
Abstract
The regioselective oxidation of the primary hydroxyl groups of cellulose, usually mediated by the (2,2,6,6-tetramethylpiperidin-1-yl)oxyl radical (TEMPO), is highly popular in the scientific literature. However, the lack of efficient monitoring techniques imposes a severe limitation to its upscaling. This work involves a portable, user-friendly near-infrared spectroscopy device, optimized preprocessing techniques, and multivariate calibration to quickly estimate the carboxyl group content of modified cellulose (i.e., the extent of the oxidation). For that, bleached pulps from eucalyptus, pine, hemp, and sisal were submitted to TEMPO-mediated oxidation, varying the dosage of spent oxidizer (NaClO) and thus attaining samples of different values of carboxyl group content. These values were related to near-infrared spectra (908–1676 nm) by Partial Least Squares regression, yielding cross-validation coefficients (RCV2) above 0.97 for wood pulps, 0.95 for sisal pulp, and 0.91 for hemp pulp. Based on the residual prediction deviation, the model for each pulp was found to show good predictability. Nonetheless, the overall regression model, comprising the four different materials, was unreliable. In light of this, spectra were submitted to principal components analysis (PCA), hinting that pulps could be classified in terms of their hemicellulose to cellulose ratio. Considering all the statistical parameters, the overall proposal presented here begins with a PCA—Linear Discriminant Analysis model to classify the sample by its fiber type, subsequently selecting a specific regression model for that class. Overall, the presented models in this work allow the determination of the extent of oxidation of different cellulosic feedstocks, expressed as carboxyl content, in a fast and simple approach using a benchtop near-infrared equipment.
Similar content being viewed by others


Introduction
The growing interest in cellulose nanomaterials, either in the form of cellulose nanofibers (CNFs), cellulose nanocrystals (CNCs), or even bacterial cellulose (BC), has opened a new paradigm on the development of value-added lignocellulosic materials for a myriad of applications (Eichhorn et al. 2010; Moon et al. 2011; Abitbol et al. 2016; Boufi et al. 2016; Li et al. 2021). CNFs are generally obtained from a top-down approach, involving different stages that suitably transform lignocellulosic fibers at the microscale to the nanoscale, particularly in terms of fiber width (Li et al. 2021). For this, lignocellulosic feedstocks are usually pretreated by means of either chemical, enzymatic, or mechanical methods, to be later fibrillated. Depending on the pretreatment (type and intensity), and the fibrillation intensity and strategy, the resulting CNFs will possess different morphological and chemical features (Sanchez-Salvador et al. 2022; Signori-Iamin et al. 2022). Among the most widely used pretreatments, TEMPO-mediated oxidation has been reported to provide highly charged and homogeneous CNFs after low-intensity fibrillation. Actually, the regioselective oxidation effectively transforms hydroxyl groups from the primary alcohol in C6 into carboxyl groups, being highly anionic and more voluminous, which promotes fibrillation in the subsequent stages (Saito and Isogai 2004; Isogai and Zhou 2019).
While TEMPO-mediated oxidation is a relatively simple process that has been successfully implemented at laboratory scale, its scaling up is still lacking on appropriate real-time monitoring systems, able to deliver the carboxyl content, for instance, as function of time in a fast and simple manner. In this line, several efforts have been paid to monitor the evolution of the carboxyl content with oxidation time and conditions, mainly based on kinetic modelling (Sun et al. 2005; Dai et al. 2011; Mazega et al. 2023). However, these models require the analysis of counter-samples by analytical methods, namely conductometric titration or methylene blue absorption (Im et al. 2018; Lin et al. 2018). These processes usually require specialized laboratory technicians to perform time-consuming analytical determinations, which generate a delay between the process and the relevant information that can be extracted from, hindering any possibility of implementing process control loops.
Near-infrared spectroscopy (NIR) has been considered as a valuable technique for providing chemical and structural information about organic molecules, because NIR spectra are influenced by characteristic vibration modes of some functional groups that have C-H, N–H, S–H and O–H bonds (Blanco and Villarroya 2002; Pu et al. 2020). This type of vibrational spectroscopy uses the region of the electromagnetic spectrum with wavelengths ranging from 780 to 2500 nm, where the absorption bands of electromagnetic energy correspond mainly to overtones and combination of fundamental vibration bands (Santos et al. 2013a, b). Therefore, the NIR spectrum generally involves overlapped and broad bands, and also weak signal without distinct signature of individual compounds. For many years, these aspects made this spectral region to be considered too difficult to interpret. However, with the advent of modern computers and advances in multivariable data analysis algorithms and chemometrics, NIR spectroscopy received a major boost being able to provide efficient correlation between spectral data and several variables linked or not to composition (Badaró et al. 2022; Simon et al. 2022). Besides, due to its capacity of being rapid and a non-destructive method which requires minimal or no sample preparation, NIR spectroscopy has become useful for process monitoring and control (Pu et al. 2020).
The application of NIR to determine the composition of cellulosic materials has been addressed in the literature with the aim of replacing time-consuming analytical characterization techniques that require a variety of chemicals. In this sense Mayr et al. (2015) and Zhou et al. (2019) determined the cellulose content in dry samples of wood pulp using information from NIR spectra. The study by Mayr et al. showed that the spectra in the NIR region were also sensitive to hemicellulose and lignin contents as well as the composition of hemicellulose (mannose and xylose). Quantitative studies were carried out by Li et al. (2015), Jin et al. (2017) and Zhang et al. (2017) correlating the NIR spectra with the content of the main lignocellulosic components (cellulose, hemicellulose and lignin) in different cellulosic biomasses (wood and plants). A few studies have investigated the quantification of specific functional groups in cellulosic samples. Thus, Simon et al. (2022) determined the degree of oxidation of dialdehyde celluloses via NIR spectroscopy. Henniges et al. (2009) developed a method for quantifying the content of carboxyl and carbonyl groups in pulp hand sheets and rag papers. In all cited studies, the NIR spectra were calibrated using experimental data from analytical characterization techniques with the help of multivariate calibration techniques such as Partial Least Squares (PLS) and Multiple Linear Regression (MLR) (Geladi and Kowalski 1986; Brereton 2003).
NIR spectroscopy has been also used together with discrimination/classification algorithms to identify samples with distinct compositional characteristics (Brereton 2003). In this sense, Cazón et al. (2022) classified cellulosic films as a function of the nature of the cellulose (vegetable or bacterial) and Diniz et al. (2019) identified samples of wood sawdust from two different species of eucalyptus.
Recently, the number of applications of NIR spectroscopy on plant products has increased significantly due to the development of portable spectrophotometers that are fast, low-cost, easy to use, and robust. Moreover, these miniature spectrophotometers can be used directly in-line and do not require a large physical laboratory space, such as benchtop equipments usually do (Dos Santos et al. 2013a, b; Beć et al. 2021).
Among the main differences between bench and portable NIR spectrophotometers, one should cite the technology used in the selection of wavelengths. For benchtop equipment, the selection is made using dispersive elements or interferograms that take up a lot of space. Commercial portable instruments have varied designs and mainly employ linear variable filter (LVF) or digital micromirror array (Beć et al. 2021). This means that portable instruments have a restricted wavelength range, lower resolution and poor signal-to-noise-ratios (Mayr et al. 2021). Due to these characteristics, the performance of portable NIR equipment for each specific application must be investigated and such studies are currently part of a very active research area. However, there are still few applications of this type of NIR spectrophotometer in determining the composition of cellulosic materials. In this sense, Chavez Lozano et al. (2023) successfully used NIR spectra collected from a portable equipment to determine the substitution content of acetate functions during the hydrolysis of these groups in cellulose acetate films. Also, Diniz et al. (2019) compared the performance of the portable instrument with benchtop equipment for identifying eucalyptus species from different sawdust. The portable equipment proved to be quite efficient in classifying the samples and the benchtop equipment only presented an accuracy between 7 and 10% greater. In both studies, the portable equipment used was the MicroNIR 1700 (Viavi Solutions) with a spectral range between 908 and 1676 nm.
As noted above, the use of portable NIR devices to evaluate the composition of cellulosic materials is still a recent topic. Worse yet, there are no studies in the literature relating the use of compact and low-cost spectrophotometers to analyze the content of carboxylic groups of cellulose fibers. In fact, the development of robust and low-cost monitoring techniques is essential for controlling the cellulose production process. Through a sensor capable of providing reliable information about the carboxylic contents, the fiber oxidation process can be correctly adjusted, ensuring the quality of the final product. For all the above, in this work, we propose the application of a portable NIR spectrophotometer to evaluate the carboxylic content of oxidized cellulose fibers, as an alternative method to traditional analytical techniques. The joint use of NIR spectra and multivariate analysis techniques allow to predict the carboxylic contents of cellulosic materials well. Moreover, calibration and validation performance of the NIR PLS models built using the portable NIR instrument were similar to those obtained from a benchtop NIR instrument. Finally, a strategy to expand the NIR spectra correlation for carboxylic contents of fibers from different sources, through the sequential use of classification algorithms and PLS models, is provided.
Experimental section
Materials
Four different commercial bleached pulps were selected, consisting of four different raw materials: (i) eucalyptus, (ii) pine, (iii) hemp, and (iv) sisal. In the case of eucalyptus and pine, both consisted of bleached kraft pulps (BKEP and BKPP, respectively) and were kindly provided by LECTA group (Zaragoza, Spain), in the case of BKEP, and by Celulosa Arauco y Constitución S.A. (Los Horcones, Chile), in the case of BKPP. Both hemp and sisal pulps were provided by Celulosa de Levante S.A. (Tortosa, Spain). In all cases, the pulps were provided in the form of dry laps with a moisture content around 10%. All the reagents required for TEMPO-mediated oxidation, sample processing and characterization, were acquired at Merck (Barcelona, Spain).
Characterization of the raw materials
The bleached pulps were characterized in terms of chemical composition and morphology. Samples were firstly dried at 105 °C until constant weight, to be later milled and sieved (40 mesh). On portion was used for ash content determination by calcination (525 °C), according to TAPPI T211 standard. In parallel, another portion was submitted to solvent extractives determination by Soxhlet extraction using ethanol-toluene mixture as solvent, as detailed in TAPPI T204 standard. Acid-insoluble lignin of the extractive-free samples was determined according to the Klason lignin method, described by TAPPI T222 standard. Finally, cellulose content was determined by high-performance anion exchange chromatography (HPAEC), as described elsewhere (Tarrés et al. 2017). Hemicellulose was then determined by difference from 100%.
The morphological features of the selected pulps were determined in a MorFi equipment (TechPap, France), equipped with a CCD video camera. The equipment is run by the MorFi v9.2 software and is able to analyzed about 30,000 fibers per test. Among other parameters, the software provides information on length, diameter, kink angle, and fines content of the analyzed fiber suspensions, both in the for of distribution and average values.
TEMPO-mediated oxidation
TEMPO-mediated oxidation was performed over the four selected pulps at different oxidizer amounts, ranging from 2 to 12 mmol of NaClO/g. In a typical experiment, 0.24 g of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and 1.5 g of NaBr were suspended in 1000 mL of deionized water and kept under stirring, at room temperature, until complete dissolution of the co-catalysts. Then, 15 g (over dry weight) were suspended in the dissolution and additional water was added to reach a total volume of 1500 mL. Once the fiber was completely suspended in water and under gentle stirring, the selected amount of NaClO was added to the suspension at once, observing an increase in the pH. The pH was maintained at 10.5 by means of the dropwise addition of a 0.1 M NaOH solution until no changes were observed in the pH. The resulting fibers were rinsed with deionized water and filtered. The oxidized fibers were stored in hermetic plastic bags at 4 °C for further use and characterization (Saito and Isogai 2004). The samples were labelled according to the raw material, indicating the first letter of the plant specie (E, P, H, or S, for eucalyptus, pine, hemp, or sisal), and the amount of added NaClO (in mmol/g). For instance, the hemp pulp oxidized with 8 mmol/g of NaClO was labelled as H8.
Carboxyl content determination of the oxidized pulps
The carboxyl content (CC) of the oxidized pulps was determined according to a previously reported methodology. Briefly, 3–5 mg of dry fiber were added in a solution containing 5 mL of methylene blue at the concentration of 300 mg/L and 5 mL of borate buffer solution at pH 8.5. The samples were kept under stirring and later centrifuged for 20 min at 3500 G-force. After centrifugation, 2 mL of the supernatant were transferred into a 25 mL flask containing 2.5 mL of HCl 0.1 N, and the volume was completed with deionized water. The absorbance at 664 nm was measured and correlated to a previously calibration curve (Mazega et al. 2023). These measurements were performed in triplicate.
Samples preparation and NIR spectra acquisition
Cellulose samples with approximately 1% fiber content involving different contents of carboxyl groups and prepared from different vegetable raw materials were freeze-dried (Liobras, model L101, São Carlos, Brazil) to completely remove water. At the end of the process, 0.35 g of dry cellulose from each sample was inserted into a glass vial and compacted in order to remove as much air as possible. Each sample was prepared in triplicate. Table S1 of the supplementary material summarizes the content of carboxyl groups and the raw material used for each sample. Note that the carboxyl content in the samples varied from 0.002 to 1.292 mmol.g−1.
Handheld spectrophotometer
NIR Spectra were collected using a portable MicroNIR 1700ES spectrophotometer (Viavi Solutions) configured to scan the spectral region ranging from 908 to 1676 nm. Each spectrum corresponded to the average of 100 scans measured using an integration time of 7 ms. The spectral resolution of the equipment was 6.2 nm. A linear variable filter (LVF) as a wavelength selector directly connected to a set of linear detectors, resulting in a spectral system without moving parts. This compact system is coupled to a tungsten lamp as a source of thermal radiation illumination. All measurements were performed using a glass vial. Before each measurement, the reference spectrum was measured using the Spectralon NIR standard with a reflection coefficient of 99% as well as the spectrum with 0% reflection (dark) with the equipment lamp turned off. Reference spectra were collected before characterizing a new sample. Spectral data were obtained with MicroNIR Pro v.2.5.1 software supplied with the instrument. For each sample, 5 spectra were taken, from which an average spectrum was determined.
Benchtop spectrophotometer
The spectral data were also taken using the FT-NIR spectrophotometer (Bruker, Vertex 70), in the spectral region from 1000 to 2500 nm, with a resolution of 0.17 nm and taking 32 scans per spectrum. Each scan consists of measuring 3111 wavelength points. The instrument was equipped with an extented InGaAs detector and a tungsten lamp as source of light radiation. Spectra acquisition was performed using the reflectance immersion probe and applying the Fourier transform to the interferogram signal. The background spectrum was acquired once at the beginning of the characterizations with the empty probe and the NIR data were obtained using OPUS 4.0 software provided with the FT-NIR instrument.
In both NIR instruments the spectral mode used was reflectance. This spectral mode is suitable for characterizing solid samples (Lohumi et al. 2015; Badaró et al. 2022). The reflectance measurements (R) were transformed into absorbance spectrum (A) through its logarithmic relationship: A = log(1/R).
Data analysis and NIR spectra preprocessing
The spectra in the NIR region obtained by the instruments' own software were transferred to the multivariate analysis software Unscrambler X 10.5.1 (Aspentech), which was used for NIR data preprocessing, calibration and validation.
In solid samples such as those used in this work, differences in spectra in the NIR region can be highly influenced not only from changes in the chemical composition of the samples but also from variations in physical properties such as compaction and surface roughness (Rinnan et al. 2009; Robert and Gosselin 2022). Changes in spectra can also be caused by undesirable variations, such as changes in temperature, environment, sampling, etc. (Brereton 2003). Thus, mathematical preprocessing techniques aim to remove the effects caused by physical properties (additive and multiplicative effects) and other unwanted variations not directly related to the property of interest. Preprocessed spectra can be better correlated with sample composition through chemometric techniques such as multiple linear calibration models. The preprocessing techniques used were Standard Normal Variate (SNV), Savitzky-Golay (SG) First and Second Derivative and Orthogonal Signal Correction (OSC) (Wold et al. 1998; Brereton 2003; Rinnan et al. 2009). These techniques were tested separately and sequentially, in such a way that the best technique (or combination of techniques) was identified using the trial and error method and evaluating the lowest error obtained in validating the quantitative calibration models (Engel et al. 2013; Jiao et al. 2020). In the current work, the Results section encompasses just those models that yielded the lowest validation errors.
The detection of outlier samples and their removal from the set of samples for calibration was performed by analyzing the residuals (Q) vs Hotelling’s T2 plots. Q is a measure of the model's error in describing each sample and T2 determines the distance from the model projection of each sample to the center of the projections (Standards 2000).
Qualitative model (PCA/LDA)
Classification algorithms were applied to classify samples with different levels of carboxyl groups depending on the vegetable raw material used to manufacture the fibers, that is, identifying whether the sample belongs to one of these four classes: eucalyptus, pine, sisal or hemp. Among the algorithms, supervised pattern recognition methods stand out for which the classification of the samples is known and this information is used to optimize the model parameters (Brereton 2003; Bakeev 2010). Model fit based on these algorithms is evaluated on a validation dataset by comparing model predictions with actual category values. Within this group of algorithms, the Linear Discriminant Analysis (LDA) Method was chosen due to its wide use in various applications involving NIR spectroscopy (Wu et al. 1996; Esteki et al. 2018; de Almeida et al. 2021; Ribeiro et al. 2021).
LDA is an algorithm based on the assumption that the population of each class can be described as a normal distribution and the covariance matrix of each class is the same. LDA is also a dimensionality reduction algorithm by maximizing the distance between samples belonging to different classes and minimizing the separation within the same class. When applying LDA to NIR spectroscopy data, in general, the number of variables is greater than the number of samples, which leads to a problem of ill-conditioned matrices. In this case, dimensionality reduction can be performed through Principal Component Analysis (PCA) by applying the LDA algorithm to the principal components (PCs). It is noteworthy that PCA reduces the dimension of the spectra by creating new variables called PCs (which are linear combinations of the first ones) in the direction of maximum data variability (Maćkiewicz and Ratajczak 1993; Brereton 2003). The combination of the two algorithms is called PCA-LDA. This study employed the LDA based on the PC scores of the spectra.
The performance of the classification model was evaluated in terms of accuracy (ACC), which represents a measure of how well the model is able to classify samples, in accordance with Eq. 1 (Ballabio and Consonni 2013; Hicks et al. 2022):
where, TP and TN are the number of true positive and true negative samples, respectively and FP and FN stand for the number of false positive and false negative samples, respectively.
Calibration model (PLS)
To build the calibration models, the Partial Least Squares multivariate calibration technique was used as it is the most popular regression method applied to instrumental NIR spectroscopy data (Geladi and Kowalski 1986; Wold et al. 2001; Pasquini 2018). In this method, the experimental data is summarized in two matrices, X and Y, which correspond to the absorbance data and the property of interest (composition), respectively. This is a method of reducing the dimension of the data space where the new subspace is determined based on a compromise between explaining the variance of X and the correct prediction of Y. The variables X and Y are decomposed into a sum of λ latent variables (LVs). The best number of latent variables for each model was estimated from the minimum values of the root mean squared error of the results predicted by cross-validation (RMSECV, from Eq. 2). This error and the coefficient of determination R2 of the validation results were both used as quality parameters of model performance.
Another useful statistics used to interpret calibration models performance is the residual prediction deviation (RPD, from Eq. 3), which is defined as the ratio between the standard deviation of the experimental data (SD) and the bias-corrected standard error of validation (SDV), given by Eq. 4:
where, \({\overline{y} }_{i}\) represents the values of property y of sample i estimated by cross-validation, \({y}_{i}\) is the corresponding reference value and n is the number of samples. Bias is calculated from Eq. 5 and stands for the average systematic error of the difference between the estimated and reference y value. Equations 2–5 comply with specific ASTM standards (Standards 2000).
The RPD was used for the first time by Williams and Sobering 1993 in order to compare PLS calibration models developed from different NIR spectrophotometers for the prediction of the same property. According to the literature, calibration models with good predictability have RPD > 2.4, while values between 1.5 and 2.4 indicate a satisfactory model prediction. Models with RPD values lower than 1.5 should not be used (Zhao et al. 2015; Baqueta et al. 2020).
In this work, due to the limited number of samples, RMSECV was calculated using the typical leave-one-out cross-validation procedure (Burns and Ciurczak 2007). In brief, a single sample is removed from the dataset, and the analysis is run on the rest of the data. This process is repeated for each sample in the dataset, allowing the model to be tested on different validation datasets.
Results and discussion
Chemical and morphological characteristics of the fibers
As described in the previous section, the selected pulps were characterized in terms of morphology and chemical composition, as revealed in Table 1, where the main characteristics of the fibers can be observed.
In terms of chemical composition, the eucalyptus pulp exhibited the lowest cellulose content, accounting for 74.0%, while the pine pulp revealed the highest value for this constituent, reaching the 87.4%. These values, together with the hemicellulose content, are of the same order of magnitude than some previous results reported in the literature for bleached kraft pulps (Syverud et al. 2011). Similarly, hemp fibers exhibited a high cellulose content (86.5%) and approximately the same hemicellulose content than pine pulp (10.6%). Finally, sisal pulp exhibited a slightly higher cellulose content than eucalyptus and, at the same time, a moderately lower hemicellulose content (Marques et al. 2010). The differences on the chemical composition will directly influence two relevant aspects from the present work. On the one hand, the MicroNIR and NIR spectra, which strongly depends on the chemical composition (Krasznai et al. 2018). On the other, the differences on the hemicellulose content and, more concretely, the xylose content due to the lack of C6 primary hydroxyls to be oxidized, affecting the selectivity of TEMPO-mediated oxidation (Saito et al. 2006; Syverud et al. 2011). The morphological analysis revealed significant differences between the selected fibers, particularly between the two woody pulps. The fiber morphology is not expected to significantly affect the oxidation process catalyzed by TEMPO, but in subsequent stages (e.g. fibrillation) it may have a direct impact, particularly in terms of energy consumption and CNF characteristics (Serra-Parareda et al. 2021; Sanchez-Salvador et al. 2022). However, effects of morphological features on the collected spectra are hypothesized and further research is required in this direction.
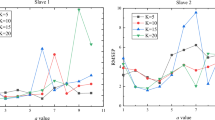
While significant differences on the chemical composition and morphology of the selected pulps were found, the CC after TEMPO-mediated oxidation did not vary significantly from one feedstock to another at certain amounts of oxidizer (Fig. 1). However, at low NaClO additions, the eucalyptus pulp exhibited a higher CC, observing an opposite behavior for the case of sisal, where the addition of high NaClO amounts resulted in remarkably higher CC. Noticeably, both pine and hemp exhibited a similar tendency of the CC with the NaClO addition, being in consonance with the similarities in the chemical composition of the starting pulps.

Evolution of the CC at different NaClO additions for the four selected pulps
Correlations between MicroNIR and NIR spectra
The use of economic, small, and portable equipment presents a series of advantages when compared to classic, large, and expensive benchtop equipment, especially for in-situ applications. Benchtop spectrophotometers present better optical properties in addition to a wider spectral range (Puig-Bertotto et al. 2019; Beć et al. 2021). In this sense, it is important to identify the potential of MicroNIR in this specific application and compare its performance with that obtained from a spectrophotometer widely applied to lignocellulosic materials.
Figure 2A, B present the collected raw spectra, in quintuplicate, for eucalyptus fiber samples at increasing oxidation degrees using the benchtop NIR and the portable MicroNIR devices, respectively. The spectrum collected in the range from 1000 to 2500 nm presented 6 absorption bands located in different sections of the NIR region, with the first two also identified in the spectra taken with the MicroNIR (from 908 to 1676 nm). The first band between 1170 and 1280 nm (B1 at 1220 nm) was found to be of relatively low intensity and could be related to the second overtone of the C − H stretching in the − CH and − CH2 structures (Burns and Ciurczak 2007; Simon et al. 2022). The second band was quite broad and covered the approximate range of 1420–1600 (B2 at 1490 nm), being one of the main bands found in cellulose and hemicellulose due to the manifestation of the first overtone of the − OH stretching (Burns and Ciurczak 2007). In the structures found in lignin, this same overtone also appears in this band for the hydroxide groups in phenols (Krongtaew et al. 2010). It is worth noting that in this region the wavelength at which the overtone appears will depend on the strength of the hydrogen bonds with the − OH groups. The third of the bands was of low intensity involving the range from 1750 to 1850 nm (B3 at 1800 nm) and can be related to the first overtone of C − H stretching including vibrations in CH3 groups in hemicelluloses (Schwanninger et al. 2011). From these wavelengths onwards vibrational combinations also become evident, with combinations between C − H and O − H stretching dominating in this range (Krongtaew et al. 2010). The next band included the range from 1900 to 2000 nm (B4 at 1940 nm) related to the second overtone of the O − H bending in water molecules (Burns and Ciurczak 2007; Simon et al. 2022). The fifth band appeared in the range between 2050 and 2180 nm (B5 at 2120 cm). From a wavelength of 2000 nm onwards, a wide variety of vibrational combinations appeared, with many possibilities for binary couplings, which made their identification difficult (Schwanninger et al. 2011). In this band, some authors pointed out the presence of combinations between O − H and C − O stretching or O − H stretching and C − O deformation (Burns and Ciurczak 2007; Simon et al. 2022) or even O − H stretching and O − H deformation (Schwanninger et al. 2011). The last band involved the range from 2300 to 2380 nm (B6 at 2340 nm) where combinations between stretching vibrations and C − H deformations occur as well as the second bending overtone in CH2 (Burns and Ciurczak 2007).

Spectra of Eucalyptus fiber samples: raw NIR spectra (A), raw MicroNIR spectra (B), SNV-pretreated NIR spectra of Ec and E12 samples (C), and SNV-pretreated MicroNIR spectra of Ec and E12 samples (D)
Figure 2C, D show the NIR and MicroNIR spectra, respectively, after the SNV treatment for the non-oxidized eucalyptus control sample (Ec) and the E12 sample, where the highest CC is found (1.097 mmol/g). It was found that the change in composition of both samples influenced the intensity and shape of the main bands. Regarding NIR spectra, the oxidation of the -OH groups resulted in a decrease in the intensity of the B2 band, related to the vibrations of this functional group, an increase in the intensity of the B4 band, related to greater moisture adsorption, and a decrease of the intensity of bands B5 and B6, related to vibration combinations. Regarding the MicroNIR spectra, the intensity of bands B1 and B2 decreased less extent than in NIR spectra, probably due to the lower optical resolution of the portable equipment. Even so, and as it will be discussed in the following calibration results using PLS, these differences in the MicroNIR spectra in the two bands, B1 and mainly B2, proved to be sufficient to correlate the spectra of the samples with the CC values.
The spectra in Fig. 2A, B were treated with different preprocessing methods and analyzed via PLS for the purpose of modeling the CC variable. Aiming at determining the most appropriate preprocessing method, the RMSECV value of the PLS model obtained from different preprocessing methods was compared. Tables S2 and S3 in the supplementary material present these results for the NIR and MicroNIR spectra, respectively. Among the best preprocessing methods, similar methods were adopted for both types of spectra in order to better compare the results with each other. In this way, the SNV method was used followed by the first SG derivative with 9 points for the NIR spectrum and 5 points for the MicroNIR spectrum followed by data mean centering. These strategies adequately minimized physical effects on both spectra from the benchtop NIR and the MicroNIR. It is worth noting that the data from the benchtop NIR device encompassed greater noise, especially in regions with shorter wavelengths, thus requiring the application of a data smoothing filter. Specifically, a Moving Average filter with 21 points was used (Brereton 2003). Table 2 summarizes the performance of the PLS models for the NIR and MicroNIR spectra based on the aforementioned preprocessing methods. Note that the results of the PLS models when employing the full range of the instrument (1000–2500 nm for NIR and 908–1676 for MicroNIR) yielded high calibration and validation R2 determination coefficients (> 0.97), indicating good sensitivity of the spectra to the carboxyl groups of the samples. Specifically, values of determination coefficient of 0.9713 and 0.9816 were obtained for the NIR and MicroNIR spectra, respectively. The slight decrease in the value obtained for the NIR device can be justified by the fact that the spectral range collected in this case is much wider, increasing the number of bands where changes occur between samples. The greater complexity in the changes in the NIR spectra can be observed by the increase in the number of LVs necessary to describe the variation in the data. In this sense, an optimal number of LVs (minimum RMSECV value) of 5 for NIR spectra and 3 for MicroNIR spectra was obtained. In order to better compare both instruments, PLS models were also applied using the same wavelength range, from 1150 to 1676 nm) for both instruments (entries 2 and 4 of Table 2), a range that includes the first two meaningful bands (B1 and B2). It is observed that the decrease in the variation of the spectral data in this new range for the NIR spectra also decreased the optimal LVs of the PLS model. Note that when applying the same spectral range and similar preprocessing, the RCV2 values are very similar for both instruments (0.9737 and 0.9779) as well as the average error in cross-validation (RMSECV, 5.70 and 5.31%). These models presented similar predictive ability according to an F-test for the RMSECV values, at 95% of confidence. It should be clear that Eq. 2 used to compute RMSECV can also be applied for calculating the RMSEC, but using the calibration dataset instead of the cross-validation one. Relative values of RMSEC and RMSECV are exhibited in Table 2 (%), which means that these errors are relative to the average value of the property (carboxy content) in a given dataset.
Figure 3 shows the loadings of the PLS model for the first latent variable that corresponds to 94% of the data variation. The modulus of this coefficient represents the weight of the absorption bands for the model, indicating the most relevant wavelengths for modeling. According to the observed peaks, special attention may be given to the following wavelengths: 1357, 1391, 1422, 1509 and 1614 nm, the last three belonging to band B2. In this sense, it can be concluded that no significant differences were observed in the results obtained from both instruments, bringing to the light the suitability of both instruments to determine CC in oxidized eucalyptus fiber samples using PLS calibration models.

Comparison of loadings as a function of wavelength for the first LV for the NIR and MicroNIR spectra, both in the range from 1150 to 1676 nm. Loadings for NIR spectra have been multiplied by a factor of 5 to facilitate their comparison
Quantitative PLS models: fibers from different sources
In this section, the application of MicroNIR for determining CC was expanded to different types of oxidized plant fibers. Thus, PLS calibration and cross-validation models were applied to pine, sisal and hemp samples, and the results were compared with those obtained for eucalyptus. Initially, 4 different models were built to correlate the CC with the MicroNIR spectra of each type of fiber, and, in a second stage, an attempt was made to generate an overall calibration model encompassing the MicroNIR data for all samples, that is, a single model for all fibers. The MicroNIR spectral data were preprocessed using the SNV method followed by first derivative SG with 9 points and mean centering of variables, already optimized for eucalyptus fibers. This preprocessing proved to be excellent in relation to obtaining minimum RMSECV values for all types of fibers. For all samples, the full MicroNIR spectral range was used (908–1676 nm).
In general, the quality of the models is characterized with the parameters RMSEC, RMSECV, RC2, RCV2 and RPD (Schwanninger et al. 2011; Hashimoto et al. 2018; Baqueta et al. 2020; Lopez et al. 2023). Table 3 summarizes the results of these parameters, as well as the LV for the individual PLS models (entry 1–4) and the overall model (entry 5). In this work, the RMSEC and RMSECV values are presented in relation to the average of the reference CC values. These parameters are a measure of the model's accuracy relative to the proximity between the reference values and the value estimated by the model and also include spectral measurement errors. However, when determining the RMSEC and RMSECV values, it is assumed that the error in the reference measurement is negligible. For those cases where the reference measurement is imprecise and the error is not negligible, the RMSEC and RMSECV values will be overestimated and will not solely reflect the quality of the model (Lopez et al. 2023).
As it can be seen in Table 3, and for the individual models, the lowest RMSECV accounted for 4.86% for the model determined for eucalyptus fibers, while the highest value was 16.21%, determined for hemp samples. The quality of the model can also be evaluated in terms of the determination coefficient, indicating that the assumption of a linear model is appropriate for values close to 1. The linear behavior of the individual models can be confirmed in Fig. 4 which presents the predicted and reference values for the calibration and validation data. Note that the RC2 values for the eucalyptus, pine and sisal models were greater than 0.97, meaning that the estimates of the calibration models were good. Moreover, as pointed out in the literature, in multivariate analysis it is quite common to obtain determination coefficients lower to 0.95 (Hashimoto et al. 2018; Baqueta et al. 2020). The lowest value of this parameter was obtained for the hemp samples. Despite that, the calibration model can be considered appropriate. The best RCV2 values were obtained for eucalyptus and pine, accounting for 0.9816 and 0.9796, respectively, followed by those obtained for sisal and hemp, being 0.9494 and 0.9096. Although these hemp results seem less accurate, the RPD values obtained for each individual model was greater than 2.4, which is an indication of the quality and predictability of these PLS calibration models (Zhao et al. 2015; Baqueta et al. 2020), including the hemp model. Also, the optimized LV value was 3 for all models, which is relatively low.

Estimated and reference data obtained by PLS calibration (full) and cross-validation (empty) models using MicroNIR spectra (908–1676 nm) for oxidized fiber samples of eucalyptus (A), pine (B), sisal (C), and hemp (D)
Considering that the evaluation of the quality parameters revealed that the individual models for each fiber are capable of correlating CC with the MicroNIR spectra, an overall model using all fiber samples oxidized was proposed. Table 3 (entry 5) and Fig. 5 present the statistical quality parameters of this model as well as the CC values predicted by the calibration and cross-validation model vs the reference CC values. Note that the correlation coefficient values showed lower RC2 and RCV2 values than the individual models and also the RPD value was close to 1.0, indicating low predictability of this overall model. Therefore, from the dataset used in this work, a single model for all fibers is not capable of correctly representing the variation in data and replacing the individual models that consider the specific compositional characteristics of each type of fiber. Considering that the performance of multivariate calibration models depends heavily on the quality of the dataset used for calibration (Lopez et al. 2023), it is expected that specific models for each type of fiber will be more accurate than those obtained from a general and unspecified model. The use of specific models for each fiber can be highly efficient in view of its better prediction of CC values, especially if the spectral data could also be employed to classify/identify the fiber type, before choosing the individual prediction model.

Estimated and reference data obtained by the PLS calibration (full) and cross-validation (empty) models using MicroNIR spectra (908–1676 nm) for all oxidized fiber samples
PCA-LDA classification models
The PLS models showed that the spectral data in the MicroNIR region are dependent on the type of fiber used. In this sense, the spectra may be sensitive to compositional changes other than variations in CC relative to the degree of oxidation of each sample. Each fiber type can then create specific spectra whose pattern can be identified using multivariate analysis methods and classification algorithms. Some differences between the spectra of the different fibers used in this study can be observed by comparing the pretreated spectra presented in Figs. S1 and S2 of the Supplementary Material. Note that spectral differences can be observed in the intensity of the peaks and shape of the main bands as well as in the shoulders between bands. Additionally, the first treatment of the spectra intensified the difference between the fiber spectra. Finally, the eucalyptus fiber sample was the one that showed the greatest spectral differences in relation to the other fibers.
The application of PCA to the MicroNIR spectral dataset of different fibers generated a new set of uncorrelated variables, of smaller dimension and identified as principal components. Such components represent the pattern of the spectra and provide information about the structure of the data. The group of samples that presents the same pattern can be identified from the graph of the scores of the first PCs. PCA was applied to the dataset of non-preprocessed and preprocessed spectra involving all fibers. In total, 111 points were processed, resulting from triplicates of 37 different samples. The first two components were sufficient to explain 99% of the data variation, 94% for PC-1 and 5% for PC-2. When applying preprocessing methods to the spectra, specifically SNV followed by first derivative (SG-5 points) and data mean centering, the number of required PCs to explain 96.5% of the data variance increased to 4. The variance explained by the first two PCs was 65% for PC-1, and 26% for PC-2. An explained variance of 99% was achieved with 12 PCs. The increase in the number of PCs from the use of preprocessed spectra can be expected since the numerical derivation of the spectra increases the variation of the data (Brereton 2003; Diniz et al. 2019). The optimal number of PCs can also be estimated from the minimization of RMSECV, as validation data can be selected for this purpose. The optimal number of calculated PCs was 2 for the raw spectra and 4 for the preprocessed spectra. Figure 6 presents the scores of the PC-1 and PC-2 components of the PCA applied to the raw or non-preprocessed spectra (A), and preprocessed spectra (B). When the raw spectrum was used, PCA was not able to identify distinct patterns for the four fibers, and the different data were mixed in the scores graph. Only the eucalyptus samples appeared to be distinguished from the rest of the samples. From the preprocessing data, a better separation into different classes depending on the type of fiber was observed, so the data from eucalyptus and sisal fibers are grouped and separated from the rest of the fibers. Apparently, using the preprocessed data allowed the generation of some feedstock-based clusters, where the origin of the fiber had a direct influence on the relationship between PC-1 and PC-2. However, pine and hemp fibers appeared in the same cluster, leading to a mixed region in the score graph. These differences could be attributed to the similarities between pine and hemp in terms of chemical composition. Indeed, it is hypothesized that the hemicellulose to cellulose ratio might have a direct influence on this classification. Considering the chemical composition of the selected fibers (Table 1), it is clear that the hemicellulose to cellulose ratio was different for all the samples, except for pine and hemp. Concretely, eucalyptus, pine, sisal, and hemp fibers exhibited a ratio of 0.31, 0.12, 0.23, and 0.12, respectively. In some previous studies, NIR has been reported to be able to provide quantitative data on the cellulose and hemicellulose content of different lignocellulosic substrates, particularly if aided by chemometrics (Li et al. 2015; Jin et al. 2017; Zhang et al. 2017; Wang et al. 2021).

PCA scores in the MicroNIR range (908–1676 nm) for raw spectra (A) and preprocessed spectra (B)
Since the analysis of the first PCs did not present a complete differentiation between all fiber classes, the LDA classification algorithm was applied to the PCA results in order to maximize the differences between samples from different groups and minimize those arising from the fiber spectra included in the same class. This is a supervised algorithm in which a set of parameters are estimated based on the information provided from the sample classification. In other words, LDA uses information from spectra in the reduced dimension of PCs in addition to information about the class to which each spectrum belongs. PCA-LDA was applied to the set of raw spectra of the various fibers and to the preprocessed spectra from the optimized methods. The number of PCs used for each spectral dataset must be optimized depending on the accuracy obtained in the classification algorithm. It is worth noting that classification algorithms are highly sensitive to PCs; depending on the dataset, omitting information included in minority PCs can cause a decrease in classification accuracy (Zheng and Rakovski 2021). Table 4 presents the PCA-LDA accuracy values as a function of PCs for the raw spectra and preprocessed spectra. For the raw spectra, the optimal number of PCs was 6, corresponding to a high accuracy of 98.2%. In this case, the model correctly classified 100% of the Eucalyptus, pine and sisal samples, and only two hemp samples were incorrectly classified as Pine. Nevertheless, for this number of PCs, the variance explained in the data by PCA is 99.98%, therefore requiring variance values very close to 100% to achieve high values of classification accuracy. In addition, for PCs values above 6, the accuracy decreased, probably due to overfitting issues. When using the preprocessed spectra again, the optimal PCs value was 6, which corresponded to an accuracy of 99.1%, even higher than that achieved from the raw spectra. In this case, the 100% of the eucalyptus, pine and sisal samples were correctly classified again, and only one hemp sample was misclassified as pine.
For PCs equal to 6 in the preprocessed spectra, the PCA model yielded an explained variation of 97.5%. For preprocessed spectra, increasing information by the inclusion of further PCs beyond 6 does not result in greater accuracy in sample classification. Figure 7 presents the results of the PCA-LDA algorithm for the preprocessed spectra and PC = 6 where the discriminant values of the different classes referring to the Eucalyptus and Pine classes are shown. Note that this figure, when compared to the graph of scores for the PCA (Fig. 6B), shows that the LDA algorithm promoted a better separation between data from different classes of fibers. Therefore, it is believed that the PCA-LDA model applied to both raw and pretreated spectra can be used as a classification algorithm for different fibers. Note that this algorithm can be used prior to PLS calibration models to predict the fiber type of a given sample and thus choose the specific calibration model for that particular fiber.

LDA discrimination in the MicroNIR range (908–1676 nm) for classes eucalyptus (x-axis) and pine (y-axis) for preprocessed spectra and PC = 6
Conclusions
This work provided a feasible strategy to estimate CC of TEMPO-oxidized cellulosic pulps from easy, quick, non-destructive measurements, that could be performed with a portable device. Remarkably, no significant differences were observed in the results obtained from this device (MicroNIR) and a benchtop instrument.
The optimal spectral preprocessing method was chosen on the basis of the mean squared error of the results predicted by cross-validation. It implied SNV plus first derivative Savitzky–Golay smoothing with 9 points and mean centering of variables. PLS regression models considering the full MicroNIR spectral range (908–1676 nm) attained satisfactory predictability, with RPD > 2.4 in all cases. That said, NIR spectra were sensitive to compositional differences other than those due to oxidation, including the hemicellulose/cellulose ratio. Therefore, an overall regression model did not succeed at predicting CC from NIR data without regard of the feedstock.
Applying PCA-LDA to both non-preprocessed and preprocessed spectra resulted in a primary classification algorithm for different cellulosic pulps. For 6 PCs, the model differentiated four clusters corresponding to eucalyptus, sisal, pine, and hemp, although there was significant overlapping between the latter two, when just PCA analysis was applied. The LDA algorithm enabled us to maximize the separation between data from different classes. Overall, the strategy involves this PCA-LDA classification to assign a certain fiber type to a given sample, giving way to the selection of the corresponding PLS model.
Data availability
Data can be made available upon request to the corresponding author.
References
Abitbol T, Rivkin A, Cao Y et al (2016) Nanocellulose, a tiny fiber with huge applications. Curr Opin Biotechnol 39:76–88
Badaró AT, Hebling e Tavares JP, Blasco J et al (2022) Near infrared techniques applied to analysis of wheat-based products: recent advances and future trends. Food Control. https://doi.org/10.1016/j.foodcont.2022.109115
Bakeev KA (2010) Process analytical technology: spectroscopic tools and implementation strategies for the chemical and pharmaceutical industries. John Wiley & Sons, Hoboken
Ballabio D, Consonni V (2013) Classification tools in chemistry. Part 1: linear models. PLS-DA Analytical Methods 5:3790–3798. https://doi.org/10.1039/c3ay40582f
Baqueta MR, Coqueiro A, Março PH, Valderrama P (2020) Quality control parameters in the roasted coffee industry: a proposal by using microNIR spectroscopy and multivariate calibration. Food Anal Methods 13:50–60. https://doi.org/10.1007/s12161-019-01503-w
Beć KB, Grabska J, Huck CW (2021) Principles and applications of miniaturized near-infrared (NIR) spectrometers. Chem A Eur J 27:1514–1532
Blanco M, Villarroya I (2002) NIR spectroscopy: a rapid-response analytical tool. TrAC Trends Anal Chem 21:240–250. https://doi.org/10.1016/S0165-9936(02)00404-1
Boufi S, González I, Delgado-Aguilar M et al (2016) Nanofibrillated cellulose as an additive in papermaking process: a review. Carbohydr Polym 154:151–166. https://doi.org/10.1016/j.carbpol.2016.07.117
Brereton RG (2003) Chemometrics: data analysis for the laboratory and chemical plant. John Wiley & Sons, Hoboken
Burns DA, Ciurczak EW (2007) Handbook of near-infrared analysis. CRC Press, Florida
Cazón P, Cazón D, Vázquez M, Guerra-Rodriguez E (2022) Rapid authentication and composition determination of cellulose films by UV-VIS-NIR spectroscopy. Food Packag Shelf Life. https://doi.org/10.1016/j.fpsl.2021.100791
Chavez Lozano MV, Catelli E, Sciutto G et al (2023) A non-invasive diagnostic tool for cellulose acetate films using a portable miniaturized near infrared spectrometer. Talanta 255:124223. https://doi.org/10.1016/j.talanta.2022.124223
Dai L, Dai H, Yuan Y et al (2011) Effect of TEMPO oxidation system on kinetic constants of cotton fibers. BioResources 6:2619–2631
de Almeida VE, de Sousa Fernandes DD, Diniz PHGD et al (2021) Scores selection via fisher’s discriminant power in PCA-LDA to improve the classification of food data. Food Chem. https://doi.org/10.1016/j.foodchem.2021.130296
Diniz CP, Grattapaglia D, de Alencar Figueiredo LF (2019) Comparative performance of bench and portable near infrared spectrometers for measuring wood samples of two eucalyptus species (E. pellita and E. benthamii). 18th international conference near infrared spectroscopy. IM Publications Open, Chichester, pp 31–38
Dos Santos CAT, Lopo M, Páscoa RNMJ, Lopes JA (2013a) A review on the applications of portable near-infrared spectrometers in the agro-food industry. Appl Spectrosc 67:1215–1233
Eichhorn SJ, Dufresne A, Aranguren M et al (2010) Review: current international research into cellulose nanofibres and nanocomposites. J Mater Sci 45:1–33
Engel J, Gerretzen J, Szymańska E et al (2013) Breaking with trends in pre-processing? TrAC Trends Anal Chem 50:96–106. https://doi.org/10.1016/j.trac.2013.04.015
Esteki M, Shahsavari Z, Simal-Gandara J (2018) Use of spectroscopic methods in combination with linear discriminant analysis for authentication of food products. Food Control 91:100–112. https://doi.org/10.1016/j.foodcont.2018.03.031
Geladi P, Kowalski BR (1986) Partial least-squares regression: a tutorial. Anal Chim Acta 185:1–17. https://doi.org/10.1016/0003-2670(86)80028-9
Hashimoto JC, Lima JC, Celeghini RMS et al (2018) Quality control of commercial cocoa beans (Theobroma cacao L.) by near-infrared spectroscopy. Food Anal Methods 11:1510–1517. https://doi.org/10.1007/s12161-017-1137-2
Henniges U, Schwanninger M, Potthast A (2009) Non-destructive determination of cellulose functional groups and molecular weight in pulp hand sheets and historic papers by NIR-PLS-R. Carbohydr Polym 76:374–380. https://doi.org/10.1016/j.carbpol.2008.10.028
Hicks SA, Strümke I, Thambawita V et al (2022) On evaluation metrics for medical applications of artificial intelligence. Sci Rep 12:5979
Im W, Rajabi Abhari A, Youn HJ, Lee HL (2018) Morphological characteristics of carboxymethylated cellulose nanofibrils: the effect of carboxyl content. Cellulose 25:5781–5789. https://doi.org/10.1007/s10570-018-1993-y
Isogai A, Zhou Y (2019) Diverse nanocelluloses prepared from TEMPO-oxidized wood cellulose fibers: nanonetworks, nanofibers, and nanocrystals. Curr Opin Solid State Mater Sci 23:101–106. https://doi.org/10.1016/j.cossms.2019.01.001
Jiao Y, Li Z, Chen X, Fei S (2020) Preprocessing methods for near-infrared spectrum calibration. J Chemom. https://doi.org/10.1002/cem.3306
Jin X, Chen X, Shi C et al (2017) Determination of hemicellulose, cellulose and lignin content using visible and near infrared spectroscopy in Miscanthus sinensis. Bioresour Technol 241:603–609. https://doi.org/10.1016/j.biortech.2017.05.047
Krasznai DJ, Champagne Hartley R, Roy HM et al (2018) Compositional analysis of lignocellulosic biomass: conventional methodologies and future outlook. Crit Rev Biotechnol 38:199–217
Krongtaew C, Messner K, Ters T, Fackler K (2010) Characterization of key parameters for biotechnological lignocellulose conversion assessed by FT-NIR spectroscopy. Part I: qualitative analysis of pretreated straw. BioResources 5:2063–2080
Li X, Sun C, Zhou B, He Y (2015) Determination of hemicellulose, cellulose and lignin in Moso bamboo by near infrared spectroscopy. Sci Rep 5:17210. https://doi.org/10.1038/srep17210
Li T, Chen C, Brozena AH et al (2021) Developing fibrillated cellulose as a sustainable technological material. Nature 590:47–56. https://doi.org/10.1038/s41586-020-03167-7
Lin C, Zeng T, Wang Q et al (2018) Effects of the conditions of the TEMPO/NaBr/NaClO system on carboxyl groups, degree of polymerization, and yield of the oxidized cellulose. BioResources 13:5965–5975
Lohumi S, Lee S, Lee H, Cho B-K (2015) A review of vibrational spectroscopic techniques for the detection of food authenticity and adulteration. Trends Food Sci Technol 46:85–98. https://doi.org/10.1016/j.tifs.2015.08.003
Lopez E, Etxebarria-Elezgarai J, Amigo JM, Seifert A (2023) The importance of choosing a proper validation strategy in predictive models. A tutorial with real examples. Anal Chim Acta. https://doi.org/10.1016/j.aca.2023.341532
Maćkiewicz A, Ratajczak W (1993) Principal components analysis (PCA). Comput Geosci 19:303–342. https://doi.org/10.1016/0098-3004(93)90090-R
Marques G, Rencoret J, Gutiérrez A, del Río JC (2010) Evaluation of the chemical composition of different non-woody plant fibers used for pulp and paper manufacturing. Open Agric J 4:93–101. https://doi.org/10.2174/1874331501004010093
Mayr G, Hintenaus P, Zeppetzauera F, Röderc T (2015) A fast and accurate near infrared spectroscopy method for the determination of cellulose content of alkali cellulose applicable for process control. J near Infrared Spectrosc 23:369–379. https://doi.org/10.1255/jnirs.1185
Mayr S, Beć KB, Grabska J et al (2021) Near-infrared spectroscopy in quality control of Piper nigrum: a comparison of performance of benchtop and handheld spectrometers. Talanta. https://doi.org/10.1016/j.talanta.2020.121809
Mazega A, Santos AF, Aguado R et al (2023) Kinetic study and real-time monitoring strategy for TEMPO-mediated oxidation of bleached eucalyptus fibers. Cellulose 30:1421–1436. https://doi.org/10.1007/s10570-022-05013-7
Moon RJ, Martini A, Nairn J et al (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Pasquini C (2018) Near infrared spectroscopy: a mature analytical technique with new perspectives–a review. Anal Chim Acta 1026:8–36
Pu Y-Y, O’Donnell C, Tobin JT, O’Shea N (2020) Review of near-infrared spectroscopy as a process analytical technology for real-time product monitoring in dairy processing. Int Dairy J. https://doi.org/10.1016/j.idairyj.2019.104623
Puig-Bertotto J, Coello J, Maspoch S (2019) Evaluation of a handheld near-infrared spectrophotometer for quantitative determination of two APIs in a solid pharmaceutical preparation. Anal Methods 11:327–335. https://doi.org/10.1039/c8ay01970c
Ribeiro JPO, Medeiros ADD, Caliari IP et al (2021) FT-NIR and linear discriminant analysis to classify chickpea seeds produced with harvest aid chemicals. Food Chem. https://doi.org/10.1016/j.foodchem.2020.128324
Rinnan Å, van den Berg F, Engelsen SB (2009) Review of the most common pre-processing techniques for near-infrared spectra. TrAC Trends Anal Chem 28:1201–1222. https://doi.org/10.1016/j.trac.2009.07.007
Robert G, Gosselin R (2022) Evaluating the impact of NIR pre-processing methods via multiblock partial least-squares. Anal Chim Acta. https://doi.org/10.1016/j.aca.2021.339255
Saito T, Isogai A (2004) TEMPO-mediated oxidation of native cellulose. The effect of oxidation conditions on chemical and crystal structures of the water-insoluble fractions. Biomacromol 5:1983–1989. https://doi.org/10.1021/bm0497769
Saito T, Nishiyama Y, Putaux J-L et al (2006) Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromol 7:1687–1691
Sanchez-Salvador JL, Campano C, Balea A et al (2022) Critical comparison of the properties of cellulose nanofibers produced from softwood and hardwood through enzymatic, chemical and mechanical processes. Int J Biol Macromol 205:220–230. https://doi.org/10.1016/j.ijbiomac.2022.02.074
Santos AF, Silva FM, Lenzi MK, Pinto JC (2013) Infrared (MIR, NIR), Raman and other spectroscopic methods. Monitoring Polymerization reactions: from fundamentals to applications. John Wiley & Sons, Hoboken, pp 107–134
Schwanninger M, Rodrigues JC, Fackler K (2011) A review of band assignments in near infrared spectra of wood and wood components. J near Infrared Spectrosc 19:287–308. https://doi.org/10.1255/jnirs.955
Serra-Parareda F, Tarrés Q, Sanchez-Salvador JL et al (2021) Tuning morphology and structure of non-woody nanocellulose: ranging between nanofibers and nanocrystals. Ind Crops Prod. https://doi.org/10.1016/j.indcrop.2021.113877
Signori-Iamin G, Santos AF, Corazza ML et al (2022) Prediction of cellulose micro/nanofiber aspect ratio and yield of nanofibrillation using machine learning techniques. Cellulose 29:9143–9162
Simon J, Tsetsgee O, Iqbal NA et al (2022) A fast method to measure the degree of oxidation of dialdehyde celluloses using multivariate calibration and infrared spectroscopy. Carbohydr Polym. https://doi.org/10.1016/j.carbpol.2021.118887
Standards AB of A (2000) Standard practices for infrared multivariate quantitative analysis-E1655–00
Sun B, Gu C, Ma J, Liang B (2005) Kinetic study on TEMPO-mediated selective oxidation of regenerated cellulose. Cellulose 12:59–66. https://doi.org/10.1007/s10570-004-0343-4
Syverud K, Chinga-Carrasco G, Toledo J, Toledo PG (2011) A comparative study of Eucalyptus and Pinus radiata pulp fibres as raw materials for production of cellulose nanofibrils. Carbohydr Polym 84:1033–1038. https://doi.org/10.1016/j.carbpol.2010.12.066
Tarrés Q, Ehman NVNVNV, Vallejos MEME et al (2017) Lignocellulosic nanofibers from triticale straw: the influence of hemicelluloses and lignin in their production and properties. Carbohydr Polym 163:20–27. https://doi.org/10.1016/j.carbpol.2017.01.017
Wang N, Li L, Liu J et al (2021) Rapid detection of cellulose and hemicellulose contents of corn stover based on near-infrared spectroscopy combined with chemometrics. Appl Opt 60:4282–4290
Williams PC, Sobering DC (1993) Comparison of commercial near infrared transmittance and reflectance instruments for analysis of whole grains and seeds. J near Infrared Spectrosc 1:25–32
Wold S, Antti H, Lindgren F, Öhman J (1998) Orthogonal signal correction of near-infrared spectra. Chemom Intell Lab Syst 44:175–185. https://doi.org/10.1016/S0169-7439(98)00109-9
Wold S, Sjöström M, Eriksson L (2001) PLS-regression: a basic tool of chemometrics. Chemom Intell Lab Syst 58:109–130. https://doi.org/10.1016/S0169-7439(01)00155-1
Wu W, Mallet Y, Walczak B et al (1996) Comparison of regularized discriminant analysis, linear discriminant analysis and quadratic discriminant analysis, applied to NIR data. Anal Chim Acta 329:257–265. https://doi.org/10.1016/0003-2670(96)00142-0
Zhang K, Xu Y, Johnson L et al (2017) Development of near-infrared spectroscopy models for quantitative determination of cellulose and hemicellulose contents of big bluestem. Renew Energy 109:101–109. https://doi.org/10.1016/j.renene.2017.03.020
Zhao N, Wu Z-S, Zhang Q et al (2015) Optimization of parameter selection for partial least squares model development. Sci Rep. https://doi.org/10.1038/srep11647
Zheng J, Rakovski C (2021) On the application of principal component analysis to classification problems. Data Sci J. https://doi.org/10.5334/dsj-2021-026
Zhou C, Han G, Gao S et al (2019) Rapid determination of cellulose content in pulp using near infrared modeling technique. Bioresources 13:6122–6132. https://doi.org/10.15376/biores.13.3.6122-6132
Acknowledgements
Authors wish to acknowledge the financial support of the agencies listed in the Funding section. Marc Delgado-Aguilar and Quim Tarrés are Serra Húnter Fellows.
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This research received funding from the Spanish Ministry of Science and Innovation (ArtInNano, CNS-2022–135789), the University of Girona and Banco Santander (IFUdG2020 and IFUdG2022), and the CNPq (312952/2021–0).
Author information
Authors and Affiliations
Contributions
AM: Investigation, Data Curation, Writing—Original Draft; GSI: Investigation, Data Curation; MF: Software, Data Curation, Formal analysis, Writing—Original Draft; RJA: Conceptualization, Writing—Review & Editing; QT: Writing—Review & Editing; AFS: Conceptualization, Writing – Review & Editing; MDA: Conceptualization, Supervision, Writing—Review & Editing, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence this work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mazega, A., Fortuny, M., Signori-Iamin, G. et al. Near-infrared spectroscopy and multivariate analysis as real-time monitoring strategy of TEMPO-mediated oxidation of cellulose fibers from different feedstocks. Cellulose 31, 3465–3482 (2024). https://doi.org/10.1007/s10570-024-05824-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-024-05824-w