacial colonization of highlands in Southeastern Brazil. An Acad Bras Ciênc 80:433–443
Stefenon VM, Gailing O, Finkeldey R (2008b) Genetic structure of plantations and the conservation of genetic resources of Brazilian pine (Araucaria angustifolia). For Ecol Manage 255:2718–2725
Stefenon VM, Steiner N, Guerra MP, Nodari RO (2009) Integrating approaches towards the conservation of forest genetic resources: a case study of Araucaria angustifolia. Biodivers Conserv 18:2433–2448
Stefenon VM, Klabunde G, Lemos RPM, Rogalski M, Nodari RO (2019) Phylogeography of plastid DNA sequences suggests post-glacial southward demographic expansion and the existence of several glacial refugia for Araucaria angustifolia. Sci Rep 9(1):2752
Steffens DL, Sutter SL, Roemer SC (1993) An alternative universal forward primer for improved automated DNA sequencing of M13. Biotechniques 15:580–582
Temunović M, Franjić J, Satovic Z, Grgurev M, Frascaria-Lacoste N, Fernández-Manjarrés JF (2012) Environmental heterogeneity explains the genetic structure of Continental and Mediterranean populations of Fraxinus angustifolia Vahl. PLoS ONE 7(8):e42764
Toniato MTZ, Leitão Filho HDF, Rodrigues RR (1998) Fitossociologia de um remanescente de floresta higrófila (mata de brejo) em Campinas, SP. Braz J Bot 21(2):197–210
Torres RB, Matthes LAF, Rodrigues RR (1994) Florística e estrutura do componente arbóreo de mata de brejo em Campinas, SP. Rev Bras Bot 17(2):189–194
Warren DL, Glor RE, Turelli M (2008) Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution 62:2868–2883
Warren DL, Glor RE, Turelli M (2010) ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33:607–611
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wiens JJ (2011) The niche, biogeography and species interactions. Philos Trans R Soc B 366:2336–2350
Wrege MS, Higa RCV, Britez RM, Garrastazu MC, De Sousa VA, Caramori P, Radin HB, Braga HJ (2009) Climate change and conservation of Araucaria angustifolia in Brazil. Unasylva (FAO)
Wrege MS, Fritzsons E, Soares MTS, Bognola IA, Sousa VA, Sousa LP, Gomes JBV, Aguiar AV, Gomes JC, Matos MFS, Scarante AG, Ferrer RS (2017) Distribuição natural e habitat da araucária frente às mudanças climáticas globais. Pesqui Florest Bras 7:331–346
Acknowledgements
First, we would like to thank for accompanying us at EMBRAPA Forestry and supporting this work, material collection, and shipment to the USA Tiago Luiz Daros (Forestry Engineer), Jacir Faber (Field Staff), Daiane Rigoni and Marianne Bernardes (analyst and laboratory assistant of the Molecular Biology Laboratory, respectively). We would like to thank Dr. Alfredo Alves (Labex Fort Collins) for his support in Fort Collins and who together with Francisco Ricardo Ferreira (EMBRAPA Recursos Genéticos e Biotecnologia) and Rosa Miriam de Vasconcelos (EMBRAPA) collaborated with the international biological material transfer. We would like to thank NCGRP/ARS (National Center for Genetic Resources Preservation)/(Agricultural Research Service) team for the technical and unconditional support, enabling this manuscript.
Funding
This work was supported by the EMBRAPA Florestas and NCGRP/ARS/USDA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest by the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Sousa, V.A., Reeves, P.A., Reilley, A. et al. Genetic diversity and biogeographic determinants of population structure in Araucaria angustifolia (Bert.) O. Ktze. Conserv Genet 21, 217–229 (2020). https://doi.org/10.1007/s10592-019-01242-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01242-9