
Abstract
This contribution focuses on understanding of paper wet-strength properties, by taking a closer look at the spatial distribution of wet-strengthening polymers inside the cellulosic fiber network deposited under different treatment conditions using confocal laser scanning microscopy as in situ imaging tool. We compare the behavior of paper samples treated with a photochemically cross-linkable copolymer using an impregnation process employing three different solvents, namely water, 2-propanol (IPA) and 1-butanol (BuOH), respectively. As these solvents swell paper fibers to quite different extents, the deposition of the polymer, on, in or in-between the cellulosic fibers varies quite strongly, as is shown by in-depth analysis using confocal laser scanning microscopy. The difference in accessibility of distinct surface sites exclusively on or also in and between the fibers controls the macroscopic tensile strength under both dry and wet conditions.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The tensile strength of paper sheets in their dry or wet state is one of the most important relevant properties for a large number of paper grades progressing from packaging, to hygienic paper, and specialty paper, such as filter or bank notes, respectively. In addition to such classical applications of paper, in recent years, fueled by the ongoing effort for environmentally friendly products, the use of paper has expanded into a variety of new sectors, such as paper for construction materials (Auslender et al. 2017) and the use in diagnostics, microfluidic and lab-on-paper devices (Credou and Berthelot 2014).
Due to its wet-layed production, the strength of the paper sheets is a complex interplay of the properties of individual lignocellulosic paper fibers (length, kink, width, degree of polymerization, fibrillation, defects) and the bonds formed between them, constituting a non-woven fiber network. Tejado and van de Ven (2010) conducted a detailed analysis of the processes in paper formation and the associated forces. They argue, that the fibers are held together via entanglement between smaller cellulose fibrils on the macroscopic fiber surfaces and the main reason for the wet-web strength of undried paper are friction forces. Belle et al. (2015) analyzed freeze-dried wet webs of different solids content with field emission-scanning electron microscopy (FE-SEM) and were able to show such entanglement between fibrils of adjacent fibers and the inter-fiber bonds produced thereby. Another mechanism leading to inter-fiber bond strength can be described by the diffusion of macromolecular cellulose chains into adjacent fibers in close contact (Lindström et al. 2005; Hubbe 2006). While inter-fiber bonding is the subject of many research projects, the exact mechanism of it is still not fully understood. However, it is widely accepted that hydrogen bonding, van der Waals and Coulomb forces, mechanical interlocking of fibrils and diffusion of cellulose chains on the surface of fibers, all play a crucial role in this complex interaction, leading to the intrinsic macroscopic tensile strength of a paper sheet (Hirn and Schennach 2015).
A first attempt describing the tensile strength of paper in a quantitative manner was done by Page (1969). Considering that the strength of a single cellulose fiber is significantly higher compared to the strength of a respective paper sheet, it becomes apparent that in order to increase the strength of a sheet, the inter-fiber bonds must be strengthened. Strengthening of the tensile properties in the dry state is traditionally done with so called dry strength agents. Additionally, pulp refining (Lindström et al. 2016) and wet pressing (He et al. 2003) of sheets can be used to adjust and control sheet tensile properties. A variety of macromolecules have been used in order to increase the dry tensile strength of paper. Examples are bio-based polymeric additives, such as starch, chitosan, or carboxy methyl cellulose (CMC), as well as synthetic polymeric systems, such as polyacrylamide (PAM), polyvinylamine, polyethyleneimine or polyelectrolyte multilayers (Lindström et al. 2005). There are at least three proposed mechanisms that explain the dry strengthening effect of additives, including increased inter-fiber bond strength by the reinforcement of existing and/or the contribution via additional bonds, decreased stress concentrations in the sheet while drying and by enhancing the consolidation of the sheet (Lindström et al. 2016).
Upon rewetting, cellulose fiber networks loose nearly their entire strength. This can be explained by the hydrophilic nature of cellulose fibers and their swelling behavior in contact with water (Lindner 2018). Water molecules penetrate the cellulose fibers and fiber walls and consecutively weaken the attractive hydrogen bonds between cellulose chains and fibrils. In addition to that, the swollen fibers are highly malleable, and the aforementioned frictional forces holding fibers together are weakened as well. It is widely discussed, that in order to increase the wet strength, the introduction of covalent bonds between adjacent fibers is necessary, however, other treatments such as wet pressing or the use of microfibrillated cellulose (MFC) have also been shown to increase the wet strength of paper. In the industrial application so called wet strength agents are used to increase the tensile strength of paper in wet conditions. The two main working models of those additives are the protection and the reinforcement mechanism, respectively (Lindström et al. 2005). With the protection mechanism, a firm non-polar polymeric sleeve wraps around fiber crossing points thereby limiting the access of water molecules to the area of fiber contact. Typically, such polymers are cross-linked hydrophobic resins such as for example phenol–formaldehyde resins, where the used polymeric precursors typically react with one another but do not significantly bind to the cellulosic fiber (homo-cross-linking additives). The other category are additives that are (also) able to form covalent bonds with the cellulose chains, so called hetero-cross-linking, like polyamidoamine epichlorohydrin (PAE). An important difference in comparison to the aforementioned formaldehyde resins is that cross-linking occurs between the polymeric additive as well as between the additive and the fiber, respectively, thereby acting towards wet-strengthening of the sheet through a reinforcement mechanism. Finally, such reinforcement additives can be of polar nature, and may even swell in water, which is necessary for applications, where paper-water contact is crucial, such as with hygienic paper grades.
Note, although there are clear evidences for these postulated mechanisms, there are still a number of open and fundamental questions that have not yet been answered to validate these models or to even pre-calculate which polymer-intrinsic and/or paper-related parameters determine the wet-strength action of (pre-)polymeric additives. For example, with respect to the latter, knowledge on the exact spatial distribution of the wet-strength agent in the non-woven sheet becomes crucial. Mangiante et al. (2018) prepared paper samples with alkyne-functionalized cellulose fibers, cross-linking them with difunctional PEG chains, leading to significantly improved wet tensile strength. They furthermore analyzed the spatial distribution via Raman confocal microscopy and found, that the fiber core had rich amounts of the alkyne. However, these observations weren’t further discussed with regard to the effect on the macroscopic wet strength.
In previous work of our own groups, we used for the first time polymers that can cross-link within paper sheets under the influence of light (Jocher et al. 2015; Bump et al. 2015). We were able to show that this treatment increases the macroscopic tensile strength, and we analyzed the binding of the polymers to the fibers by Raman spectroscopy and imaging via confocal laser scanning microscopy, respectively. However, a systematic analysis of the spatial distribution of strengthening agents in the cellulosic fiber network and their impact on dry and wet strengths is still missing. Finally, it is not yet understood how fiber swelling may affect the interaction of the fiber with the polymeric additive and how the latter leads to different deposition scenarios of the polymer on/in the fiber. The latter may also influence the macroscopic wet-strength.
As we will show in this contribution, taking a closer look at the spatial distribution inside the fiber network and the fibers themselves can improve our understanding of the mechanisms involved in dry and wet strengthening of paper. In particular, as will be shown, fiber swelling leads to very different deposition mechanisms of a polymeric wet-strength agent and therefore affects largely the macroscopic tensile strength in wet conditions. We compare the treatment of paper samples with a light-sensitive copolymer (Toomey et al. 2004; Janko et al. 2015; Jocher et al. 2015; Bump et al. 2015) using an impregnation process with three different solvents. The solvents were chosen based on literature-data of cellulose fiber swelling (El Seoud et al. 2008; Karppinen et al. 2004) because we were particularly interested in the use of solvents, that don’t lead to significant fiber swelling, in comparison to water. The latter is known for its capability to significantly swell paper fibers thereby altering the morphology. The paper topology includes pores or grooves in which surrounding polymer molecules can be taken up during impregnation. On this basis water, 2-propanol (IPA) and 1-butanol (BuOH) were used to apply the copolymer to lab-made paper samples. By using confocal laser scanning microscopy in combination with tensile measurements, we study how this difference in swelling leads to different spatial deposition of the copolymer inside the paper samples and, ultimately, controls dry and wet paper sheet tensile strength.
Experimental section
Materials
All chemicals and solvents were purchased from Merck, Alfa Aesar, Alberdingk Boley, Fisher Scientific, Fluka, Covestro and TIB Chemicals, respectively, and were used as received, unless otherwise specified. For impregnation and extraction pure distilled water was used, which is denoted as H2O throughout this work.
Pulp and handsheet preparation
All paper samples, modified with the copolymer as described below, where lab-engineered using bleached eucalyptus-sulfate pulp [median fiber length (length-weighted): 0.76 mm; curl: 15.9%; fibrillation degree: 5.1%; fines content: 9.1%]. The paper samples having a grammage of 80 ± 1.6 g m−2 where prepared using a Rapid-Köthen sheet former according to DIN 54358 and ISO 5269/2. In order to prevent any influence on the physical properties, no additives or filler materials other than the copolymer were used. Prior to impregnation, the paper was conditioned for at least 24 h under standard conditions (23 °C, 50% r.h.).
Polymer synthesis
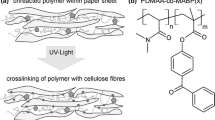
The photo-cross-linking monomer 4-benzoylphenyl methacrylate (MABP) was synthesized according to the procedure developed in one of our groups and described by Toomey et al. (2004). First, the fluorescent monomer rhodamine B methacrylamide (RhBMa) was prepared in two steps from the fluorescing rhodamine B base according to previously published literature (Schäfer et al. 2013). The copolymer poly(dimethylacrylamide-co-4-benzoylphenyl-2-methacrylate-co-rhodamine B-methacrylic acid (P(DMAA-co-MABP-co-RhBMA)) carrying the fluorescent monomer, the photo-cross-linking monomer and the matrix monomer, respectively, was synthesized using free radical copolymerization according to the procedure described by Janko et al. (2015). The structure is schematically illustrated in Fig. 1. The chemical analysis of the molar composition of the copolymer was done using 1H-NMR (see supporting information), which showed that the copolymer consists of about 96.5 mol% of the matrix DMAA, and 3.5 mol% of MABP and RhBMA, respectively. Using size exclusion chromatography (SEC) with a GRAM VS/GRAM linear 10 HS/100μL (DMF 0.002 LiCl) column and a narrow dispersed poly(methyl methacrylate) standard, the molar mass was determined to be around Mn = 27 000 g mol−1 (Đ ~ 5.7). The copolymer was stored inside a plastic container in a refrigerator at 6 °C until further use.

Molecular structure of the synthesized copolymer poly(dimethylacrylamide-co-4-benzoylphenyl-2-methacrylate-co-rhodamine B-methacrylic acid (P(DMAA-co-MABP-co-RhBMA)) carrying the fluorescent monomer (m), the photo-cross-linking monomer (o) and the matrix monomer (n), respectively. Molar composition as determined by 1H-NMR (see supporting information): m + o = 3.5%; n = 96.5%
Copolymer treatment of the paper samples
Before impregnation, the lab-engineered paper was cut into samples with the dimensions of 15 × 120 mm. Those dimensions were chosen because tensile testing was carried out according to DIN ISO 1924-2, where a gauge length of 100 mm is specified. To determine the amount of cross-linked copolymer in the samples, a gravimetric approach was chosen. Before each weighing/measurement on a scale under climate controlled conditions (23 °C and 50% r.h.), the samples were conditioned for at least 24 h. Three measurements were carried-out, once before impregnation (m before), after drying the impregnated samples (m after) and finally after the extraction and consecutive drying (m extraction), in order to calculate the amount (wt%) of copolymer in relation to the paper weight as well as the amount of extractable (unbound) copolymer. For the three solvents the amount of copolymer in the paper samples was 11.4 (H2O), 8.0 (IPA) and 7.3 wt% (1-BuOH). For the reference experiments the paper samples were impregnated in H2O without any copolymer, but otherwise the procedure was carried out in the same way as described before. Here, the measurement after swelling and drying (m delta) was used to calculate the wt% change induced by the treatment without copolymer. Complete details of these calculations and results can be found in the supplementary information. The copolymer was dissolved in H2O, in 2-propanol, or in 1-butanol, respectively, at a concentration of 25 mg mL−1 and stored in flasks in a refrigerator at 6 °C under exclusion of light until use. The polymer-modification of the paper samples is illustrated in Fig. 2 and was conducted as follows.

Schematic illustration of the impregnation process of paper samples with the copolymer dissolved in three different solvents H2O, 2-propanol (IPA) and 1-butanol (BuOH)
In brief, (1) a PTFE (Teflon)-dish was filled with the given copolymer solution (H2O, IPA or BuOH) and the pre-weighed paper samples were submerged one at a time. A petri dish on top (of the Teflon-dish) was used to prevent large amount of solvent evaporating and thus changing the concentration over time. After at least 60 s, the paper samples were pulled out and laid flat on a 3D-printed frame with Teflon yarn (see supporting information for details) for drying overnight under climate-controlled conditions. (2) After equilibration and consecutive weighing of the impregnated paper samples, ambient dried samples were pressed with a defined weight (~ 15 kg) for about four hours to ensure flat samples and a uniform UV-excitation. By illumination of the paper samples with UV-light, the benzophenone groups were excited and thus the cross-linking of the copolymer was induced. The UV source used was a Newport 1000 W Oriel Flood Exposure Source with a wavelength of λ = 365 nm. Five samples were illuminated at a time, each with an energy density of E = 16 J cm−2, at which approx. 94% of the present benzophenone groups reacted, as reported previously (Toomey et al. 2004; Jocher et al. 2015). (3) After the UV-excitation, extraction with cold H2O was carried out, in order to remove any unbound copolymer from the paper samples. For this, the paper samples were extracted in an extraction apparatus for four cycles, which is sufficient for complete removal of unbound copolymer, after which the paper samples were dried on the Teflon-sieve under climate-controlled conditions. After equilibration the dried paper samples were weighed one more time, before they were pressed again to guarantee flat samples for the tensile measurements. Note, for the reference experiments, paper samples were impregnated in H2O, IPA or BuOH for at least 60 s and dried afterwards, all other steps were carried out as described above.
Preparation of thin slices for confocal microscopy
To improve the visibility of cellulose fibers under fluorescence microscopic investigation, paper sheets were stained in a 100 μM aqueous Calcofluor White (CW) solution for ten minutes, washed in H2O for another 10 min to remove unbound CW and dried overnight. A commercially available polyurethane system consisting of an aliphatic polyisocyanate (Desmodure 3200), a polyol (Albodur 956), and a catalyst (TIB Kat 318) in a ratio of 1:1:5 × 10–4 was used to embed the samples. After a few vacuum cycles at room temperature to improve the penetration of the embedding medium inside the paper samples, they were cured overnight and consecutively cut into 120 μm slices using a microtome.
This technique has been used to analyze the spatial modification of paper and the functionalization of fiber surfaces, where details can be found in recent publications (Bump et al. 2015; Janko et al. 2015; Nau et al. 2019).
Despite an increased resolution and contrast that confocal detection provides over standard widefield microscopy, light scattering limits axial resolution, especially with highly scattering samples, such as paper. The latter to some extent can be circumvented, by imaging thin cross-sections of paper to assess the distribution of chemical modifications across the paper width. As lateral resolution of microscopy applies, which, for confocal detection, exceeds the axial resolution by a factor of around 3, this method becomes highly suitable for distinguishing, e.g. whether the copolymer has adsorbed onto or within a fiber wall, this resolution gain is required. Additionally, image stacks of paper samples were made with traditional confocal detection and combined into 3D-images in order to analyze the spatial distribution of the copolymer between the fibers and inside the whole fiber network. It should be noted, that this approach reintroduces some of the challenges, which were avoided by using thin cross-sections.
For confocal investigation, the prepared slices or the paper samples were mounted between two round coverslips with Type F Immersion liquid from Leica or H2O, respectively, as immersion medium. Using a Leica TCS SP8 (Leica Microsystems, Mannheim, Germany) CW fluorescence was excited at 405 nm and fluorescence emission was detected between 420 and 480 nm. Rhodamine B labelled copolymer, in turn, was excited at 552 nm and detected between 570 and 650 nm. Image stacks of ~ 50 slices with widths of 0.6 to 1 μm were acquired and processed within the Leica Application Suite X to render 3D-stacks of paper samples for further analysis.
It should be noted, that during image acquisition the settings for laser intensity, detector sensitivity (gain and offset) and later on during image processing, the brightness and contrast of the images were adjusted, in order to achieve the highest detail and images where the spatial distribution of the copolymers can best be compared. Thus, no quantitative information about copolymer amount in the different samples can be derived from the images provided/shown in this work. However, quantitative analysis through calibration via a concentration series can be done as was shown previously by our group (Bump et al. 2015), however it was not in focus in this work here.
Tensile strength analysis
Dry tensile strength of the paper samples was determined as an average of at least five samples according to DIN ISO 1924-2 with a Zwick Z1.0 with a 1 kN load cell using the software testXpert II V3.71 (ZwickRoell GmbH & Co. Kg) in a controlled environment with 23 °C and 50% r.h. Wet tensile strength measurements were conducted in analogy, after the paper samples had been submerged in H2O for at least five minutes. To remove any excess water, the paper samples were sandwiched between tissue paper and slightly pressed. A 20 N load cell was used for those measurements, to improve the precision of the tensile tests in the wet state. Along the measured dry and wet tensile indices, the relative wet strength is a relevant quantity, which is used in the paper formation industry and can be calculated according to this equation:
The definition of the dry/wet tensile index is shown in the following equation:
Here Fmax is the maximum force at break in N, b is the width of the sample in mm and the grammage of the paper is given in g m−2.
Setup for fiber failure analysis via Zwick-videography
A commercially available full frame mirrorless camera from Panasonic (DC S1) with a macro lens from Canon (MP-E 65 mm f/2.8 1–5 × Macro Photo) and an adapter from Novoflex (SL/EOS) was used. The camera was mounted on a manual x/y/z-stage on a table that was decoupled from vibrations of the tensile testing equipment. The aperture was set to 5.6, the shutter speed was 1/30 s and the ISO was set to 800. The videos were recorded with a resolution of 3840 × 2160 px at a frame rate of 29.97 frames/second. A UV-lamp (365 nm) was used from the backside of the paper samples to excite the fluorophore (cationic dye—Pergasol Yellow F6-GZ) of the labelled fibers (0.1 wt% of all fibers).
Results and discussion
As water is by far the predominant processing solvent used in the paper formation industry, we used this solvent in conjunction with copolymer treatments of paper in previous studies (Jocher et al. 2015). However, while it was possible to prepare paper samples with high wet tensile indices with the copolymer dissolved in H2O, we also became interested in the use of other solvents in combination with copolymer immobilization. We were particularly interested in the use of solvents, that don’t lead to significant fiber swelling, in comparison with water, which is known to swell paper fibers. As model paper sheets eucalyptus-sulfate paper samples with a grammage of 80 g m−2 were prepared and impregnated with the respective copolymer solution in the chosen solvent. In order to prevent any mechanical agitation of the cellulose fibers during treatment with the copolymer solution, an impregnation approach was used in this work. This also enables the analysis of the diffusion process of the copolymer in the different solvents throughout the fiber network and the fibers themselves, without any influence from external forces. A sieve with thin Teflon-yarn was designed for non-contact drying, to prevent further influence on the spatial distribution by capillary or gravitational forces. The copolymer modified paper sheets were illuminated by UV-light in order to cross-link the copolymer inside the sheet. Non-bound macromolecules were removed by solvent extraction. The amount of paper sheet attached, cross-linked polymer was determined by gravimetric analysis and was typically on the order of 10 wt% in relation to untreated paper samples (see supporting information for details). After cross-linking and extraction, respectively, we first investigated possible failure-mechanisms by time-resolved tensile microscopy. Secondly, the macroscopic tensile properties in the dry and the wet state were determined via tensile strength analysis and the spatial distribution of the macromolecules inside the fiber network was finally analyzed in detail using confocal laser scanning microscopy (CLSM).
Tensile properties
The tensile properties of paper sheets are very sensitive to a wide variety of environmental factors, e.g. humidity, temperature, UV-exposure, swelling and drying. Since the copolymer-application includes multiple swelling and drying cycles in addition to significant UV-exposure, the influence of this treatment without the copolymer on the tensile and wet tensile strength was analyzed first as a reference. It should be noted that paper without any wet strengthening is intrinsically very weak in the presence of water and it is therefore challenging to analyze such wet paper sheets in a conventional tensile testing device, as such measurements generally lead to significant errors in the range of 10–13% (SD) for eucalyptus-sulfate paper with a grammage of 80 g m−2. While the influence of grammage or fiber type was not the focus of the present study, this may well be addressed in a follow-up work.
Figure 3 shows examples of tensile curves in (a) the dry and (b) the (aqueous) wet-state, respectively, for paper samples that have been modified and cross-linked with the same copolymer from different solvents mentioned in the figure. The dry and wet tensile index, as derived from these measurements using Eq. (2) is shown in Fig. 4.

Exemplary stress–strain curves measured during dry (a) and wet (b) tensile testing, from which the maximum force at break (Fmax) and subsequently the tensile indices were calculated

Dry and wet tensile indices of eucalyptus-sulfate paper samples with a grammage of 80 g m−2 shown side by side with the amount of copolymer inside the paper samples, as determined by gravimetric analysis for: pure cellulose paper samples not subjected to any treatment (Ref), samples subjected to the procedure for copolymer application, without any copolymer in the solution (RefSwell), and samples where the copolymer was applied out of H2O, IPA and BuOH (for more details, see supplementary information)
Failure mechanisms of functionalized paper sheets in the wet state
It is believed that the main failing mechanism of cellulose paper in the wet state is governed by fiber pull out. This would be intuitive, because in the wet state the fiber–fiber-bonds are weakened, and the fibers themselves become more flexible provided that the fibers do swell significantly. The failure of single fibers, on the other hand, should occur less frequently, but should be observable nevertheless (Siqueira 2012).
In order to get a better understanding of the failure-mechanisms for the eucalyptus polymer-modified paper samples used in this study here, an experimental setup for observing single cellulose fibers during the tensile straining was assembled. Paper sheets, where 0.1 wt% of all fibers have been stained with fluorophore Pergasol Yellow F6-GZ, were analyzed in a time-resolved fashion.
Figure 5 shows snapshots of tensile-test videos taken from polymer-modified paper sheets. Note, the complete video sections can be found in the supporting information. As shown in the first example in Fig. 5a, we frequently observed tearing of single fibers, while being under significant bending load. By looking at the combined length of the fiber fragments and comparing it to the initial fiber length, it becomes apparent, that the fiber-wall delaminates (Fig. 5a). Delamination of the fiber cell wall has been shown to occur in other eucalyptus paper samples with the help of electron microscopy by other groups as well. (Siqueira 2012) In addition, fiber pull-out was observed as a second typical failure mechanism, shown in Fig. 5b, respectively. Both mechanisms, fiber pull-out and fiber delamination driven breakage, have been reported before by others, studying the tensile properties of wet-strengthened paper sheets. (Siqueira 2012) Studying single-fiber behavior in more depth is of high interest, however, also requires an extensive statistical analysis. Note, at present we did not intend to draw such quantitative information from the tensile-test videos in order to judge which kind of failure mechanism determines the breakage of the wet paper samples most. We instead moved our attention to a quantitative macroscopic tensile behavior of the paper sheets, and in particular, of polymer-functionalized paper sheets, which allows for higher throughput of various samples.

Examples of two frequently observed mechanisms (a fiber failure and b fiber pull-out) during the failure of polymer-modified paper sheets during tensile straining in the wet state, captured with a self-made Zwick-videography setup
Impact of swelling-drying-cycles combined with UV-treatment
In order to compare the tensile strength of paper samples treated with the copolymer, we first analyzed the influence of the solvent treatment without any copolymer. When comparing the tensile-straining behavior of the Ref- and RefSwell-samples in the dry state in Fig. 3, it is apparent, that the treatment without copolymer leads to a significantly lower maximum force at break, while the elongation (strain at break) is increased. In the wet state, however, differences in the elongation are not as significant. In Fig. 4 the dry and wet tensile index, as derived from the stress–strain curves for the different paper samples are shown. If a reference paper sheet (Ref) is analyzed with respect to the tensile index, we observe typical values of about 17.6 N m g−1 and 0.3 N m g−1 for the dry and wet state, respectively. If the same paper is now being treated with one of the solvents without any copolymer and a similar swelling/drying protocol in combination with UV-exposure is used as with the copolymer impregnation process, we observe a decrease of both, dry and wet tensile index, to 9.8 and 0.2 N m g−1 (RefSwell—H2O), 12.1 and 0.2 N m g−1 (RefSwell—IPA) and 12.2 and 0.2 N m g−1 (RefSwell—BuOH), respectively.
There are multiple factors which have to be taken into account, when analyzing the effect of such a swelling/drying treatment with UV-exposure. Gravimetric experiments show, that drying under climate controlled conditions (23 °C, 50% r.h.) leads to a significant increase in moisture content (for all three solvents—detailed information in table S4), which can weaken the forces described earlier, being responsible for the intrinsic strength of paper, and thus the tensile strength. Further on, it has been found that unrestrained drying, which is comparable to drying on a mesh, significantly lowers the tensile strength (Strand et al. 2017; McKenzie and Higgins 1955), which further explains our results with the reference paper. Another factor is the extraction of fiber fragments known as “fines” of the fiber network, which is also known to decrease the tensile strength. Although the paper samples in this study haven’t been dry or wet pressed after paper making, the additional swelling and drying cycles during the treatment of paper samples can also lead to hornification, which could explain the observed decrease in measured tensile strength. In addition, it is safe to assume, that the intensive UV-exposure also has an influence on the tensile properties. It is known, that such UV-exposure significantly reduces the degree of polymerization (DP) of cellulose fibers (Kolar et al. 2000), and the group of Fang et al. (2020) observed that such a decrease of cellulose DP leads to a significant decrease of the tensile strength of nanocellulose films prepared thereof. Considering, that for the three reference experiments without any copolymer addition, the extraction process was carried out in H2O and the UV-treatment was identical, the observed similar values for dry and wet tensile index can be explained.
Impregnation using H2O versus IPA versus BuOH
We next investigated the stress–strain behavior of paper sheets modified with the copolymers in water, 2-propanol and 1-butanol, respectively. Examples of the data are shown in Fig. 3. It can be inferred that the dry tensile behavior for all solvents chosen are (almost) similar, whereas the stress–strain behavior of the respective samples in the wet state differs significantly. In particular, the maximum force at break progresses as BuOH < IPA < H2O. Dry and wet tensile index were further calculated from the measurements and are shown for all samples in Fig. 4. As can be inferred from the figure, the dry tensile index was increased by all three impregnation-treatments from 17.6 and 9.8 N m g−1 for the Ref and RefSwell, respectively, to 36.8, 46.3 and 44.5 N m g−1 for H2O-, IPA- and BuOH-impregnation, respectively. Comparing the wet tensile indices, it becomes clear that the H2O-impregnated paper samples possessed significantly higher wet strength. The latter increased from 0.3 and 0.2 N m g−1 for RefSwell and Ref, respectively, to 8.9 N m g−1, while IPA and BuOH-impregnation led to less increased values of 2.2 N m g−1 and 1.4 N m g−1 respectively. Considering, that the coefficients of variation are all below 5% (see supplementary information), these results are reproducible over many test samples. Only measurements for the wet tensile index of IPA-impregnated samples have a significantly higher coefficient of variation, indicating that IPA introduces a broader range of slightly different impregnation results, even though this can’t be observed for tensile tests in the dry state where the coefficient of variation is significantly lower.
The amount of cross-linked copolymer inside the paper samples was evaluated by a gravimetric approach under norm climate conditions. The results (see supplementary information) show that more copolymer adsorbs during impregnation with H2O (11.4 wt%) if compared to IPA- (8.0 wt%) and BuOH-impregnation (7.3 wt%). However, given the low absolute difference of the impregnated amount combined with the similar dry tensile values, we do not believe that this parameter is the determining factor for the observed difference in tensile properties.
In Fig. 6 the calculated values for relative wet strength of the different paper samples are shown. It is immediately evident, that applying the copolymer from H2O yields the highest relative wet strength paper samples. The high relative wet strength of 24.2% is directly related to the significant increase in wet tensile index, while at the same time the dry tensile index increases only by a significantly lower factor. Because the relative wet strength (Eq. 1) is calculated from the ratio of wet to dry tensile index, this overstates/exaggerates the wet strength of the paper samples, in comparison to the IPA- and BuOH-impregnated variants, where the increase in wet tensile strength was lower, while the increase in dry tensile strength was significantly higher. Therefore, the values for relative wet strength should always be treated with caution, when comparing paper samples and the data used for the calculation should be taken into account.

Relative wet strength of eucalyptus-sulfate paper samples with a grammage of 80 g m−2 shown side by side for: pure cellulose paper samples not subjected to any treatment (Ref), samples subjected to the procedure for copolymer application, without any copolymer in the solution (RefSwell), samples where the copolymer was applied out of H2O, IPA and BuOH (for more results, see supplementary information)
Spatial distribution—impregnation in H2O versus IPA versus BuOH
Next, we addressed the question why different solvents lead to significant differences in tensile strength. To this, the spatial distribution of copolymer inside the cellulose fiber network was analyzed using CLSM. By incorporating a fluorescing group, Rhodamine B, into the copolymer and subsequently staining the treated cellulose fibers with the fluorescent brightening agent CW, before embedding them in an epoxy-resin and preparing thin cross-sections, the spatial distribution throughout the fiber network was analyzed.
Cellulose fibers were stained after the copolymer-treatment, in order to enhance their visibility while fluorescence imaging. Although cellulose fibers from eucalyptus-sulfate pulp contain considerable amounts of lignin detectable with fluorescence imaging, the importance of the staining procedure can be seen in Fig. S4 in the supplementary information. Staining cellulose fibers after impregnation was done in order to rule out any effect of the staining on the spatial distribution of the copolymer and on the development of tensile properties of the samples. Due to this, some images may show inhomogeneous staining (e.g. Fig. 7g), probably caused by interactions between the copolymer and the dye.

CLSM cross-sectional images of an embedded paper sample, with the fluorescently labelled copolymer applied out of H2O (a–c), IPA (d–f) and BuOH (g–i), respectively, beforehand. The images show the cellulose fibers stained by CW (a, d, g), PDMAA labelled by RhB (b, e, h) and an overlay of the latter (c, f, i). The insets each show a magnification to highlight the spatial distribution of the RhB-labeled PDMAA across the fiber width and inside the fiber lumen. Scale bars are 100 and 20 µm for the overlay and insets, respectively. Overlays of both fluorescent signals appear as white
In consideration of the ability of H2O to swell cellulose fibers, it can be assumed, that the dissolved copolymer is able to penetrate the cellulose fiber network as a whole, which includes the fiber wall and the lumen, respectively. Taking a closer look at the fluorescence-channel from the fluorescently-labelled copolymer in Fig. 7b, this hypothesis can be confirmed. The fluorescence can be observed across the whole paper width, pointing towards the complete and homogeneous penetration on the scale of the fibers. Furthermore, we find copolymer inside the fiber lumen and upon higher magnification (see Fig. 7c) even within the fiber walls. In contrast, as both IPA and BuOH do not account for high degrees of swelling of the paper fibers, polymers dissolved in these solvents and brought into contact with the fiber are not expected to access all spaces within the non-woven sheets. Taking a closer look at the superimposed image of the fluorescence channels of the IPA- and BuOH-impregnated cellulose fibers (CW—cyan) and the copolymer (RhB—magenta) in Fig. 7f, i, respectively, this hypothesis can be confirmed. The copolymer seems to be mainly accumulated on the outside of the fibers and scattered rather inhomogeneously across the paper width. Upon higher magnification, Fig. 7f, i, respectively, show, that the copolymer is not able to penetrate inside the fiber or into the fiber walls and only a few of the fiber lumens are partially filled.
To further analyze the spatial distribution between the fibers, image-stacks of the cellulose fiber network were taken with the CLSM, and combined to 3D-images. The fluorescent brightening agent CW was once again used to stain the cellulose fibers.
The stacked images of the cellulose fiber network in Fig. 8 reveal the distribution of the copolymer in between the fibers. In analogy to the observations in the cross-sectional images, the overlay of the fluorescence channels in Fig. 8c shows, that the copolymer dissolved in H2O seems to be distributed homogeneously across the paper width. In comparison, applying the copolymer out of IPA and BuOH seems to lead to a more scattered, inhomogeneous distribution across the paper width, as can be inferred from Fig. 8f, i, respectively. However, BuOH-impregnation seems to be more homogeneous, especially when comparing the fluorescence channels of the copolymer for IPA- and BuOH-impregnated samples in Fig. 8e, h, respectively. It is also apparent, that in particular with H2O as a solvent there are almost no sleeves around fiber crossing points, which could yield a significant reinforcement of these fiber crossing-points, if in contact with water. Note, the use of dimethylacrylamide as the matrix monomer leads to a highly hydrophilic polymer, which can readily swell in water, even in the cross-linked state. Thus, we do see clear evidence for action of the copolymer/H2O system by the reinforcement mechanism, rather than by a protection mechanism. However, as is also evident from the structure analysis, the copolymers do not act purely on the reinforcement of fiber-crossing points but rather on the complete paper fiber. In the case of IPA and BuOH, the copolymer can be observed as sleeve-like-aggregates both at as well as away from fiber–fiber crossings, as can be seen in Fig. 8f, i, respectively. However, as they are not fully wrapping around the fiber crossing points and due to the hydrophilic nature of the cross-linked polymer, the protection of those crossing points from moisture according to a protection mechanism can be neglected. While those aggregates are able to form cross-links between adjacent fibers, the remaining fiber surface, without any fibers in close proximity, is virtually free of adsorbed copolymer—at least as far as fluorescence imaging allows to state this. In contrast, when comparing the fluorescence channels of the fibers (cyan) and the copolymer (magenta) of the H2O-impregnated paper samples (Fig. 8a, b), it is difficult to see significant differences. It seems as though the fluorescence of the CW-labelled fibers is at the outermost part and the fluorescence of the copolymer is beneath it, which can be observed in the cross-sectional images in Fig. 7c, too. This suggests, that the copolymer is not outside of the fibers or on the surface, but rather inside the fiber wall and the lumen, respectively. The presence of the copolymer inside the fibers can also be observed when looking at the in-plane projection of the stacked images (see supplementary information).

CLSM image stacks of paper samples combined to 3D-images, with the fluorescently labelled copolymer applied out of H2O (a–c), IPA (d–f) and BuOH (g–i), respectively, beforehand. The images show a top view of cellulose fibers stained by CW (a, d, g), PDMAA labelled by RhB (b, e, h) and overlays of the latter (c, f, i). For H2O-impregnation the images show the homogeneous spatial distribution of copolymer across the whole fiber network. Overlays of both fluorescent signals appear as blue in 3D-renderings. In contrast, the IPA -impregnated samples show an inhomogeneously distributed copolymer with agglomeration at fiber–fiber crossings, while the BuOH-impregnated samples show a more homogeneous distribution, but not on the single fiber scale comparable to H2O-impregnation. Scale bar is 100 µm
When comparing different solvents, the hydrodynamic properties, and, in particular, size exclusion effects as well as thermodynamic behavior of the polymers in solution can become critical and can impact the deposition and resulting spatial distribution of the adsorbed copolymers in/on the fibers. In order to learn more about the thermodynamic behavior of the polymers in dilute and concentrated solutions, we analyzed the polymer solutions by dynamic light scattering (at concentrations of 5 mg mL−1) and additional turbidity measurements at higher concentrations (5–45 mg mL−1), in order to determine the phase behavior at variable temperatures (5–50 °C). Data can be found in the supporting information. In brief, hydrodynamic radii of the copolymers in H2O, and IPA, show very similar values in the range of 7–11 nm, respectively.
The turbidity experiments (see supplementary information) do not show any aggregation for the solvents IPA and BuOH over a wide temperature-range even at highest chosen concentrations of up to 45 mg mL−1. However, for the copolymer-solution in H2O, clouding can be observed at around 13 °C which becomes more pronounced with increasing temperature. Lowering the concentration to 25 mg mL−1 (impregnation) and further to 5 mg mL−1 (DLS), the temperature, where clouding is first observed, increases significantly to 21 and 36 °C, respectively. In particular, in alcoholic solutions under impregnation-conditions chosen, no aggregates are present.
To elucidate how accessible the adsorption sites are for the copolymer in the cellulose fiber network, information on the pore size (distribution) is important. The pore sizes of the paper sheets used in our study range from tens of nanometers to 30 µm and was analyzed by mercury-intrusion (for an example of the latter see supporting information). With respect to the adsorption of macromolecules on/in paper fibers, smaller pores in the cell walls were studied by various methods and values ranging from 10 to 100 nm were reported (Wu et al. 2009). Pores and grooves that are on the order of the size of an unperturbed polymer chain may not be trivial to be accessed by the macromolecules due to confinement effects (i.e. the polymer chain can only enter the pore if segments are stretching, which is thermodynamically not favored).
However, polymers may easily enter pores that are larger than the molecular size of the macromolecule. With respect to this, it therefore makes a large difference, if fibers are pre-swollen in the solvent or if the polymer solution is transferred onto a dry paper sample. Submerging a dried paper sample in a given solvent leads to sudden imbibition into the sample, due to strong capillary forces. (Alava and Niskanen 2006) The work of El Seoud et al. (2008) allows for a quantitative comparison of the used solvents in this regard, by determining the extent of swelling using a simple gravimetric approach. The results show a significant difference of the ability to swell cellulose fibers: H2O ~ 62.7%, IPA ~ 4.7% and BuOH ~ 7.2%, respectively. From these values it can be inferred, that impregnating the paper with the copolymer dissolved in H2O, a significantly higher volume of the copolymer solution, i.e. around ten-fold more, is spontaneously pulled inside the fiber network and inside the fibers by capillary forces and fiber swelling, as compared to impregnation of paper with IPA and BuOH solutions.
Without the possibility of fiber swelling, IPA and BuOH can only transport the copolymer to the outer surface of fibers, or inside the fiber lumen through larger defects and pinholes in the fiber wall. While drying, the solvent accumulates at fiber–fiber crossings, due to increased capillary forces, leading to precipitation of the copolymer at such spots. In addition, IPA has a lower boiling point and a higher vapor pressure [42.6 hPa (20 °C) (Lide 2004)] compared to H2O and BuOH, leading to a more sudden evaporation of the solvent and hence yielding an inhomogeneous distribution of the copolymer across the paper width, which again is in accordance with our structure analysis.
The copolymer inside the fibers of H2O-impregnated samples can form homo-cross-links with its own backbone (Toomey et al. 2004; Körner et al. 2016; Prucker et al. 2018), but is also able to yield covalent hetero-cross-links with CH-groups of cellulose chains (Jocher et al. 2015). In this way, cellulose chains, penetrating into the fiber wall from adjacent fibers, can covalently link with each other and with the fibers under the influence of UV-light, thereby also strengthening fiber–fiber bonds. Hence, the latter, may be one explanation for the increase in dry and wet tensile strength, even though most of the copolymer is located inside the fibers, ostensibly not contributing to the reinforcement of fiber–fiber bonds. Another explanation, how a cross-linking polymer within the fiber wall may contribute to a higher wet strength can be deduced from our video observations. Single fiber failure and in particular delamination would directly benefit from a strengthened fiber wall, i.e. reinforced by the cross-linked copolymer on and in the fiber wall. While the slipping mechanism may strongly be affected by fiber–fiber connections, it is reasonable to assume, that fiber flexibility in the dry and the wet state plays a significant role if fibers glide/slide past one another during tensile load.
Combining the results of the spatial analysis and the tensile tests, it may be assumed, that the copolymer works as a bulking wet strength agent. Luner and Zhou (1993) reported that depositing or introducing chemicals of molecular size, small enough to penetrate the cell wall, is a method to achieve wet strengthening. By doing this, the moisture regain is reduced and therefore the dimensional stability is increased, inhibiting swelling of the cellulose fibers. Swelling of cellulose fibers has a major effect on the morphology especially the length and width (Lindner 2018). A possible explanation for the significant impact decreased swelling and increased dimensional stability of single cellulose fibers have on the wet tensile strength of the fiber network inside paper sheets is, that the relative bonded area (RBA) isn’t decreased when the paper comes into contact with moisture. The RBA is one of the parameters of the Page equation and is a key factor influencing the dry tensile strength, next to the single fiber bond strength (Page 1969). Even though introducing moisture inside a fiber network changes the situation and probably the failure mechanism under tensile load, one may still consider the RBA as an important factor, if describing the tensile strength of paper in the wet state.
Studies looking at the enhancement of dry strength by using carboxymethylcellulose-grafted (CMC) pulp, cationic starch (C-starch) and microfibrillated cellulose (MFC) found, that the enhancement was mainly due to the increase in RBA on the microscale (Lindström et al. 2016). These results are supported by experiments on the influence of the application-method of polyacrylamide (PAM) on the enhancement of dry tensile strength of paper (Mihara et al. 2008). They found, that an external application method by impregnating finished paper samples in an aqueous PAM solution yielded significantly higher dry tensile index values, compared to the mass application during paper formation. Additionally, the spatial adsorption of PAM was analyzed by controlled etching of the cellulose and concurrent ATR-FTIR measurements, which showed, that the external application method led to the deposition of PAM mainly on the fiber surface and around fiber–fiber bonds, respectively.
This can explain the difference in the observed dry tensile indices of H2O-, IPA- and BuOH-impregnated samples, since impregnation from H2O yields densely homo- and hetero-cross-linked networks of copolymer inside the cellulose fibers. Therefore, the amount of copolymer contributing to cross-linking outside and in between fibers, possibly increasing the RBA and thus the dry tensile strength, can’t be as high.
Conclusions
The preparation of paper samples impregnated with a photo-cross-linkable fluorescent copolymer P(DMAA-co-MABP-co-RhBMA) was achieved using three different solvents H2O, IPA and BuOH, respectively. Concurrently the spatial distribution of the copolymer inside the fiber network and the fibers themselves was analyzed with confocal microscopy (CLSM) of thin cross-sections and 3D-images. This method, in contrast to commonly used imaging methods in paper science and technology, does not require sophisticated sample preparation nor any specialized very expensive equipment and would thus be of interest for many more analytical tasks in the context of paper structure analysis.
Using either of the three solvents for impregnation, we were able to significantly increase the dry and wet tensile indices, by using the aforementioned copolymer yielding relative wet strengths of up to 25%.
Because H2O has the ability to readily swell cellulose fibers inside the paper network, a homogeneous distribution of the copolymer in and on the fibers can be observed. This reinforces both the fiber crossing points and the fibers themselves, which increases the tensile index, especially in the wet state. By using solvents which do not swell the cellulose fibers, e.g. IPA or BuOH, the copolymer can’t penetrate into the fibers, the distribution of the copolymer on the surface of the fibers is less homogeneous and wet tensile (relative wet-) strength is much lower.
Overall, we can thus conclude that impregnation with IPA and BuOH solutions of the copolymer is preferable for the dry tensile strength, while impregnation with H2O dissolved macromolecules followed by photocrosslinking allows for increased wet tensile strength of paper samples.
To gain an even deeper understanding of the underlying mechanisms, in particular when using the photo-reactive wet-strength agents, in future, we will focus in more detail on the influence of paper-extrinsic parameters, such as the source of light, illumination conditions and solvent content during illumination, as well as paper/paper technology intrinsic parameters such as a comparison of the here reported impregnated papers vs. the use of (charged) copolymers in wet end processes.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request. All data generated or analysed during this study are included in this published article (and its supplementary information files).
References
Alava M, Niskanen K (2006) The physics of paper. Rep Prog Phys 69(3):669–723. https://doi.org/10.1088/0034-4885/69/3/R03
Auslender A, Biesalski M, Groche P, Knaack U, Schneider J, Schabel S (2017) Building with Paper New Applications for a diverse Material. Wochenbl Pap 145(12):822–824
Bump S, Böhm A, Babel L, Wendenburg S, Carstens F, Schabel S, Biesalski M, Meckel T (2015) Spatial, spectral, radiometric, and temporal analysis of polymer-modified paper substrates using fluorescence microscopy. Cellulose 22(1):73–88. https://doi.org/10.1007/s10570-014-0499-5
Credou J, Berthelot T (2014) Cellulose: from biocompatible to bioactive material. J Mater Chem B 2(30):4767–4788. https://doi.org/10.1039/C4TB00431K
El Seoud OA, Fidale LC, Ruiz N, D’Almeida MLO, Frollini E (2008) Cellulose swelling by protic solvents: which properties of the biopolymer and the solvent matter? Cellulose 15(3):371–392. https://doi.org/10.1007/s10570-007-9189-x
Fang Z, Li B, Liu Y, Zhu J, Li G, Hou G, Zhou J, Qiu X (2020) Critical role of degree of polymerization of cellulose in super-strong nanocellulose films. Matter 2(4):1000–1014. https://doi.org/10.1016/j.matt.2020.01.016
He J, Batchelor WJ, Johnston RE (2003) The behavior of fibers in wet pressing. Tappi J 2:27–31
Hirn U, Schennach R (2015) Comprehensive analysis of individual pulp fiber bonds quantifies the mechanisms of fiber bonding in paper. Sci Rep 5(1):10503. https://doi.org/10.1038/srep10503
Hubbe MA (2006) Bonding between cellulosic fibers in the absence and presence of dry-strength agents–A review. BioResources 1(2):281–318
Janko M, Jocher M, Boehm A, Babel L, Bump S, Biesalski M, Meckel T, Stark RW (2015) Cross-linking cellulosic fibers with photoreactive polymers: visualization with confocal Raman and fluorescence microscopy. Biomacromol 16(7):2179–2187. https://doi.org/10.1021/acs.biomac.5b00565
Jocher M, Gattermayer M, Kleebe H-J, Kleemann S, Biesalski M (2015) Enhancing the wet strength of lignocellulosic fibrous networks using photo-crosslinkable polymers. Cellulose 22(1):581–591. https://doi.org/10.1007/s10570-014-0477-y
Karppinen T, Kassamakov I, Hæggström E, Stor-Pellinen J (2004) Measuring paper wetting processes with laser transmission. Meas Sci Technol 15(7):1223–1229. https://doi.org/10.1088/0957-0233/15/7/001
Körner M, Prucker O, Rühe J (2016) Kinetics of the generation of surface-attached polymer networks through C. H-Insertion React Macromol 49(7):2438–2447. https://doi.org/10.1021/acs.macromol.5b02734
Kolar J, Strlic M, Pentzien S, Kautek W (2000) Near-UV, visible and IR pulsed laser light interaction with cellulose. Appl Phys A 71(1):87–90. https://doi.org/10.1007/s003390000491
Lide DR (2004) CRC handbook of chemistry and physics, 85th edn. CRC Press
Lindner M (2018) Factors affecting the hygroexpansion of paper. J Mater Sci 53(1):1–26. https://doi.org/10.1007/s10853-017-1358-1
Lindström T, Fellers C, Ankerfors M, Nordmark GG (2016) On the nature of joint strength of paper—Effect of dry strength agents—Revisiting the Page equation. Nord Pulp Pap Res J 31(3):459–468. https://doi.org/10.3183/npprj-2016-31-03-p459-468
Lindström T, Wågberg L, Larsson T (2005) On the nature of joint strength in paper—a review of dry and wet strength resins used in paper manufacturing. In: Proceedings of 13th fundamental research symposium. Cambridge, Surrey, UK, pp 457–562
Luner P, Zhou YJ (1993) Wet reinforcing of paper and board by novel crosslinking chemicals. In: Products of papermaking. Trans. of the X Fund. Res. Symp., Oxford, pp 1045–1072
Mangiante G, Alcouffe P, Gaborieau M, Zeno E, Petit-Conil M, Bernard J, Charlot A, Fleury E (2018) Biohybrid cellulose fibers: toward paper materials with wet strength properties. Carbohydr Polym 193:353–361. https://doi.org/10.1016/j.carbpol.2018.04.009
McKenzie A, Higgins H (1955) The structure and properties of paper. III. Significance of swelling and hydrogen bonding in interfiber adhesion. Aust J Appl Sci 6:208–217
Mihara I, Sakaemura T, Yamauchi T (2008) Mechanism of paper strength development by the addition of dry strength resin and its distribution within and around a fiber wall: effect of application method. Nord Pulp Pap Res J 23(4):382–388. https://doi.org/10.3183/npprj-2008-23-04-p382-388
Nau M, Herzog N, Schmidt J, Meckel T, Andrieu-Brunsen A, Biesalski M (2019) Janus-type hybrid paper membranes. Adv Mater Interfaces 6(18):1900892. https://doi.org/10.1002/admi.201900892
Page DH (1969) A theory for the tensile strength of paper. Tappi J 52(4):674–681
Prucker O, Brandstetter T, Rühe J (2018) Surface-attached hydrogel coatings via C, H-insertion crosslinking for biomedical and bioanalytical applications (review). Biointerphases 13(1):010801. https://doi.org/10.1116/1.4999786
Schäfer CG, Gallei M, Zahn JT, Engelhardt J, Hellmann GP, Rehahn M (2013) Reversible light-, thermo-, and mechano-responsive elastomeric polymer Opal films. Chem Mater 25(11):2309–2318. https://doi.org/10.1021/cm400911j
Siqueira EJ (2012) Polyamidoamine epichlorohydrin-based papers: mechanisms of wet strength development and paper repulping. Dissertation, Université de Grenoble
Strand A, Sundberg A, Retulainen E, Salminen K, Oksanen A, Kouko J, Ketola A, Khakalo A, Rojas O (2017) The effect of chemical additives on the strength, stiffness and elongation potential of paper. Nord Pulp Pap Res J 32(3):324–335. https://doi.org/10.3183/npprj-2017-32-03-p324-335
Tejado A, van de Ven TGM (2010) Why does paper get stronger as it dries? Mater Today 13(9):42–49. https://doi.org/10.1016/S1369-7021(10)70164-4
Toomey R, Freidank D, Rühe J (2004) Swelling behavior of thin, surface-attached polymer networks. Macromolecules 37(3):882–887. https://doi.org/10.1021/ma034737v
Wu N, Hubbe M, Rojas O, Park S (2009) Permeation of polyelectrolytes and other solutes into the pore spaces of water-swollen cellulose: a review. BioRes 4(3):1222–1262. https://doi.org/10.15376/biores.4.3.1222-1262
Acknowledgments
The authors thank Heike Herbert and Martina Ewald for technical support. Funding by the DFG under Grants #405422473, and #406026418 is gratefully acknowledged. This study was in part also supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC-2193/1–390951807 (livMatS).
Funding
Open Access funding enabled and organized by Projekt DEAL. Funding of this work is provided by the German Research Council, DFG under grants #405422473 and #406026418 is gratefully acknowledged. This study was in part also supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC-2193/1–390951807 (livMatS).
Author information
Authors and Affiliations
Contributions
J-LS and SS have contributed equally to this work. SS contributed the expertise and synthesized and provided the materials for photochemically cross-linkable copolymers and performed the measurements of the hydrodynamic radii. J-LS contributed the expertise in cellulose (paper) modification and prepared the functionalized paper samples, carried-out all tensile tests as well as confocal laser scanning microscopy measurements. ARvS prepared sample paper sheets as well as he provided specifications of the used lab-paper. OP and TB planned and supervised the synthesis of the copolymers, and TM planned and supervised the measurements by various microscopy techniques. JR and MB planned and supervised the whole project and are equally responsible for any correspondence. All authors contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
There are no conflicts of interest nor competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 (MP4 350 kb)
Supplementary file3 (MP4 2147 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schäfer, JL., Schölch, S., Prucker, O. et al. Accessibility of fiber surface sites for polymeric additives determines dry and wet tensile strength of paper sheets. Cellulose 28, 5775–5791 (2021). https://doi.org/10.1007/s10570-021-03817-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-021-03817-7