
Abstract
Platinum and ruthenium nanoparticles stabilised by an amine modified polymer immobilised ionic liquid (MNP@NH2-PEGPIILS, M = Pt, Ru) catalyse the hydrolytic liberation of hydrogen from dimethylamine borane (DMAB), ammonia borane (AB) and NaBH4 under mild conditions. While RuNP@NH2-PEGPIILS and PtNP@NH2-PEGPIILS catalyse the hydrolytic evolution of hydrogen from NaBH4 with comparable initial TOFs of 6,250 molesH2.molcat−1.h−1 and 5,900 molesH2.molcat−1.h−1, respectively, based on the total metal content, RuNP@NH2-PEGPIILS is a markedly more efficient catalyst for the dehydrogenation of DMAB and AB than its platinum counterpart, as RuNP@NH2-PEGPIILS gave initial TOFs of 8,300 molesH2.molcat−1.h−1 and 21,200 molesH2.molcat−1.h−1, respectively, compared with 3,050 molesH2.molcat−1.h−1 and 8,500 molesH2.molcat−1.h−1, respectively, for PtNP@NH2-PEGPIILS. Gratifyingly, for each substrate tested RuNP@NH2-PEGPIILS and PtNP@NH2-PEGPIILS were markedly more active than commercial 5wt % Ru/C and 5wt% Pt/C, respectively. The apparent activation energies of 55.7 kJ mol−1 and 27.9 kJ mol−1 for the catalytic hydrolysis of DMAB and AB, respectively, with RuNP@NH2-PEGPIILS are significantly lower than the respective activation energies of 74.6 kJ mol−1 and 35.7 kJ mol−1 for its platinum counterpart, commensurate with the markedly higher initial rates obtained with the RuNPs. In comparison, the apparent activation energies of 44.1 kJ mol−1 and 46.5 kJ mol−1, for the hydrolysis NaBH4 reflect the similar initial TOFs obtained for both catalysts. The difference in apparent activation energies for the hydrolysis of DMAB compared with AB also reflect the higher rates of hydrolysis for the latter. Stability and reuse studies revealed that RuNP@NH2-PEGPIILS recycled efficiently as high conversions for the hydrolysis of DMAB were maintained across five runs with the catalyst retaining 97% of its activity.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The limited reserves of fossil fuels coupled with the negative environmental impact resulting from our continued reliance as a major source of energy is placing increasing pressure on developing an alternative clean and sustainable energy vector [1,2,3,4]. To this end, green hydrogen has attractive credentials as a clean and sustainable energy carrier for use in stationary and transport applications as it has a suitable gravimetric energy density (142 MJ kg−1 vs 54 MJ kg−1 for natural gas) and can be generated either from the electrolysis of water or from water splitting, generating oxygen as the only by-product [5,6,7,8,9,10,11,12,13,14]. However, the safe large-scale storage and transportation of such a highly flammable gas and its low volumetric energy density are major challenges that must be addressed if this technology is to become a practical reality and viable [15, 16]. One potential solution is to store the hydrogen either in the form of a low molecular weight solid or liquid hydride or to absorb it within the channels of a porous material [17,18,19,20,21,22,23,24]. In this regard, in addition to sodium borohydride [25,26,27,28,29,30], amine boranes have attracted significant interest as possible hydrogen storage materials because they are highly stable, have a high hydrogen content and are non-toxic and water soluble [31,32,33,34,35,36,37,38,39]. While ammonia borane (AB) has the highest hydrogen content (19.6 wt%), dimethylamine borane (DMAB) is ca. 30 times less expensive and as such considerable effort is currently being committed to developing cost effective catalysts for the facile and controlled liberation of hydrogen from this substrate [40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61].
The dehydrogenation of one mole of DMAB in organic media only liberates 1 mol of H2 (Eq. 1) and while high rates of hydrogen production have been achieved with homogeneous catalysts under relatively mild conditions, they suffer from poor long-term stability, often require expensive ligands and are difficult to recover after use [62,63,64,65,66,67,68,69,70,71,72,73,74,75,76]. Heterogeneous catalysts offer several advantages in terms of good thermal stability, range of operating temperatures, ease of recovery and integration into a device [77,78,79,80,81,82]. To the end, nanoparticle-based catalysts have attracted particular attention as their activity can be controlled through their size, and thereby surface area to volume ratio and number of active sites, as well as their morphology, however, the high surface area of metal NPs can lead to self-aggregation under the conditions of catalysis, forming larger species that are less active [83,84,85,86,87,88,89,90,91,92,93]. One approach with the potential to prevent aggregation has been to stabilize the nanoparticles by encapsulation into a suitable support such as a metal oxide or zeolite, a porous carbon material e.g. graphene oxide or reduced graphene oxide, polymers or porous organic cages and MOF [94]. To this end, there have been several recent examples of NPs that catalyse the dehydrogenation of DMAB with encouraging performance profiles either solventless or in organic media including mono-, bi and trimetallic systems encapsulated in PVP [41, 42, 55, 56, 95, 96], graphene oxide and reduced graphene oxide [44,45,46, 58, 97,98,99], Vulcan carbon [49], ceria, alumina or titania [100,101,102,103], MOFs [104,105,106,107,108,109], hydrophilic polymers [110], and MW carbon nanotubes [111]. Moreover, it is becoming increasingly more evident that the encapsulation of NPs has additional benefits for catalysis; these include control of the growth and morphology due to confinement [112,113,114,115,116,117], dramatic enhancements in catalyst performance arising from strong metal support interactions [118,119,120,121,122,123], and significant improvements in activity and selectivity by incorporating surface functionality or organic modifiers/ligands to modulate the surface electronic structure and/or steric environment as well as the solubility of the reactants [124,125,126,127]. In contrast to the dehydrogenation in organic media, the hydrolytic release of hydrogen from DMAB liberates three moles of H2 (Eq. 2), however, there are surprisingly few reports of this hydrolysis catalysed by ruthenium nanoparticles and as such there is still a need to identify cost effective catalysts for the rapid and controlled hydrolysis of DMAB under mild conditions and to develop an understanding of the factors that influence catalyst efficacy.
While ionic liquids have also been used for the stabilisation of metal nanoparticles [128,129,130] and their applications in a broad range of fundamentally important transformations explored [131,132,133,134], there do not appear to be any reports of the use of ionic liquid stabilised NPs for the catalytic release of hydrogen from DMAB, which is somewhat surprising as amine boranes have been widely used as hydrogen donors for catalytic transfer hydrogenations [135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151]. This is even more surprising considering that ionic liquids promote the dehydrogenation of ammonia borane and as such might be expected to show a cooperative or synergistic effect with NPs [152,153,154,155,156,157]. Ionic liquids have also been grafted to supports such as polymers [158,159,160,161,162,163,164,165,166,167], mesoporous silica [168,169,170,171] and MOFs [172,173,174,175,176,177,178,179] to combine their favourable characteristics, such as the stabilization of nanoparticles through weak electrostatic interactions in the same manner as an IL, with covalent attachment to a support which would immobilise the ionic liquid, facilitate recovery of the catalyst and reduce the amount of ionic liquid as the catalyst would be confined within the support. Moreover, the tuneable physicochemical properties and modular synthesis of PIILs should enable the ionic environment, charge density and distribution to be modified, additional surface functionality to be introduced and the stoichiometry of the metal precursor to be controlled to facilitate the synthesis of synergetic bi- and multimetallic nanoparticles and thereby improve catalyst efficacy and develop new processes and technology to meet the criteria required for commercial applications. Although support-grafted ionic liquids are finding wide ranging applications for the stabilisation of NPs and molecular catalysts there are only a few reports of their use as supports to stabilise NPs for the hydrolytic release of hydrogen from storage materials. In the first of these, highly dispersed PdAuNPs stabilized by an imidazolium-modified porous organic polymer catalyses the hydrolytic release of H2 from AB more efficiently than either of its monometallic counterparts [180], while RuNPs supported on a polymeric ionic liquid catalyses the synthesis of benzimidazole from CO2 and 1,2-diamines by reductive dehydrogenation of DMAB as well as the DMAB-mediated reduction of olefins and nitroarenes [181, 182]. We have been developing heteroatom donor functionalised PIILs as supports for the stabilisation of NPs to determine whether the heteroatom donor can supplement the weak stabilisation provided by the ionic liquid fragment, influence the growth of the NPs or modify the surface electronic structure and steric properties and thereby exploit the beneficial influence that ligands can impart on the performance of heterogeneous nanocatalysts [183,184,185,186,187,188,189]. To this end, we recently reported that platinum and ruthenium NPs stabilised by phosphine or amine-modified PIILs catalyse the hydrolytic dehydrogenation of NaBH4 [190, 191]. In a subsequent study to explore the efficacy of amino-modified PIIL stabilised Pt and Ru nanoparticles as catalysts for the DMAB-mediated reduction of quinoline, we discovered that their disparate efficiency may be associated with the different rates of hydrogen evolution from the DMAB as this would influence the availability of surface hydride species [192]. We have now conducted a thorough and detailed comparison of the performance of amino-functionalised PEG-modified PIIL stabilised ruthenium and platinum NPs as catalysts for the hydrolytic dehydrogenation of DMAB, AB and NaBH4 and herein report the results of this study. Surprisingly, RuNP@NH2-PEGPIILS is a markedly more active catalyst for the hydrolysis of DMAB and AB than its platinum counterpart whereas both catalysts gave comparable rates for the hydrolysis of NaBH4. Moreover, the TOF of 8,300 h−1 for the aqueous phase dehydrogenation of DMAB using RuNPs stabilised by PEG-modified amine-decorated polymer immobilized ionic liquid is among the highest to be reported under mild conditions but lower than the 14,926 h−1 obtained with a PtRu nanocatalyst stabilised by Vulcan carbon; currently the most efficient state-of-the-art catalyst for the aqueous phase dehydrogenation of this substrate [49]. In addition, regardless of the catalyst, the hydrolytic dehydrogenation of ammonia borane was substantially faster than DMAB. Preliminary, kinetic studies with deuterium isotope effects were undertaken to probe the mechanism of hydrolysis and possible pathways are discussed.
2 Results and Discussion
2.1 Synthesis and Characterisation of Precatalyst 1a and Nanoparticle Catalysts 2a-b
The composition of the tetrachloroplatinate loaded precursor [PtCl4]@NH2-PEGPIILS (1a) and the polymer immobilised ionic liquid stabilised platinum and ruthenium nanoparticles 2a-b required for this study are summarised in Fig. 1. Precursor 1a was prepared by impregnation of the corresponding polymer NH2-PEGPIIL with the tetrachloroplatinate anion such that complete exchange of all the bromide and chloride would afford an amine to platinum ratio of one; the resulting precursor 1a was isolated as a dark red solid in near quantitative yield. Quantitative analysis of the Cl 2p and Br 3d components of the XPS spectrum of 1a revealed that the exchange was not complete as evidenced by the presence of both bromide and chloride, although as expected impregnation of NH2-PEGPIIL with [PtCl4]2− resulted in a significant reduction of the bromide content as evidenced by a comparison of the bromide to chloride ratio of 1:0.22 for NH2-PEGPIIL and 1:1.75 for 1a; this was supported by ICP OES analysis of the precursor which gave a bromide to chloride ratio of 1:2.2. The corresponding amine decorated polymer immobilised ionic liquid stabilised platinum nanoparticles PtNP@NH2-PEGPIILS (2a) was obtained by sodium borohydride mediated reduction of 1a in ethanol and isolated as a free-flowing black powder and its ruthenium counterpart 2b was prepared as previously described [191]. The solid state 13C NMR spectrum of 1a contains a series of signals associated with the carbon atoms of the imidazolium ring and the aromatic carbons at δ 124 and δ 146 ppm, high field signals at δ 11–49 ppm for the methyl group on the imidazolium ring and the methylene units of the polymer backbone and the benzylamine as well as an intense signal at δ 71 ppm for the methylene units of the PEG and a weaker signal at δ 58 ppm for the terminal OMe. The solid-state 13C NMR spectra of 2a-b were identical that of the polymer support. Comparison of the solid state 15N NMR spectrum for the polymer support, NH2-PEGPIIL, with that for 1a provided convincing evidence for a Pt–-N interaction as the former contains three signals at δ -195/-207 and δ -332 ppm for the nitrogen atoms in the imidazolium ring and the amine, respectively, while the latter contains an additional signal at δ -291 ppm; the 41 ppm shift to low field is indicative of a platinum coordinated amine (Fig. S13 in the supporting information). The IR spectra of the polymer, NH2-PEGPIIL, precatalyst 1a and catalysts 2a-b each contain bands at ca. 1580 cm−1 and 1605 cm−1 characteristic of C = C and C = N stretching vibrations of the imidazolium ring and a band at ca. 1450 cm−1 due to the C-N(imidazolium or amine) stretching vibration confirming that the ionic liquid monomer was incorporated into the polymer. The thermal stability of the NH2-PEGPIIL support was investigated by thermogravimetric analysis which showed an initial weight loss just below 100 °C associated with removal of a minor amount of physiosorbed ethanol and/or water; this was followed by three major degradation pathways between 260–650 °C, confirming its suitability as a support for the stabilisation of nanoparticles for use in catalysis. Moreover, the TGA profiles for 2a-b revealed that these catalysts begin to decompose close to 210 °C, which is well below the temperatures typically required for the release of hydrogen from storage materials. The platinum and ruthenium loadings in 1a and 2a-b were determined using ICP-OES. In addition, since NH2-PEGPIIL is a mixed bromide/chloride salt and 2a and 2b were generated by reduction of the corresponding metal chloride impregnated precursors, ICP-OES analysis was also undertaken to establish the chloride and bromide composition of 2a-b as the amount and type of halide is likely to influence their efficacy. To this end, the chloride to bromide ratios of 6.25:1 and 7.0:1 in 2a and 2b, respectively, confirm that both contain similar amounts of residual bromide. This was supported by quantitative analysis of the Cl 2p and Br 3d components of 2a and 2b which was used to determine the relative amounts of chloride and bromide retained after reduction of the metal halide impregnated precursor; reassuringly, the ratios of 7.2:1 and 7.9:1 for 2a and 2b, respectively, support those determined by ICP-EOS.

Composition of [PtCl4]2− impregnated amine-decorated PIIL, PtCl4@NH2-PEGPIILS (1a) and the corresponding PIIL-stabilised platinum nanoparticles PtNP@NH2-PEGPIILS (2a) and corresponding ruthenium nanoparticles RuNP@NH2-PEGPIILS (2b)
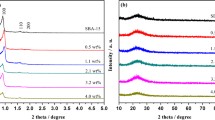
Surface characterisation of the polymer, precatalyst 1a and catalysts 2a-b was undertaken by X-ray photoelectron spectroscopy (XPS). Due to the limitations associated with using advantageous carbon as the references [193], peak assignments were mainly based on the characteristic BE separations between the elements. The local nitrogen environment of the polymer was fitted to two peaks at 397.4 eV and 395.6 eV characteristic of nitrogen in an imidazolium ring and an amine, respectively (see Fig. S15 in the support information). The C 1 s region was also consistent with the polymer composition as it was fitted to three peaks, one at 280.6 eV for C–C/C-H species and two at 282.3 eV and 281.2 eV characteristic of C-O and C-N species, respectively [189]. The N 1 s region of the tetrachloroplatinate loaded precatalyst 1a was also fitted to two major peaks at 397.4 eV and 395.6 eV corresponding to imidazolium and amine species, respectively (see Fig. S21 in the supporting information) [189]; both peaks do not appear to shift relative those in the polymer suggesting that the chemical environment is unchanged. An additional minor component was also fitted at 395.0 eV, which may be indicative of a Pt–-N interaction. The Pt 4f region of the tetrachloroplatinate-loaded precursor was fitted to two pairs of 4f7/2 and 4f5/2 spin orbit doublets, consistent with two Pt 4f electronic environments (Fig. 2a). The binding energies of the major 4f5/2 and 4f7/2 peaks at 71.5 eV and 68.2 eV correspond to Pt(II) coordinated to chloride as the Pt 4f5/2 and Cl 2p3/2 and Pt 4f7/2 and Cl 2p3/2 separation of 121.3 eV and 124.6 eV, respectively, are comparable to literature reported values for a Pt(II)-Cl species [194,195,196,197,198,199], while the minor 4f5/2 and 4f7/2 doublet at 72.8 eV and 69.5 eV is shifted to higher binding energy and most likely corresponds to platinum coordinated to amine, as the separation of 3.3 eV between these peaks and the difference on the BE scale of 326.7 eV between the 4f7/2 state and the N 1 s is comparable to values previously reported for a Pt–-N interaction [200,201,202,203]. Such a peak shift to higher binding energy indicates that the formation of a Pt–-N interaction renders the platinum more electron deficient, which may be a result of the lower negative charge at platinum due to substitution of a chloride in [PtCl4]2− for a neutral amine. The peak with a binding energy centred at 64.2 eV corresponds to residual bromide, derived from the PEG-modified imidazolium monomer, and was fitted to two pairs of 3d5/2 and 3d3/2 doublets, which mostly likely correspond to bromide anion and Pt-coordinated bromide [197]. The Pt 4f region of the XPS spectrum of catalyst 2a (Fig. 2b) was fitted to two pairs of 4f7/2 and 4f5/2 doublets and the separation between the BEs of the Pt 4f7/2 peaks and the O 1 s peak of 465.4 eV and 463.8 eV is consistent with Pt metal and PtO2, respectively [204,205,206]. In the case of catalyst 2b, the Ru 3p region was analysed due to overlap of the C 1 s and Ru 3d regions and the broad peak at 459.5 eV was fitted to two Ru 3p3/2 peaks; the peak at higher BE was assigned to RuO2 while the one at lower BE was attributed to Ru(0) (Fig. 2c). This assignment was based on the separation between these peaks and the aliphatic C 1 s peak as the values of 177.7 eV and 180.2 eV for this difference are consistent with previously reported values for metallic ruthenium and RuO2, respectively [207,208,209,210,211]. The peaks assigned to RuO2 species are probably due to surface oxidation of the preformed ruthenium nanoparticles. Finally, while Na and B 1 s peaks in the XPS spectra for 2a and 2b confirmed the presence of a borate salt such as NaBO2 or NaB(OH)4 these were subsequently removed by extraction with water. The powder XRD pattern for 2b contained diffraction peaks at 2θ = 28.1°, 35.2°, 40.3°, 54.5°, 58.1°, 59.6°, 65.8°, 67.2°, 69.8° and 74.3° which index to the (110), (101), (200), (210), (211), (220), (002), (310), (112), (301) and (202) lattice planes of the tetragonal phase of RuO2 with the P42/mmm space group (ICDD No. 00–040-1290), consistent with the reported literature. The absence of diffraction peaks for metallic ruthenium suggest that the nanoparticles are highly dispersed with sizes < 2.5 nm, which was supported by TEM analysis. Similarly, the powder XRD pattern for 2a contained characteristic diffraction peaks at 2θ = 27.9°, 34.7°, 34.8°, 39.6°, 40.2°, 40.3°, 53.8°, 57.7°, 58.8°, 65.0°, 65.6° which index to (110), (011), (101), (020), (200), (111), (121), (220), (002), (130), (221) lattice planes for the orthorhombic phase of β-PtO2 (ICDD No. 01–073-2361) and the absence of diffraction peaks associated with metallic PtNPs is also consistent with their small size and high dispersion, as described below. TEM micrographs of 2a and 2b showed that the platinum and ruthenium nanoparticles were ultrafine and near monodisperse with average diameters of 1.70 ± 0.5 nm and 2.46 ± 0.4 nm, respectively; illustrative micrographs and the corresponding distribution histograms resulting from sizing of > 100 particles are shown in Fig. 3 a-f.

Pt 4f core level XPS spectra of (a) 1a and (b) 2a and (c) Ru 3p XPS spectrum of 2b

TEM images and corresponding sizing histograms of (a-c) PtNPs for 2a and (d-f) RuNPs for 2b determined by counting > 100 particles
2.2 PtNP and RuNP Catalysed Hydrogen Evolution from Dimethylamine Borane, Ammonia Borane and NaBH4
Having recently reported that PtNPs and RuNPs stabilised by heteroatom donor decorated polymer immobilised ionic liquids are efficient catalysts for the hydrolytic evolution of hydrogen from sodium borohydride, this project was extended to compare the catalytic efficacy of amino-decorated PIIL-stabilized Pt and Ru nanoparticles for the hydrolysis of DMAB, an inexpensive model substrate for ammonia borane, and discovered that RuNP@NH2PEGPIILS (2b) was a markedly more efficient catalyst than its platinum counterpart 2a. Preliminary testing was conducted using 0.25 mol% of 2a-b to catalyse the hydrolytic liberation of hydrogen from 0.27 M DMAB at 313 K (Fig. 4a). Reactions were monitored by measuring the amount of hydrogen liberated with time by displacement of water from an inverted burette; the data was subsequently corrected by accounting for the background volume of hydrogen liberated under the same conditions at the same time in the absence of catalyst. The data in Fig. 4a shows that RuNP@NH2-PEGPIILS (2b) is a markedly more efficient catalyst for the hydrolysis of DMAB than its platinum counterpart (2a) as the former reached ca. 99% completion after 20 min with an initial total turnover frequency (TOF) of 8,300 moleH2.molRu−1.h−1, whereas the latter only reached 22% conversion after the same time and 38% conversion when the reaction time was extended to 60 min, with an initial total TOF of 3,050 moleH2.molPt−1.h−1. This difference in efficiency between 2a and 2b is even more evident when the initial TOFs are determined based on the estimated surface metal atoms (Table 1). For comparison, a catalytic hydrolysis conducted with 0.25 mol% Ru/C (5 wt%) only achieved 46% conversion after 20 min with an initial TOF of 2,800 moleH2.molRu−1.h−1 while 0.25 mol% Pt/C (5 wt%) only reached 24% conversion after 35 min with an initial TOF of 2,200 moleH2.molPt−1.h−1 (Fig. S1 in the supporting information). Moreover, pre-stirring the Ru/C and Pt/C with a homogeneous solution of NH2-PEGPIIL in water for 12 h prior to performing the hydrolysis only resulted in a marginal improvement in the initial TOF to 3,050 moleH2.molRu−1.h−1 and 2,300 moleH2.molPt−1.h−1, respectively. A reference hydrolysis conducted by replacing the catalyst with 0.25 mol% NH2PEG-PIIL confirmed that the metal nanoparticles were necessary for catalysis.

Hydrolytic liberation of hydrogen from a 0.27 M solution of (a) dimethylamine borane (b) ammonia borane and (c) NaBH4 as a function of time at 313 K catalysed by 0.25 mol% 2a and 2b
Having established that 2a-b catalyse the hydrolysis of DMAB, the same protocol was applied to the hydrolysis of 0.27 M ammonia borane (AB) to assess their relative efficacy and compare their performance as a function of the substrate. Under the same conditions, 2b was also significantly more active than 2a, as measured by the volume of hydrogen released at the onset of the reaction, and the initial total TOFs of 21,200 moleH2.molRu−1.h−1 and 8,500 moleH2.molPt−1.h−1, respectively, and the relative difference in these TOFs as measured by their ratio, is comparable to that for the hydrolysis of DMAB (Table 1). Moreover, the initial rates of hydrolysis of AB for catalysts 2a and 2b are considerably faster than those for the hydrolysis of DMAB and near complete conversions were obtained in much shorter reaction times (Table 1). Reassuringly, 2a and 2b are both markedly more active as catalysts for the hydrolytic dehydrogenation of AB than commercially available 5 wt% Ru/C and Pt/C, which both reached 93% conversion, with initial TOFs of 7,750 moleH2.molRu−1.h−1 and 5,700 moleH2.molPt−1.h−1, respectively; the higher activity obtained with Ru/C is in keeping with its efficacy for the hydrolysis of DMAB described above. A review of the literature revealed that initial TOFs for the PtNP-catalysed hydrolysis of AB have been reported from 1,962 h−1 up to 28,800 h−1 whereas those for RuNP based catalysts range from 2,400 h−1 to 43,000 h−1; gratifyingly, 2a and 2b clearly compete with the more active of these. Although a comparison of the relative rates of hydrolysis of DMAB and AB described here with those reported in the literature should be treated with a degree of caution, this survey revealed the same trend in rates in that the hydrolysis of AB is typically much faster than DMAB with both ruthenium and platinum nanoparticles [97, 105, 212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239]. However, while there do not appear to be any direct comparisons between the efficacy of platinum and ruthenium nanoparticles stabilised or confined on the same support, ruthenium nanoparticle-based catalysts appear to give higher initial rates than platinum nanoparticles.
Finally, as NaBH4 is also a promising hydrogen storage material, the efficacy of 2a and 2b as catalysts for the hydrolytic liberation of hydrogen from aqueous 0.27 M sodium borohydride was examined. In stark contrast to DMAB and AB, the initial total TOFs of 5,900 moleH2.molRu−1.h−1 and 6,250 moleH2.molPt−1.h−1 for the hydrolysis of NaBH4, catalysed by 2a and 2b, respectively, are comparable to each other and any difference in catalyst efficacy only manifested itself in the final conversions which reached 92% for 2b and 68% for 2a. The corresponding comparison for the hydrolysis of NaBH4 catalysed by 0.25 mol% Pt/C and Ru/C is consistent with the performance of 2a and 2b as reactions reached 45% and 67% conversion, respectively, and the initial total TOFs of 5,050 moleH2.molPt−1.h−1 and 5,400 moleH2.molRu−1.h−1, respectively, were comparable to each other. In addition, these initial rates are only slightly lower than those obtained with the same catalyst loading of 2a and 2b; whereas, in contrast, 2a and 2b were distinctly more active as catalysts for the hydrolytic dehydrogenation of DMAB and AB than their Ru/C and Pt/C counterparts.
While evaluation of the efficiency of 2a-b as catalysts for the hydrolysis of DMAB against literature reports of other ruthenium nanoparticle-based systems may have limited credibility or value due the disparate protocols and conditions, a review of the most recent literature revealed that 2b appears to be among the most active ruthenium nanoparticle-based catalysts for the aqueous phase dehydrogenation of DMAB. For example, the initial TOF of 8,300 h−1 is a considerable improvement on 896 h−1, 403 h−1 362 h−1, obtained with RuNPs stabilised by a PVP-graphene hybrd support, PVP and GO, respectively [240], 812 h−1 with ceria supported RuNPs [52], 51 h−1 and 56 h˗1 for Ru(acac)3 derived nanoclusters generated in the presence of Al2O3 and PVP, respectively [56], 282 h−1 with a graphite-supported ruthenium nanocatalyst [241], 59 h−1 for RuNPs stabilized by confinement in the metal organic framework ZIF-8 [109], 173 h−1 for starch stabilised RuNPs [242], and 46 h−1 for the solventless dehydrogenation of DMAB with RuNPs loaded on cellulose [54], as well as a host of bi- and trimetallic nanoparticles stabilised by reduced graphene oxide [45], PVP [41, 42, 95, 96], multi-walled carbon nanotubes [111], graphene oxide [58, 97,98,99, 243], polymer hydrogels [244], and silica [50]. While it is also slightly greater than the 7,500 h−1 obtained with PtNPs immobilised onto Ni(OH)2 colloid [40] it is significantly lower than 14,926 h−1 for platinum-ruthenium nanoparticles decorated on Vulcan carbon, which is currently the most efficient supported nanoparticle catalyst for the hydrolytic evolution of hydrogen from DMAB [49]. To the best of our knowledge, the initial TOF of 8,300 h−1 obtained with 2b is the highest to be reported for the aqueous phase hydrolytic dehydrogenation of DMAB catalysed by a monometallic RuNP-based catalyst, which may be due to a combination of the small size of the nanoparticles and the hydrophilic environment created by the PEGylated support which would affect dispersion of the catalyst in the aqueous phase as well as facilitate formation of the surface-coordinated hydrogen bonded array [NMe2HBH2-H–-H-OH] responsible for activation of one of the H-OH bonds in water. While there are relatively few literature reports of the hydrolysis of DMAB using PtNP based catalysts, the total TOF of 3,050 h−1 obtained with 2a is markedly higher than 49.2 h−1 with PtNPs decorated with a polyaniline-reduced graphene oxide composite [97] and 59 h−1 with platinum supported on Vulcan carbon [49] but lower than that of 4,151 h−1 obtained with Pt/C alloyed with nickel and 7,500 h−1 for PtNPs immobilised onto Ni(OH)2 colloid [40].
The disparate performance of 2a and 2b as catalysts for the hydrolysis of DMAB and AB compared with their comparable performance for the hydrolysis of NaBH4 prompted us to conduct a comparison of the kinetics on each of the reactions to investigate the rates as a function of temperature to determine the activation energies and to explore the effect of the catalyst and the DMAB/AB/NaBH4 concentration on the rate of the hydrolysis. A series of hydrolytic reactions were performed over a range of temperatures between 294 and 318 K and the initial rate of hydrogen evolution monitored as a function of time. The apparent activation energies (Ea) for the hydrolysis of 0.27 M DMAB using a 0.25 mol% loading of 2a and 2b were calculated to be 74.6 kJ mol−1 and 55.7 kJ mol−1, respectively, from the Arrhenius plot of ln(k) against 1/T shown in Fig. 5c-d; the initial rates were obtained from the linear region of the graphs in Fig. 5a-b. Such disparate activation energies are entirely consistent with the relative catalytic efficacy of 2a and 2b, as measured by the initial TOFs of 3050 moleH2.molPt−1.h−1 and 8,300 moleH2.molRu−1.h−1, respectively (vide supra). The corresponding activation energies for the hydrolysis of aqueous 0.27 M ammonia borane under otherwise identical conditions and across a similar range of temperatures, were determined to be 35.7 kJ mol−1 and 27.9 kJ mol−1 for 2a and 2b, respectively; these values are substantially lower than those for the hydrolysis of DMAB and reflect the higher rates of hydrolysis of AB under similar conditions. For comparison, the apparent activation energies of 44.1 kJ mol−1 and 46.5 kJ mol−1 for the hydrolysis of 0.27 M sodium borohydride catalysed by 2a and 2b, respectively, are similar and consistent with their comparable initial TOFs of 6,200 moleH2.molRu−1.h−1 and 5,900 moleH2.molPt−1.h−1, respectively. The Arrhenius plots and the graphs showing the volume of hydrogen against time associated with the hydrolytic dehydrogenation of AB and NaBH4 are available in the supporting information (Figs. S2-S3).

a Plots of volume of hydrogen liberated against reaction time for the hydrolysis of 2 mL of 0.27 M DMAB across a range of temperatures catalysed by (a) 0.25 mol% 2a and (b) 0.25 mol% 2b and the corresponding Arrhenius plots for the hydrolysis of DMAB catalysed by (c) 2a and (d) 2b; the initial rates (k) were calculated from the slopes of the fitted lines. Each hydrolysis was conducted in triplicate. Initial rate (k) = molH2.min−1
The hydrolytic dehydrogenation of DMAB was further explored by investigating the activity as a function of the concentration of 2a and 2b across various catalyst concentrations in 0.27 M DMAB. The logarithmic plot of the initial hydrogen generation rate against the catalyst loading were straight lines with slopes of 1.04 for 2a and 0.96 for 2b, confirming that the hydrolysis of DMAB is first order with respect to both catalysts (Fig. 6c-d). Similarly, kinetic studies for the hydrolysis of AB conducted with different concentrations of 2a and 2b were consistent with first order kinetics as evidenced from the slopes of 0.98 and 0.91, respectively, for the plot of the hydrogen generation rate against the catalyst concentration on the logarithmic scale (Fig. S4). A review of the recent literature revealed that this data is in line with related reports of noble metal nanoparticle catalysed hydrolysis of DMAB and AB including slopes of 1.08 for ceria supported RuNPs [52], 1.03 for RuNPs loaded on cellulose [54], 0.86 for RuNP stabilised by a graphene oxide-PVP hybrid support [240], 1.01 for RuCu alloy NPs immobilised on reduced graphene oxide [45], 0.98 for RuPd@GO and 1.04 for RuNiPd nanoclusters immobilised on horse chestnut seed [51]. The corresponding study for the hydrolysis of a 0.27 M solution of sodium borohydride as a function of the concentration of 2a and 2b also gave straight line plots for the logarithm of the initial hydrogen generation rate against catalyst loading with slopes of 1.07 and 0.99, respectively, again these slopes indicate first order kinetics; details of which are available in Figures S4-S5 of the supporting information.

a and (b) Volume of hydrogen generated as a function of time for the hydrolytic dehydrogenation of 2 mL of aqueous 0.27 M DMAB at 303 K catalysed by various concentrations of 2a and 2b, respectively; c and (d) plots of initial hydrogen generation rate versus logarithm of the catalyst loading for 2a and 2b, respectively. The initial rates (k) were determined from the slopes of the fitted lines. Each hydrolysis was conducted in triplicate. Initial rate (k) = molH2.min−1
A study of the effect of the initial substrate concentration on the rate of hydrolysis of DMAB was also performed with 17 μmol of 2a and 2b and changing the initial concentration of DMAB ([DMAB]0 = 0.085 mM to 0.51 mM) to obtain comparative kinetic data across catalyst/substrate ratios between 1:1 and 1:5 (Fig. 7); such low catalyst to substrate mole ratios were used to avoid saturation of the surface active sites, which would otherwise give zero order kinetics. The plot of the volume of hydrogen generated against time for each substrate concentration (Fig. 7a-b) was used to determine the initial rates. The corresponding logarithmic plots of the hydrogen generation rate against DMAB concentration gave slopes of 0.99 and 1.12 for 2a and 2b, respectively, confirming that the hydrolysis is first order in substrate and that activation of DMAB is integral to the rate limiting step (Fig. 7c-d). Similar first order kinetics for the hydrolysis of DMAB or ethylenediamine bisborane have been reported for RuNPs supported on graphene oxide [58], cellulose [54], 2-hydroxyethyl starch-p-(2-acrylamide-2-methyl-1-propanesulfonic acid) hydrogel network [61], monodisperse graphite [241], a graphene oxide PVP hybrid [240], and oleylamine [245], as well as PtRu nanocatalysts supported on Vulcan carbon [49], graphene oxide stabilised PdCoNPs [44], monodispersed palladium-ruthenium alloy nanoparticles assembled on PVP [42], silica-based gold-nickel nanohybrid [50], a MOF decorated with monodisperse palladium-cobalt nanohybrids [104] and RuCu nanomaterials on reduced graphene oxide [45]. The corresponding study on the kinetics of hydrolysis of AB as a function of the substrate concentration also gave straight line plots for the logarithm of the initial hydrogen generation rate versus the concentration of AB with slopes of 1.08 and 1.03 for 2a and 2b, respectively; consistent with activation of AB in the rate limiting step (Fig. S6 in the supporting information). However, interestingly, while there have been reports of first order kinetics for the hydrolysis AB [232], the majority of studies appear to report that the rate of hydrolysis of AB does not depend on the substrate concentration, i.e. activation of AB is not involved in the rate determining step; in such cases the rate limiting step has been proposed to involve activation of an O–H bond of water, which may well be facilitated by a hydrogen bonding interaction between a hydrogen atom of water and either a hydridic B-H bond of a surface coordinated AB [246,247,248,249] or a hydridic NP-H [250, 251]. Under the same conditions, the corresponding kinetic studies for the hydrolytic dehydrogenation of NaBH4 using 26 μmol of catalysts 2a and 2b and changing the initial concentration of NaBH4 to afford catalyst:substrate ratios ranging from 1:1 to 1:6 gave slopes of 1.01 and 0.97 for 2a and 2b, respectively, for the logarithmic plot of the initial hydrogen generation rate versus concentration of NaBH4, meaning that this hydrolysis is also first order in NaBH4; complete details are available in Figure S7 of the supporting information.

a-b Plots of volume of hydrogen liberated against time for the hydrolytic dehydrogenation of DMAB at 303 K catalysed by 2a and 2b (17 μmol) in water (200 mL), initial concentrations of DMAB ([DMAB]0 = 0.085, 0.17, 0.25, 0.34, 0.42, 0.51 mM); c-d plots of the hydrogen generation rate against initial concentration of DMAB in logarithmic scale. The initial rates (k) were determined from the slopes of the fitted lines. Each hydrolysis was conducted in triplicate. Initial rate (k) = molH2.min−1
Kinetic isotope effect studies have proven to be an informative tool for elucidating information about the key rate limiting step (RLS) of the nanoparticle catalysed hydrolytic liberation of hydrogen from ammonia borane and sodium borohydride. While several mechanistic scenarios have been proposed for the hydrolysis of ammonia boranes including (1) rate limiting formation of an activated surface coordinated ammonia borane followed by attack of water to cleave the B-N bond and hydrolysis of the BH3 [252], (2) formation of BH3OHNH4 by attack of water held proximal to a surface coordinated AB [253], and (3) dissociation of the B-N bond by attack of water at an activated surface bound ammonia borane followed by release of H2 through attack of water on a transient hydridic NP-H, in much the same manner as the metal catalysed hydrolysis of NaBH4 [254], and (4) rate limiting activation of one of the O–H bonds of water in a hydrogen-bonded array involving a hydridic B-H bond of a surface-coordinated ammonia borane and a water proton [246,247,248], it is clear that ammonia borane and NaBH4 both act as sources of hydride and provide one hydrogen atom to the derived hydrogen while water provides the other in the form of a proton.
The role of water in the hydrolysis of DMAB catalysed by 2a and 2b was investigated by performing reactions in H2O and D2O and monitoring the amount of hydrogen liberated with time to compare the initial rates and thereby determine the KIE. Reactions were conducted under the same conditions described above using 0.25 mol% 2a and 2b to catalyse the hydrolysis of 0.27 M DMAB at 303 K. A comparison of the initial rates of hydrolysis of DMAB in H2O and D2O catalysed by 2a and 2b showed that reactions were faster in H2O than in D2O with primary kinetic isotope effects (kH/kD) of 2.5 and 3.8, respectively (Fig. 8a-b); similar values of kH/kD were also obtained for the catalytic hydrolysis of a 0.27 M solution of AB in H2O and D2O using 0.25 mol% 2a (kH/kD = 2.6) and 2b (kH/kD = 4.0) and the associated data is presented in Figure S8a-b while that for the hydrolysis of 0.27 M NaBH4 in H2O and D2O is presented in Figure S9a-b. These values for the KIE are comparable to that of 4.95 reported for the Ni2Pt@ZIF-8 catalysed hydrolysis of ammonia borane [255] as well as atomically disperse platinum on the surface of Ni particles (kH/kD = 4.0) [224], CoNPs supported on a covalent triazene framework (kH/kD = 2.8) [256], ultrasmall Pt nanoclusters on heterostructured NiO/Ni (kH/kD = 2.8) [257], CoPt nanocatalysts encapsulated in click dendrimer (kH/kD = 2.46) [249] and graphene oxide supported RhNPs (kH/kD = 2.6) [258] and is consistent with a mechanism involving rate limiting activation of one of the O–H (or O-D) bonds in water, which is activated/weakened by hydrogen bonding to one of the hydridic hydrogen atoms in DMAB.

Hydrogen evolution from 2 mL of a 0.27 M solution of DMAB in H2O (red) and D2O (blue) at 303 K catalysed by 0.25 mol% (a) 2a and (b) 2b
While these KIE values are consistent with previously reported mechanisms in which a surface-coordinated hydrogen bonded array of the type Me2NH-BH2-H–––H––OH, involving a hydridic B-H hydrogen and a proton on water, activates the O–H bond toward oxidative addition to generate a water derived M-H and a surface-coordinated DMAB, as shown in Fig. 9, it does not distinguish between oxidative addition of an O–H bond coupled with concerted hydride transfer from the DMAB (Fig. 9 path a) and a double oxidative addition of O–H and B-H bonds (Fig. 9 path c). The former pathway would liberate the hydrogen via reductive elimination of a water derived M-H and a DMAB derived M-H and generate NMe2HBH2-OH as an intermediate via electrophilic abstraction of a surface coordinated M-OH by the [NMe2HBH2]+ cation (Fig. 9 path b), while the latter pathway would liberate the hydrogen and Me2HBH2-OH via reductive elimination (Fig. 9 path d). The remaining two B-H bonds in the monohydroxylated boron intermediate would subsequently activate in a similar manner to ultimately afford [NMe2H]2[BO2]. While the above pathways are initiated by rate limiting oxidative addition of an O–H bond of water in a cooperative surface-coordinated hydrogen bonded array, we cannot unambiguously exclude the possibility that an O–H bond could be activated by hydrogen bonding between a surface adsorbed water and a DMAB-derived transient metal hydride, as described by Jagidar for the copper and Cu@Cu2O core shell nanoparticle catalysed hydrolysis of sodium borohydride [254] and Guella for the palladium catalysed hydrolysis of NaBH4 [259]. Alternatively, the hydrogen may be liberated via rate limiting activation of an O–H bond in water followed by a concerted σ-bond metathesis-type process between the resulting water-derived M-H and the σ-bonded B-H of a surface-coordinated DMAB (Fig. 9 path e–f).

Proposed pathways for the metal nanoparticle catalysed hydrolytic evolution of hydrogen from the hydrogen bonded array Me2NH-BH2-H–––H––OH via; a-b rate limiting oxidative addition of an O–H bond and hydride transfer followed by reductive elimination of H2 and abstraction of a surface hydroxide (c-d) double oxidative addition involving O–H and B-H bonds and subsequent reductive elimination of H2 and [HO-BH2NMe2H] and (e–f) activation of an O–H bond followed by a σ-bond metathesis type process between the water derived M-H and the weakly σ-coordinated B-H of a DMAB
Recycle studies were conducted using 2b to catalyse the catalytic hydrolytic evolution of DMAB to assess its activity profile as a function of reuse and thereby determine its longevity and potential for integration into a flow-based system. The practical challenges encountered with filtering and recovering the small quantity of catalyst required for these reactions (2.0 mg, 0.25 mol%) meant that it was not feasible to conduct a recycle study on this scale. Thus, a catalytic reaction using 2 mol% 2b to hydrolyse 20 mL of 0.027 M DMAB was monitored until gas evolution ceased after which the aqueous solution was charged with an additional portion of DMAB, and the progress of the reaction monitored; this sequence was repeated to chart the hydrogen evolution against time as a function of run number. The resulting data in Fig. 10a shows the hydrogen evolution time profile as a function of run number while the initial TOFs in Fig. 10b were determined from the first 3 min of the hydrolysis to provide a reliable comparison of the initial rates between successive runs. While high conversions were obtained across five reuses (84–88%), the initial TOF increased from 505 h−1 in the first run to 781 h−1 in the second run but then remained relatively constant in subsequent runs (Fig. 10b), retaining 97% of its initial activity across the next 4 runs. Such an increase in activity during the first run could be due either to reduction of surface ruthenium oxide species increasing the number of active sites or a structural change. Good conversions and stable activity profiles during reuse have also been reported for the catalytic hydrolytic evolution of hydrogen from DMAB using supported nanoparticles including ultrafine RuNPs anchored on porous g-C3N4, [230], cellulose [54], PVP [56] and Amberlyst-15 [226,227,228,229,230,231,232,233,234,235,236,237,238,239], nickel-polymer nanogel hybrid particles [47], reduced graphene oxide decorated with monodisperse RuCu alloy nanoparticles [45] whereas the initial activity of ceria supported RuNPs [52], PVP supported Ru and RuNi nanoparticles [41, 57] and monodisperse PdNPs as well as bimetallic PdCo and PdRu nanoparticles anchored on graphene oxide [44, 46, 58] all showed a significant reduction in activity across four or five reuses.

a Plot of volume of hydrogen liberated against time for the catalytic hydrolysis of 0.027 M DMAB using 2 mol% 2b to map catalyst performance during a reusability study across 5 runs; b initial TOF in moleH2.molRu−1.h−1 for each run. Volumes are an average of two runs. Conditions: 0.54 mmol DMAB (0.032 g), 2 mol% 2b (0.0149 g, 0.0103 mmol), water (20 mL), 303 K
The heterogenous nature of the active species was investigated with a hot filtration study in which a catalytic hydrolysis of 0.27 M DMAB using 0.25 ml% 2b was allowed to reach ca. 50% conversion after which the reaction mixture was then filtered through a syringe filter and the evolution of hydrogen from the recovered filtrate monitored for a further 30 min. Figure 11a shows that there was no evidence of post filtration gas evolution which is a strong indication that the active ruthenium species had been removed by the filtration. In a separate parallel hot filtration test, a catalytic hydrolysis of 0.27 M DMAB was allowed to reach completion and the reaction mixture subsequently filtered through a syringe filter, the filtrate was then re-charged with an additional portion of DMAB, and the gas evolution monitored. The amount of hydrogen liberated after filtration was quantified and found to correspond to background hydrolysis, providing additional support that the active species was heterogeneous. Moreover, the ruthenium content of the aqueous phase retrieved after the hot filtration was below the detection limit of the ICP-OES (< 0.1 mg L−1), which also suggests that leaching of the ruthenium to generate inactive or less active homogeneous species is unlikely. However, these experiments do not differentiate between heterogeneous catalysis at defect sites on the surface of the NP and homogeneous catalysis via a process involving rapid leaching to generate active homogeneous species that are subsequently redeposited. While the PEG-modified imidazolium component was designed with a C2-methyl substituent to prevent the formation of metal carbenes, we cannot unequivocally exclude the formation of an abnormal N-heterocyclic carbene (aNHC) species [260] from the leached ruthenium which would then be supported and retained by the polymer; as such there would be no apparent leaching of the metal as it would all be immobilised in the form of RuNPs and molecular carbene species. To this end, there have been numerous reports of the serendipitous formation of ruthenium-based abnormal carbenes during the attempted synthesis of normal N-heterocyclic carbene complexes as well as targeted synthesis for applications in catalysis as their strong σ-donor character is expected to improve performance [261,262,263,264,265,266]. Finally, TEM analysis of the catalyst recovered after the 5th run revealed that the nanoparticles remained near monodisperse with a mean diameter of 1.96 ± 0.4 nm, comparable to that of 2.46 ± 0.4 nm for freshly prepared 2b, confirming that agglomeration does not occur and that leaching and re-deposition is unlikely as this would probably result in an obvious change in the size and/or distribution of the NPs (Fig. 11b-d).

a Hot filtration test for the hydrolysis of 0.27 M DMAB catalysed by 0.25 mol% 2b showing that the activity is quenched after filtering at t = 12 min. Red line – reaction in the presence of catalyst; blue line – reaction catalysed by 2b and filtered at t = 12 min. b Sizing histogram of RuNPs for 2b after 5 hydrolysis cycles. c-d TEM images of the material
3 Conclusions
In conclusion, a systematic comparison of the efficacy of amino-modified PIIL stabilised platinum and ruthenium nanoparticles as catalysts for the hydrolytic evolution of hydrogen from DMAB and AB and NABH4 has been undertaken. Interestingly, while the RuNP@NH2-PEGPIILS is significantly more active than its platinum counterpart for the hydrolysis of DMAB and AB, both catalysts gave comparable initial TOFs and conversions for the hydrolysis of NaBH4. Kinetic studies revealed that the hydrolysis of DMAB, AB and NaBH4 were all first order in catalyst and substrate. The apparent activation energies of 55.7 kJ mol−1 and 27.9 kJ mol−1 for the hydrolysis of DMAB and AB, respectively, with RuNP@NH2-PEGPIILS as the catalyst are significantly lower than those of 74.6 kJ mol−1 and 35.7 kJ mol−1 for its platinum counterpart, which is consistent with the markedly higher initial TOFs obtained with the RuNPs for both substrates. Moreover, both activation energies for the hydrolysis of AB are markedly lower than the respective activation energies for the hydrolysis of DMAB which is consistent with the markedly higher rates of hydrogen evolution from the former. In stark contrast to the hydrolysis of DMAB and AB, both catalysts gave comparable initial rates for the hydrolysis of NaBH4 which was supported by their similar apparent activation energies. One of the remaining challenges hampering the implementation of sodium borohydride for hydrogen storage is the need to close the loop by effecting its regeneration from the hydrolysis product, NaBO2. To this end, facile regeneration of NaBH4 has recently been achieved by reacting NaBO2 with CO2 to form Na2B4O7.10H2O and Na2CO3 both of which can be ball-milled with magnesium under ambient conditions to afford NaBH4 in high yield [267]. This is an efficient, low-cost procedure compared to previously reported methods which either require an expensive reducing agent such as MgH2, an energy intensive dehydration step or a high pressure of hydrogen [26]. As the modular synthesis of the PIIL support will facilitate further diversification we will next explore the influence of varying the amine and its loading as well as the ionic environment, porosity, and hydrophilicity on catalyst efficacy. Catalyst modifications coupled with in operando surface studies and the synthesis of multimetallic nanoparticles will also be the focus of future endeavours to develop a detailed understanding of this system and to identify more active and robust catalysts for use in scale-up.
References
Nejat P, Jomehzadeh F, Taheri MM, Gohari M, Abd. Majid MZ (2015) A global review of energy consumption, CO2 emissions and policy in the residential sector (with an overview of the top ten CO2 emitting countries). Renew Sustain Energy Rev 43:843–862
Davis SJ, Caldeira K (2010) Consumption-based accounting of CO2 emissions. Proc Nat Acad Sci 107:5687–5692
CO2.earth are we stabilizing yet? Prooxygen web site https://www.co2.earth/. Accessed 20 Sept 2021
Olivier JGJ, Peters JAHW (n.d.) Trends in global CO2 and total greenhouse gas emissions 2019 report. PBL Netherlands Environmental Assessment Agency The Hague, PBL publication number: 4068
Schlapbach L, Züttel A (2001) Hydrogen-storage materials for mobile applications. Nature 414:353–358
Rodríguez-Lugo RE, Trincado M, Vogt M, Tewes F, Santiso-Quinones G, Grützmacher H (2013) A homogeneous transition metal complex for clean hydrogen production from methanol–water mixtures. Nat Chem 5:342–347
He T, Pachfule P, Wu H, Xu Q, Chen P (2016) Hydrogen carriers. Nat Rev Mater 1:16059
Ball M, Wietschel M (2009) The future of hydrogen – opportunities and challenges. Int J Hydrogen Energy 34:615–627
https://www.energy.gov/eere/fuelcells/doe-technical-targets-onboard-hydrogen-storage-light-duty-vehicles. Accessed 24 Oct 2023
Mazloomi K, Gomes C (2012) Hydrogen as an energy carrier: prospects and challenges. Renew Sustain Energy Rev 16:3024–3033
Abdin Z, Zafaranloo A, Rafiee A, Mérida W, Lipiński W, Khalilpour KR (2020) Hydrogen as an energy vector. Renew Sustain Energy Rev 120:109620
Hirscher M, Yartys VA, BariccoJose M, von Colbe B, Blanchard D, Bowman RC, Broom DP, Buckley CE, Chang F, Chen P, Cho YW, Crivello JC, Cuevas F, David WIF, de Jongh PE, Denys RV, Dornheim M, Felderhoff M, Filinchuk Y, Froudakis GE, Grant DM et al (2020) Materials for hydrogen-based energy storage – past, recent progress and future outlook. J Alloys Compd 827:153548
Abbasi R, Setzler BP, Lin S, Wang J, Zhao Y, Xu H, Pivovar B, Tian B, Chen X, Wu G, Yan Y (2019) A Roadmap to low-cost hydrogen with hydroxide exchange membrane electrolyzers. Adv Mater 31:1805876
Vincent I, Bessarabov D (2018) Low-cost hydrogen production by anion exchange membrane electrolysis: a review. Renew Sustain Energy Rev 81:1690–1704
Ren J, Musyoka NN, Langmi HW, Mathe M, Liao S (2017) Current research trends and perspectives on materials-based hydrogen storage solutions: a critical review. Int J Hydrogen Energy 42:289–311
Rivard E, Trudeau M, Zaghib K (2019) Hydrogen storage for mobility: a review. Materials 12:1973
Demirci UB, Miele P (2011) Chemical hydrogen storage: ‘material’ gravimetric capacity versus ‘system’ gravimetric capacity. Energy Environ Sci 4:3334–3341
Schneemann A, White JL, Kang SY et al (2018) Nanostructured metal hydrides for hydrogen storage. Chem Rev 118:10755–10839
Yu X, Tang Z, Sun D, Ouyang L, Zhu M (2017) Recent advances and remaining challenges of nanostructured materials for hydrogen storage applications. Progress Mater Sci 88:1–48
Wang C, Astruc D (2021) Recent developments of nanocatalyzed liquid-phase hydrogen generation. Chem Soc Rev 50:3437–3484
Eberle U, Felderhoff M, Schueth F (2009) Chemical and physical solutions for hydrogen storage. Angew Chem Int Ed 48:6608–6630
Sun Q, Wang N, Xu Q, Yu J (2020) Nanopore-supported metal nanocatalysts for efficient hydrogen generation from liquid-phase chemical hydrogen storage materials. Adv Mater 32:2001818
Lang C, Jia Y, Yao X (2020) Recent advances in liquid-phase chemical hydrogen storage. Energy Storage Mater 26:290–312
Wang C, Wang Q, Fu F, Astruc D (2020) Hydrogen generation upon nanocatalyzed hydrolysis of hydrogen-rich boron derivatives: recent developments. Acc Chem Res 53:2483–2493
Abdelhamid HN (2021) A review on hydrogen generation from the hydrolysis of sodium borohydride. Int J Hydrog Energy 46:726–765
Muir SS, Yao X (2011) Progress in sodium borohydride as a hydrogen storage material: development of hydrolysis catalysts and reaction systems. Int J Hydrogen Energy 36:5983–5997
Santos DMG, Sequeira CAC (n.d.) Sodium borohydride as a fuel for the future. Rev Sustain Energy Rev 15:3980–4001
Liu BH, Li ZP (2009) A review: hydrogen generation from borohydride hydrolysis reaction. J Power Sources 187:527–534
Brack P, Dann SE, Wijayantha KGU (2015) Heterogeneous and homogenous catalysts for hydrogen generation by hydrolysis of aqueous sodium borohydride (NaBH4) solutions. Energy Sci Eng 3:17–88
Patel N, Miotello A (2015) Progress in Co–B related catalyst for hydrogen production by hydrolysis of boron-hydrides: a review and the perspectives to substitute noble metals. Int J Hydrog Energy 40:1429–1464
Staubitz A, Robertson APM, Manners I (2010) Ammonia-borane and related compounds as dihydrogen sources. Chem Rev 110:4079–4124
Hamilton CW, Baker RT, Staubitz A, Manners I (2009) B-N compounds for chemical hydrogen storage. Chem Soc Rev 38:279–293
Yadav M, Xu Q (2012) Liquid-phase chemical hydrogen storage materials. Energy Mater Sci 5:9698–9725
Zhan WW, Zhu QL, Xu Q (2016) Dehydrogenation of ammonia borane by metal nanoparticle catalysts. ACS Catal 6:6892–6905
Akbayraka S, Özkar S (2018) Ammonia borane as hydrogen storage materials. Int J Hydrogen Energy 43:18592–18606
Yüksel Y, Senem A, Gülbay K, Colpan CO (2020) A review on the catalysts used for hydrogen production from ammonia borane. Int J Hydrog Energy 45:3414–3434
Navlani-García M, Salinas-Torres D, Cazorla-Amorós D (2021) Hydrolytic dehydrogenation of ammonia borane attained by Ru-based catalysts: an auspicious option to produce hydrogen from a solid hydrogen carrier molecule. Energies 14:2199
Demirci U (2020) Ammonia borane: an extensively studied, though not yet implemented. Hydrog Carrier Energies 13:3071
Huo J, Zhang K, Wei H, Fu L, Zhao C, He C, Hu X (2023) A review on hydrogen production from ammonia borane: experimental and theoretical studies. Chin Chem Lett 34:108280
Zhang Q, Xu F, Huang H, Wang Y, Liu X (2022) Hydrogen production upon the hydrolysis of dimethylamine borane over Pt/Ni(OH)2 nanocomposite. Fuel 324:124695
Sen B, Kuyuldar E, Demirkan B, Okyay TO, Şavk A, Sen F (2018) Highly efficient polymer supported monodisperse ruthenium-nickel nanocomposites for dehydrocoupling of dimethylamine borane. J Colloid Interface Sci 526:480–486
Sen B, Aygün A, Fellah MF, Calimli MH, Sen F (2019) Highly monodispersed palladium-ruthenium alloy nanoparticles assembled on poly(N-vinyl-pyrrolidone) for dehydrocoupling of dimethylamine–borane: an experimental and density functional theory study. J Colloid Interface Sci 546:83–91
Günbatar S, Aygun A, Karataş Y, Gülcan M, Şen F (2018) Carbon-nanotube-based rhodium nanoparticles as highly-active catalyst for hydrolytic dehydrogenation of dimethylamine borane at room temperature. J Colloid Interface Sci 530:321–327
Şen B, Aygün A, Şavk A, Çalımlı MH, Gülbay SK, Şen F (2020) Bimetallic palladium-cobalt nanomaterials as highly efficient catalysts for dehydrocoupling of dimethylamine borane. Int J Hydrog Energy 45:3569–3576
Sen B, Kuyuldar E, Şavk A, Calimli MH, Duman S, Sen F (2019) Monodisperse rutheniumcopper alloy nanoparticles decorated on reduced graphene oxide for dehydrogenation of DMAB. Int J Hydrog Energy 44:10744–10751
Şen B, Aygün A, Okyay TO, Şavk A, Kartop R, Şen F (2018) Monodisperse palladium nanoparticles assembled on graphene oxide with the high catalytic activity and reusability in the dehydrogenation of dimethylamine-borane. Int J Hydrog Energy 43:20176–20182
Cai H, Liu L, Chen Q, Lu P, Dong J (2016) Ni-polymer nanogel hybrid particles: a new strategy for hydrogen production from the hydrolysis of dimethylamine-borane and sodium borohydride. Energy 99:129–135
Geniş D, Filiz BC, Depren SK, Figen AK (2020) Reusable hybrid foam catalyst for hydrolytic dehydrogenation of amine adducts of borane: porous PVA-Immobilized Co–Ru nanoparticles. Microporous Mesoporous Mater 305:110363
Karatas J, Acidereli H, Gulcan M, Sen F (2020) A novel highly active and reusable carbon-based platinum-ruthenium nanocatalyst for dimethylamine-borane dehydrogenation in water at room conditions. Sci Reports 10:7149
Alptekin O, Sen B, Savk A, Ercetin U, Mustafov SD, Fellah MF, Sen F (2020) Use of silica-based homogeneously distributed gold nickel nanohybrid as a stable nanocatalyst for the hydrogen production from the dimethylamine borane. Sci Rep 10:7215
Ulutas K, Alshawesh M, Duman S (2022) Eco-friendly dehydrogenation of dimethylamine-borane catalyzed by core-shell-looking tri-metallic RuNiPd nanoclusters loaded on white-flowering horse-chestnut seed. Int J Hydrog Energy 47:38198–38218
Karaboga S, Özkar S (2019) Ceria supported ruthenium nanoparticles: remarkable catalyst for H2 evolution from dimethylamine borane. Int J Hydrog Energy 44:26296–26307
Unlu D, Hilmioglu ND (2020) Application of aspen plus to renewable hydrogen production from glycerol by steam reforming. Int J Hydrog Energy 45:3509–3576
Ozhava D, Duman S (2023) Solventless dimethylamine borane dehydrogenation in the presence of transition metal(0) nanoparticles loaded on cellulose. ChemCatChem 15:e202201563
Şen B, Demirkan B, Levent M, Şavk A, Şen F (2018) Silica-based monodisperse PdCo nanohybrids as highly efficient and stable nanocatalyst for hydrogen evolution reaction. Int J Hydrog Energy 43:20234–20242
Bukan B, Duman S (2018) Green dehydrogenation of dimethylamine-borane catalyzed by in situ generated ruthenium nanoclusters in presence of various supporters and its comparison with classical methods. Int J Hydrog Energy 43:8278–8289
Karataş Y, Aygun A, Gülcan M, Şen F (2019) A new highly active polymer supported ruthenium nanocatalyst for the hydrolytic dehydrogenation of dimethylamine-borane. J Taiwan Chem Eng 99:60–65
Acidereli H, Cellat K, Calimli MH, Sen F (2021) Palladium/ruthenium supported on graphene oxide (PdRu@GO) as an efficient, stable and rapid catalyst for hydrogen production from DMAB under room conditions. Renew Energy 161:200–206
Xu F, Liu X (2021) Synergistically promoted H2 evolution from dimethylamine-borane and hydrazine monohydrate by simply alloying of Pt/C with Ni. Fuel 304:121433
Al-Mahamad GLL (2020) Gold nanoparticles as a catalyst for dehydrogenation reaction of dimethylamine borane at room temperature. Int J Hydrog Energy 45:11916–11922
Ozay H, Ilgin P, Sezgintürk MK, Ozay O (2020) Ruthenium nanoparticles supported in the network of HES-p(AMPS) IPN hydrogel as efficient catalyst for hydrogen production from the hydrolysis of ethylenediamine bisborane. Int J Hydrog Energy 45:9892–9902
Friedrich A, Drees M, Schneider S (2009) Ruthenium-catalyzed dimethylamine borane dehydrogenation: stepwise metal-centered dehydrocyclization. Chem Eur J 15:10339–10342
Sloan ME, Staubitz A, Clark TJ, Russell CA, Lloyd-Jones GC, Manners I (2010) Homogeneous catalytic dehydrocoupling/dehydrogenation of amine−borane adducts by early transition metal, group 4 metallocene complexes. J Am Chem Soc 132:3831–3841
Kawano Y, Uruich M, Shimoi M, Taki S, Kawaguchi T, Akizawa T, Ogino H (2009) Dehydrocoupling reactions of borane−secondary and −primary amine adducts catalyzed by group-6 carbonyl complexes: formation of aminoboranes and borazines. J Am Chem Soc 131:14946
Sloan ME, Clark TJ, Manners I (2009) Homogeneous catalytic dehydrogenation/dehydrocoupling of amine-borane adducts by the Rh(I) Wilkinson’s complex analogue RhCl(PHCy2)3 (Cy = cyclohexyl). Inorg Chem 48:2429–2435
Cory A, Jaska CA, Temple K, Lough AJ, Manners I (2003) Transition metal-catalyzed formation of boron−nitrogen bonds: catalytic dehydrocoupling of amine-borane adducts to form aminoboranes and borazines. J Am Chem Soc 125:9424–9434
Keaton RJ, Blacquiere JM, Baker RT (2007) Base metal catalyzed dehydrogenation of ammonia−borane for chemical hydrogen storage. J Am Chem Soc 129:1844–1845
Li Y, Bastakoti BP, Malgras V, Li C, Tang J, Kim JH, Yamauchi Y (2015) Polymeric micelle assembly for the smart synthesis of mesoporous platinum nanospheres with tunable pore sizes. Angew Chem Int Ed 54:11073–11077
Robertson APM, Suter R, Chabanne L, Whittell GR, Manners I (2011) Heterogeneous dehydrocoupling of amine-borane adducts by skeletal nickel catalysts. Inorg Chem 50:12680–12691
Jaska CA, Manners I (2004) Heterogeneous or homogeneous catalysis? Mechanistic studies of the rhodium-catalyzed dehydrocoupling of amine-borane and phosphine-borane adducts. J Am Chem Soc 126:9776–9785
Pun D, Lobkovsky E, Chirik PJ (2007) Amine-borane dehydrogenation promoted by isolable zirconium sandwich, titanium sandwich and N2 complexes. Chem Commun 3297–3299
Jiang Y, Berke H (2007) Dehydrocoupling of dimethylamine-borane catalysed by rhenium complexes and its application in olefin transfer-hydrogenations. Chem Commun 3571–3573
Clark TJ, Russell CA, Manners I (2006) Homogeneous, titanocene-catalyzed dehydrocoupling of amine−borane adducts. J Am Chem Soc 128:9582–9583
Alcaraz G, Vendier L, Clot E, Sabo-Etienne S (2010) Ruthenium Bis(σ-B-H) aminoborane complexes from dehydrogenation of amine–boranes: trapping of H2B-NH2. Angew Chem Int Ed 49:918–920
Ramachandran PV, Mistry H, Kulkarnia SA, Gagarea PD (2014) Ammonia-mediated, large-scale synthesis of ammonia borane. Dalton Trans 43:11404–11408
Barın EU, Masjedi M, Özkar S (2015) A new homogeneous catalyst for the dehydrogenation of dimethylamine borane starting with Ruthenium(III)Acetylacetonate. Materials 8:3155–3167
Astruc D, Lu F, Aranzaes JR (2005) Nanoparticles as recyclable catalysts: the frontier between homogeneous and heterogeneous catalysis. Angew Chem Int Ed 44:7852–7872
Sir Thomas JM (2010) The advantages of exploring the interface between heterogeneous and homogeneous catalysis. ChemCatChem 2:127–132
Copéret C in green catalysis, Vol. 2, Heterogeneous catalysis (Ed. R.H. Crabtree). Wiley-VCH, 2014, 117
Chen F, Jiang X, Zhang L, Lang R, Qiao B (2018) Single-atom catalysis: bridging the homo- and heterogeneous catalysis. Chin J Catal 39:893–898
Farnetti E, Monte MD, Kašpar J (2009) Homogeneous and heterogeneous catalysis. In: Bertini I (ed) Inorganic and bio‐inorganic chemistry. Encyclopedia of life support systems, vol II. pp 50–87
Copéret C, Chabanas M, Saint-Arroman RP, Basset JM (2003) Homogeneous and heterogeneous catalysis: bridging the gap through surface organometallic chemistry. Angew Chem Int Ed 42:156–181
Yang XF, Wang A, Qiao B, Li J, Liu J, Zhang T (2013) Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc Chem Res 46:1740–1748
Wang N, Sun Q, Yu J (2019) Ultrasmall metal nanoparticles confined within crystalline nanoporous materials: a fascinating class of nanocatalysts. Adv Mater 31:1803966
Somorjai GA, Contreras AM, Montano M, Rioux RM (2006) Clusters, surfaces, and catalysis. PNAS 103:10577–10583
Cao A, Lu R, Veser G (2010) Stabilizing metal nanoparticles for heterogeneous catalysis. PhysChemPhysChem 12:13499–13510
Li Y, Somorjai GA (2010) Nanoscale advances in catalysis and energy applications. Nano Lett 10:2289–2295
Narayanan R, El-Sayed MA (2005) Catalysis with transition metal nanoparticles in colloidal solution: nanoparticle shape dependence and stability. J Phys Chem B 109:12663–12676
Moreno-Mañas M, Pleixats R (2003) Formation of carbon−carbon bonds under catalysis by transition-metal nanoparticles. Acc Chem Res 36:638–643
Gommes CJ (2019) Ostwald ripening of confined nanoparticles: chemomechanical coupling in nanopores. Nanoscale 11:7386–7393
Cheong S, Watt JD, Tilley RD (2010) Shape control of platinum and palladium nanoparticles for catalysis. Nanoscale 2:2045–2053
Goesmann H, Feldmann C (2010) Nanoparticulate functional materials. Angew Chem Int Ed 49:1362–1395
Schmid G (2004) Nanoparticles: from theory to applications. Wiley-VCH, Weinheim, pp 1–359
Gao C, Lyu F, Yin Y (2021) Encapsulated metal nanoparticles for catalysis. Chem Rev 121:834–881 (and references therein)
Çelik B, Yıldız Y, Sert H, Erken E, Koşkun Y, Şen F (2016) Retracted article: monodispersed palladium–cobalt alloy nanoparticles assembled on poly(N-vinyl-pyrrolidone) (PVP) as a highly effective catalyst for dimethylamine borane (DMAB) dehydrocoupling. RSC Adv 6:24097–24102
Tiri RNE, Aygün A, Gülbay SK, Sen F, Cheng CK, Jafarzadeh H, Mehrizi AA, Vasseghian Y (2022) Improving hydrogen generation from dehydrogenation of dimethylamine borane using polyvinylpyrrolidone stabilized platinum-rhodium nanoclusters as highly efficient and reusable catalysts: development of ANN model. Chem Eng Res Des 182:305–311
Sen B, Kuzu S, Demir E, Akocak S, Sen F (2017) Polymer-graphene hybride decorated Pt nanoparticles as highly efficient and reusable catalyst for the dehydrogenation of dimethylamine–borane at room temperature. Int J Hydrog Energy 42:23284–23291
Sen B, Kuzu S, Demir E, Akocak S, Sen F (2017) Monodisperse palladium–nickel alloy nanoparticles assembled on graphene oxide with high catalytic activity and reusability in the dehydrogenation of dimethylamine–borane. Int J Hydrog Energy 42:23276–23283
Sen B, Demirkan B, Şavk A, Gülbay SK, Sen F (2018) Trimetallic PdRuNi nanocomposites decorated on graphene oxide: a superior catalyst for the hydrogen evolution reaction. Int J Hydrog Energy 43:17984–17997
Tanyildizi S, Morkan İ, Özkar S (2017) Nanotitania-supported Rhodium(0) nanoparticles: superb catalyst in dehydrogenation of dimethylamine borane. Chem Select 2:5751–5759
Karaboga S, Özkar S (2019) Nanoalumina supported palladium (0) nanoparticle catalyst for releasing H2 from dimethylamine borane. Appl Surf Sci 487:433–441
Mori K, Taga T, Yamashita H (2015) Synthesis of a Fe–Ni alloy on a ceria support as a noble-metal-free catalyst for hydrogen production from chemical hydrogen storage materials. ChemCatChem 7:1285–1291
Tanyıldızı S, Morkan İ, Özkar S (2017) Ceria supported copper(0) nanoparticles as efficient and cost effective catalysts for the hydrogenation of dimethylamine borane. Mol Catal 434:57–68
Şen B, Aygün A, Şavk A, Yenikaya X, Cevik S, Şen F (2019) Metal-organic frameworks based on monodisperse palladium-cobalt nanohybrids as highly active and reusable nanocatalysts for hydrogen generation. Int J Hydrog Energy 44:2988
Aijaz A, Karkamkar A, Choi AJ, Tsumori N, Rönnebro E, Autrey T, Shioyam H, Xu Q (2012) Immobilizing highly catalytically active Pt nanoparticles inside the pores of metal–organic framework: a double solvents approach. J Am Chem Soc 134:13926–13929
Li Q, Kim H (2012) Hydrogen production from NaBH4 hydrolysis via Co-ZIF-9 catalyst. Fuel Process Technol 100:43–48
Srinivas G, Travis W, Ford J, Wu H, Guo ZX, Yildirim T (2013) Nanoconfined ammonia borane in a flexible metal–organic framework Fe–MIL-53: clean hydrogen release with fast kinetics. J Mater Chem A 1:4167–4172
Srinivas G, Ford J, Zhou W, Yildirim T (2012) Zn-MOF assisted dehydrogenation of ammonia borane: enhanced kinetics and clean hydrogen generation. Int J Hydrog Energy 37:3633–3638
Yurderi M, Bulut A, Zahmakiran M, Gulcan M, Ozkar S (2014) Ruthenium(0) nanoparticles stabilized by metal-organic framework (ZIF-8): highly efficient catalyst for the dehydrogenation of dimethylamine-borane and transfer hydrogenation of unsaturated hydrocarbons using dimethylamine-borane as hydrogen source. Appl Catal B 160–161:534–541
Caliskan S, Zahmakiran M, Durapc F, Özkara S (2012) Hydrogen liberation from the hydrolytic dehydrogenation of dimethylamine–borane at room temperature by using a novel ruthenium nanocatalyst. Dalton Trans 14:4976–4984
Sen B, Kuzu S, Demir E, Akocak S, Sen F (2017) Highly monodisperse RuCo nanoparticles decorated on functionalized multiwalled carbon nanotube with the highest observed catalytic activity in the dehydrogenation of dimethylamine−borane. Int J Hydrog Energy 42:23292
Lu G, Li S, Guo Z, Farha OK, Hauser BG, Qi X, Wang Y, Wang X, Han S, Liu X, DuChene JS, Zhang H, Zhang Q, Chen X, Ma J, Loo SCJ, Wei DW, Yang Y, Hupp JT, Huo F (2012) Imparting functionality to a metal–organic framework material by controlled nanoparticle encapsulation. Nat Chem 4:310–316
Choi KM, Na K, Somorjai GA, Yaghi OM (2015) Chemical environment control and enhanced catalytic performance of platinum nanoparticles embedded in nanocrystalline metal–organic frameworks. J Am Chem Soc 137:7810–7815
Na K, Choi M, Yaghi OM, Somorjai GA (2014) Metal Nanocrystals embedded in single nanocrystals of MOFs give unusual selectivity as heterogeneous catalysts. Nano Lett 14:5979–5983
Rungtaweevoranit B, Baek B, Araujo JR, Archanjo BS, Choi KM, Yaghi OM, Somorjai GA (2016) Copper nanocrystals encapsulated in Zr-based metal–organic frameworks for highly selective CO2 hydrogenation to methanol. Nano Lett 16:7645–7649
Zha M, Yuan K, Wang Y, Li G, Guo J, Gu GL, Hu W, Zhao H, Tang Z (2016) Metal–organic frameworks as selectivity regulators for hydrogenation reactions. Nature 539:76–80
Choi KM, Kim D, Rungtaweevoranit B, Trickett CA, Barmanbek JTD, Alshammari AS, Yang P, Yaghi OM (2017) Plasmon-enhanced photocatalytic CO2 conversion within metal–organic frameworks under visible light. J Am Chem Soc 139:356–362
Wang X, Jiang XF, Liu YN, Xu AW (2018) Erbium oxide as a novel support for palladium nanocatalysts with strong metal–support interactions: remarkable catalytic performance in hydrogenation reactions. New J Chem 42:19901–19907
Sankar M, He Q, Engel RV, Sainna MA, Logsdail AJ, Roldan A, Willock DJ, Agarwal N, Kiely CJ, Hutchings GJ (2020) Role of the support in gold-containing nanoparticles as heterogeneous catalysts. Chem Rev 120:3890–3938
Wu C, Cheng D, Wang M, Ma D (2021) Understanding and application of strong metal-support interactions in conversion of CO2 to methanol: a review. Energy Fuels 35:19012–19023
Pu T, Zhang W, Zhu M (2023) Engineering heterogeneous catalysis with strong metal-support interactions: characterization, theory and manipulation. Angew Chem Int Ed 62:e202212278
van Deelen TW, Mejía CH, de Jong KP (2019) Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat Catal 2:955–970
Luo Z, Zhao G, Pan H, Sun W (2022) Strong metal-support interaction in heterogeneous catalysts. Adv Energy Mater 12:2201395
Lu L, Zou Z, Fang B (2021) The critical impacts of ligands on heterogeneous nanocatalysis: a review. ACS Catal 11:6020–6058
Rossi LM, Fiorio JL, Garcia MAS, Ferraz CP (2018) The role and fate of capping ligands in colloidally prepared metal nanoparticle catalysts. Dalton Trans 47:5889–5915
Campisi S, Schiavoni M, Chan-Thaw CE, Villa A (2016) Untangling the role of the capping agent in nanocatalysis: recent advances and perspectives. Catalysts 6:185
Liu K, Qin R, Zheng N (2021) Insights into the interfacial effects in heterogeneous metal nanocatalysts toward selective hydrogenation. J Am Chem Soc 143:4483–4499
Janiak C (2013) Ionic liquids for the synthesis and stabilization of metal nanoparticles. Z Naturforsch 68b:1059–89
Pensando AS, Pádua AAH (2011) Solvation and stabilization of metallic nanoparticles in ionic liquids. Angew Chem Int Ed 50:8683–8687
Kraynov A, Müller TE (2011) Concepts for the stabilization of metal nanoparticles in ionic liquids. Applications of ionic liquids in science and technology. Handy S IntechOpen. https://doi.org/10.5772/22111
Scholten JD, Leal BC, Dupont J (2012) Transition metal nanoparticle catalysis in ionic liquids. ACS Catal 2:184–200
Doherty S (2014) Homogeneous catalysis in ionic liquids. In: Hardacre C, Parvulescu V (eds) Catalysis in Ionic liquids: from catalyst synthesis to applications, catalysis series, Royal Society of Chemistry, pp 44–308
Migowski P, Luska KL, Leitner W (2016) In: Prechtl MGH (ed) Nanocatalysts in ionic liquids. Wiley VCH, Weinheim
Luska KL, Moores A (2012) Functionalized ionic liquids for the synthesis of metal nanoparticles and their application in catalysis. ChemCatChem 4:1534–1546
Lau S, Gasperini D, Webster RL (2021) Amine-boranes as transfer hydrogenation and hydrogenation reagents: a mechanistic perspective. Angew Chem Int Ed 60:14272–14294
Lara P, Philippot K, Suárez A (2019) Phosphane-decorated platinum nanoparticles as efficient catalysts for H2 generation from ammonia borane and methanol. ChemCatChem 11:766–771
Vermaak V, Vosloo HCM, Swarts AJ (2020) Fast and efficient nickel (II)-catalysed transfer hydrogenation of quinolines with ammonia borane. Adv Synth Catal 362:5788–5793
Yun R, Ma W, Hong L, Hu Y, Zhan F, Liu S, Zheng B (2019) Ni@ PC as a stabilized catalyst toward the efficient hydrogenation of quinoline at ambient temperature. Catal Sci Technol 9:6669–6672
Gao C, Xuan Q, Song Q (2021) Cu-catalyzed chemoselective reduction of N-heteroaromatics with NH3·BH3 in aqueous solution. Chin J Chem 39:2504–2508
Zeng YF, Li YN, Zhou MX, Han S, Guo Y, Wang Z (2022) Metal-free hydrogenation of N-heterocycles with trimethylamine borane and TFA in aqueous solution. Adv Synth Catal 364:3664–3669
Vasilikogiannaki E, Titilas I, Vassilikogiannakis G, Stratakis M (2015) cis-Semihydrogenation of alkynes with amine borane complexes catalyzed by gold nanoparticles under mild conditions. Chem Commun 51:2384–2387
Yang Q, Chen YZ, Wang ZU, Xu Q, Jiang HL (2015) One-pot tandem catalysis over Pd@ MIL-101: boosting the efficiency of nitro compound hydrogenation by coupling with ammonia borane dehydrogenation. Chem Commun 51:10419–10422
Goksu H, Ho SF, Metin O, Korkmaz K, Garcia AM, Gultekin MS, Sun S (2014) Tandem dehydrogenation of ammonia borane and hydrogenation of nitro/nitrile compounds catalyzed by graphene-supported NiPd alloy nanoparticles. ACS Catal 4:1777–1782
Goksu H, Can H, Sendil K, Gultekin MS, Metin O (2014) Tandem dehydrogenation of ammonia borane and hydrogenation of nitro/nitrile compounds catalyzed by graphene-supported NiPd alloy nanoparticles. Appl Catal A 488:176–182
Vasilikogiannaki E, Gryparis C, Kotzabasak V, Lykakis IN, Stratakis M (2013) Facile reduction of nitroarenes into anilines and nitroalkanes into hydroxylamines via the rapid activation of ammonia⋅ borane complex by supported gold nanoparticles. Adv Synth Catal 355:907–911
Wu J, Lang W, Li H, Du K, Deng J, Zhao S, Zhang Q, Peng Z, Liu Z (2023) Interfacial effect of CNT-supported ultrafine Ru nanoclusters on efficient transfer hydrogenation of nitroaromatic compounds. ACS Sustain Chem Eng 41:14960–14968
Wang G, Wang C, Zhang H, Liu Y, Xu X (2021) Facile preparation of Cu–Fe oxide nanoplates for ammonia borane decomposition and tandem nitroarene hydrogenation. RSC Adv 11:29920–29924
Zhang L, Pan J, Liu L, Zhang S, Wang X, Song S, Zhang H (2022) Photothermal-driven high-performance selective hydrogenation system enabled by delicately designed IrCo nanocages. Small 18:2201271
Lu BB, Chen XY, Feng CF, Chang J, Ye F (2021) Palladium nanoparticles immobilized on a Resorcin[4]arene-based metal-organic framework for hydrogenation of nitroarenes. ACS Appl Nano Mater 4:2278–2284
Kinik FP, Nguyen TN, Mensi M, Ireland CP, Stylianou KC, Smit B (2020) Sustainable hydrogenation of nitroarenes to anilines with highly active in-situ generated copper nanoparticles. ChemCatChem 12:2833–2839
Du J, Chen J, Xia H, Zhao Y, Wang F, Liu H, Zhou W, Wang B (2020) Commercially available CuO catalyzed hydrogenation of nitroarenes using ammonia borane as a hydrogen source. ChemCatChem 12:2426–2430
Kundu D, Pugazhenthi G, Banerjee T (2020) Low- to room-temperature dehydrogenation of dimethylamine borane facilitated by ionic liquids: molecular modelling and experimental studies. Energy Fuels 34:13167
Patni AN, Mantri AS, Kundu D (2021) Ionic liquid promoted dehydrogenation of amine boranes: a review. Int J Hydrog Energy 46:11761–11781 (and references therein)
Himmelberger DW, Alden LR, Bluhm ME, Sneddon LG (2009) Ammonia borane hydrogen release in ionic liquids. Inorg Chem 48:9883–9889
Bluhm ME, Bradley MG, Butterick R, Kusari U, Sneddon LG (2006) Amineborane-based chemical hydrogen storage: enhanced ammonia borane dehydrogenation in ionic liquids. J Am Chem Soc 128:7748–7749
Kundu D, Chakma S, Pugazhenthi G, Banerjee T (2018) Ionic liquid facilitated dehydrogenation of tert-butylamine borane. ACS Omega 3:273–281
Sahler S, Konnerth H, Knoblauch N, Prechtl MHG (2013) Hydrogen storage in amine boranes: ionic liquid supported thermal dehydrogenation of ethylene diamine bisborane. Int J Hydrog Energy 38:3283–3290
Giacalone F, Gruttadauria M (2016) Covalently supported ionic liquid phases: an advanced class of recyclable catalytic systems. ChemCatChem 8:664–684
Bordet A, Leitner W (2021) Metal nanoparticles immobilized on molecularly modified surfaces: versatile catalytic systems for controlled hydrogenation and hydrogenolysis. Acc Chem Res 54:2144–2157
Qian W, Texter J, Yam F (2017) Frontiers in poly(ionic liquid)s: syntheses and applications. Chem Soc Rev 46:1124–1159
Li H, Bhadury PS, Song B, Yang S (2012) Immobilized functional ionic liquids: efficient, green, and reusable catalysts. RSC Adv 2:12525–12551
Manojkumar K, Sivaramakrishna A, Vijayakrishna KA (2016) Short review on stable metal nanoparticles using ionic liquids, supported ionic liquids, and poly(ionic liquids). J Nanopart Res 18:103
Montolio S, Vicent C, Aseyev V, Alfonso I, Burguete MI, Tenhu H, García-Verdugo E, Luis SV (2016) AuNP–polymeric ionic liquid composite multicatalytic nanoreactors for one-pot cascade reactions. ACS Catal 6:7230–7237
Salvo AMP, Giacalone F, Gruttadauria M (2016) Advances in organic and organic-inorganic hybrid polymeric supports for catalytic applications/. Molecules 21:1288
Xie Y, Zhang Z, Jiang T, He J, Han B, Wu T, Ding K (2007) CO2 cycloaddition reactions catalyzed by an ionic liquid grafted onto a highly cross-linked polymer matrix. Angew Chem Int Ed 46:7255–7258
Wang X, Zhou Y, Guo Z, Chen G, Li J, Shi Y, Liu Y, Wang J (2015) Heterogeneous conversion of CO2 into cyclic carbonates at ambient pressure catalyzed by ionothermal-derived meso-macroporous hierarchical poly(ionic liquid)s. Chem Sci 6:6916–6924
Wang X, Li J, Chen G, Guo Z, Zhou Y, Wang J (2015) Hydrophobic mesoporous poly(ionic liquid)s towards highly efficient and contamination-resistant solid-base catalysts. ChemCatChem 7:993–1003
Wolny A, Chrobok A (2022) Silica-based supported ionic liquid-like phases as heterogeneous catalysts. Molecules 18:5900
Ziarani GM, Javadi F, Mohajer F, Badiei A (2022) The synthesis and application of ionic liquid functionalized mesoporous silica SBA-15 for organic synthesis. Curr Org Synth 19:874–904
Kim DW, Kim HG, Cho DH (2016) Application of silica-supported ionic liquid catalysts to cycloaddition of CO2. Appl Chem Eng 27:239–244
Han L, Park SW, Park DW (2009) Silica grafted imidazolium-based ionic liquids: efficient heterogeneous catalysts for chemical fixation of CO2 to a cyclic carbonate. Energy Environ Sci 2:1286–1292
Karimi S, Gholinejad M, Khezri R, Sansano JM, Nájera C, Yus M (2023) Gold and palladium supported on an ionic liquid modified Fe-based metal-organic framework (MOF) as highly efficient catalysts for the reduction of nitrophenols, dyes and Sonogashira-Hagihara reactions. RSC Adv 13:8101–8113
Wu Y, Xiao Y, Yuan H, Zhang Z, Shi S, Wei R, Gao L, Xiao G (2021) Imidazolium ionic liquid functionalized UiO-66-NH2 as highly efficient catalysts for chemical fixation of CO2 into cyclic carbonates. Microporous Mesoporous Mater 310:110578
Ali AAQ, Siddiqui ZN (2023) Ionic liquid functionalized metal-organic framework ([DEIm][PF6]@MOF-5): synthesis, characterization, and catalytic application in the reduction of 4-nitrophenol. ACS Omega 8:3785–3797
Ding M, Jiang HL (2018) Incorporation of imidazolium-based poly(ionic liquid)s into a metal-organic framework for CO2 capture and conversion. ACS Catal 8:3194–3201
Fujie K, Kitagawa H (2016) Ionic liquid transported into metal–organic frameworks. Coord Chem Rev 307:382–390
Liang J, Chen RP, Wang XY, Liu TT, Wang XS, Huang YB, Cao R (2017) Postsynthetic ionization of an imidazole–containing metal–organic framework for the cycloaddition of carbon dioxide and epoxides. Chem Sci 8:1570–1575
Tharun J, Bhin KM, Roshan R, Kim DW, Kathalikkattil AC, Babu R, Ahn HY, Won YS, Park DW (2016) Ionic liquid tethered post functionalized ZIF-90 framework for the cycloaddition of propylene oxide and CO2. Green Chem 18:2479–2487
Ding LG, Yao BW, Jiang WL, Li JT, Fu QJ, Li YA, Liu ZH, Ma JP, Dong YB (2017) Bifunctional imidazolium-based ionic liquid decorated UiO-67 type MOF for selective CO2 adsorption and catalytic property for CO2 cycloaddition with epoxides. Inorg Chem 56:2337–2344
Gong Y, Zhong H, Lu W, Zhang B, Hu S, Wang R (2018) General synthetic route toward highly dispersed ultrafine Pd–Au alloy nanoparticles enabled by imidazolium-based organic polymers. ACS Appl Mater Interfaces 10:776–786
Saptal VB, Sasaki T, Bhanage BM (2018) Ru@PsIL-catalyzed synthesis of N-formamides and benzimidazole by using carbon dioxide and dimethylamine borane. ChemCatChem 10:2593–2600
Patil MN, Sasaki T, Bhanage BM (2016) Immobilized ruthenium metal-containing ionic liquid-catalyzed dehydrogenation of dimethylamine borane complex for the reduction of olefins and nitroarenes. RSC Adv 6:52347–52352
Doherty S, Knight JG, Backhouse T et al (2017) Highly efficient aqueous phase chemoselective hydrogenation of α, β-unsaturated aldehydes catalysed by phosphine-decorated polymer immobilized IL-stabilized PdNPs. Green Chem 19:1635–1641
Doherty S, Knight JG, Backhouse T et al (2018) Highly efficient aqueous phase reduction of nitroarenes catalyzed by phosphine-decorated polymer immobilized ionic liquid stabilized PdNPs. Catal Sci Technol 8:1454–1467
Doherty S, Knight JG, Backhouse T et al (2018) Heteroatom donor-decorated polymer-immobilized ionic liquid stabilized palladium nanoparticles: efficient catalysts for room-temperature suzuki-miyaura cross-coupling in aqueous media. Adv Synth Catal 360:3716–3731
Doherty S, Knight JG, Backhouse T et al (2022) Highly efficient and selective aqueous phase hydrogenation of aryl ketones, aldehydes, furfural and levulinic acid and its ethyl ester catalyzed by phosphine oxide-decorated polymer immobilized ionic liquid-stabilized ruthenium nanoparticles. Catal Sci Technol 12:3549–3567
Doherty S, Knight JG, Backhouse T et al (2019) Highly selective and solvent-dependent reduction of nitrobenzene to N-Phenylhydroxylamine, azoxybenzene, and aniline catalyzed by phosphino-modified polymer immobilized ionic liquid-stabilized AuNPs. ACS Catal 9:4777–4791
Doherty S, Knight JG, Paterson R et al (2023) Highly efficient and selective partial reduction of nitroarenes to N-arylhydroxylamines catalysed by phosphine oxide-decorated polymer immobilized ionic liquid stabilized ruthenium nanoparticles. J Catal 417:74–88
Paterson R, Fahy LF, Arca E, Dixon C, Wills C, Yan H, Griffiths A, Collins SM, Wu KJ, Bourne RA, Chamberlain TW, Knight JG, Doherty S (2023) Amine-modified polyionic liquid supports enhance the efficacy of PdNPs for the catalytic hydrogenation of CO2 to formate. Chem Commun 59:13470–13473
Doherty S, Knight JG et al (2022) Efficient hydrolytic hydrogen evolution from sodium borohydride catalyzed by polymer immobilized ionic liquid-stabilized platinum nanoparticles. ChemCatChem 14:e202101752
Doherty S, Knight JG, Paterson R et al (2022) Heteroatom modified polymer immobilized ionic liquid stabilized ruthenium nanoparticles: efficient catalysts for the hydrolytic evolution of hydrogen from sodium borohydride. Mol Catal 528:112476
Alharbi AA, Wills C, Chamberlain TW, Bourne RA, Griffiths A, Collins SM, Wu KJ, Mueller P, Knight JG, Doherty S (2023) Amino-modified polymer immobilized ionic liquid stabilized ruthenium nanoparticles: efficient and selective catalysts for the partial and complete reduction of quinolines. ChemCatChem 15:e202300418
Greczynski G, Hultman L (2020) X-ray photoelectron spectroscopy: towards reliable binding energy referencing. Prog Mater Sci 107:100591
Han KN, Li CA, Bui MPN, Pham XH, Kim BA, Choa YH, Seong GH (2012) Development of Pt/TiO2 nanohybrids-modified SWCNT electrode for sensitive hydrogen peroxide detection. Sens Actuators B 174:406–413
Romanchenko A, Likhatski M, Mikhlin Y (2018) X-ray Photoelectron Spectroscopy (XPS) study of the products formed on sulfide minerals upon the interaction with aqueous platinum (IV) chloride complexes. Minerals 8:578
Kolbeck C, Taccardi N, Paape N, Schulz PS, Wasserscheid P, Steinrück HP, Maier F (2014) Redox chemistry, solubility, and surface distribution of Pt(II) and Pt(IV) complexes dissolved in ionic liquids. J Mol Liquids 192:103–113
Klauke K, Gruber I, Knedel TO, Schmolke L, Barthel J, Breitzke H, Buntkowsky G, Janiak C (2018) Silver, gold, palladium, and platinum N-heterocyclic carbene complexes containing a selenoether-functionalized Imidazol-2-ylidene moiety. Organometallics 37:298–308
Fiekkies JTR, Fourie E, Erasmus E (2021) Cisplatin-functionalized nanodiamonds: preparation and characterization, with potential antineoplastic application. Appl Nanoscience 11:2235–2245
Vakili R, Gibson EK, Chansai S, Xu S, Al-Janabi N, Wells PP, Hardacre C, Walton A, Fan X (2018) Understanding the CO oxidation on Pt nanoparticles supported on MOFs by operando XPS. ChemCatChem 10:4238–4242
Xiao F, Luo X, Fu X, Zheng Y (2013) Cleavage enhancement of specific chemical bonds in DNA by cisplatin radiosensitization. J Phys Chem 117:4893–4900
Alwin E, Wojcieszak R, Kočí K, Edelmannová M, Zieliński M, Suchora A, Pędziński T, Pietrowski M (2022) Reductive modification of carbon nitride structure by metals—the influence on structure and photocatalytic hydrogen evolution. Materials 15:710
Zhang L, Long R, Zhang Y, Duan D, Xiong Y, Zhang Y, Bi Y (2020) Direct observation of dynamic bond evolution in single-atom Pt/C3N4 catalysts. Angew Chem Int Ed 59:6224–6229
Alwin E, Zieliński M, Suchora A, Gulaczyk I, Piskuła Z, Pietrowski M (2022) High surface area, spongy graphitic carbon nitride derived by selective etching by Pt and Ru nanoparticles in hydrogen. J Mater Sci 57:15705–15721
Alderucci V, Pino L, Antonucci PL, Roh W, Cho J, Kim H, Cocke DL, Antonucci V (1995) XPS study of surface oxidation of carbon-supported Pt catalysts. Mat Chem Phys 41:9–14
Maksic A, Rakocevic Z, Smiljanic M, Nenadovic M, Strbac S (2015) Methanol oxidation on Pd/Pt(poly) in alkaline solution. J Power Sources 273:724–734
Fu S, Zhang B, Hu H, Zhang Y, Bi Y (2018) ZnO nanowire arrays decorated with PtO nanowires for efficient solar water splitting. Catal Sci Technol 8:2789–2793
Deka JR, Saikia D, Hsia KS, Kao HM, Yang YC, Chen CS (2020) Ru nanoparticles embedded in cubic mesoporous silica SBA-15 as highly efficient catalysts for hydrogen generation from ammonia borane. Catalysts 10:267
Chen B, Cao ZZ, Diao ZJ, Huang Q, Zhao SJ, Yuan H, He JM (2022) Hydrogenolysis of lignin and C-O linkages containing lignin-related compounds over a macroporous silicalite-1 array-supported Ru-Ni phosphide composite. Catalysts 12:1625
Zhang Y, Jiang H, Lia G, Zhang M (2016) Controlled synthesis of highly dispersed and nano-sized Ru catalysts supported on carbonaceous materials via supercritical fluid deposition. RSC Adv 6:16851–16858
Morgan DJ (2015) Resolving ruthenium: XPS studies of common ruthenium materials. Surf Interfaces Anal 47:1072–1079
Cai B, Zhang Y, Feng J, Huang C, Ma T, Pan H (2021) Highly efficient g-C3N4 supported ruthenium catalysts for the catalytic transfer hydrogenation of levulinic acid to liquid fuel γ-valerolactone. Renew Energy 177:652–662
Khalily MA, Eren H, Akbayrak S, Susapto HH, Biyikli N, Özkar S, Guler MO (2016) Facile synthesis of three-dimensional Pt-TiO2 nano-networks: a highly active catalyst for the hydrolytic dehydrogenation of ammonia-borane. Angew Chem Int Ed 55:12257–12261
Yao Q, Shi Y, Zhang X, Chen X, Lu ZH (2016) Facile synthesis of Platinum–Cerium(IV) oxide hybrids arched on reduced graphene oxide catalyst in reverse micelles with high activity and durability for hydrolysis of ammonia borane. Chem Asian J 11:3251–3257
Zhou Q, Xu C (2016) Nanoporous PtRu alloys with unique catalytic activity toward hydrolytic dehydrogenation of ammonia borane. Chem Asian J 11:705–712
Uzundurukan A, Devrim Y (2019) Carbon nanotube-graphene hybrid supported platinum as an effective catalyst for hydrogen generation from hydrolysis of ammonia borane. Int J Hydrog Energy 44:26773–26782
Zhang J, Chen C, Chen S, Hu Q, Gao Z, Lia Y, Qin Y (2017) Highly dispersed Pt nanoparticles supported on carbon nanotubes produced by atomic layer deposition for hydrogen generation from hydrolysis of ammonia borane. Catal Sci Technol 7:322–329
Zhang SY, Kochovski Z, Lee HC, Lu Y, Zhang H, Zhang J, Sun JK, Yuan J (2019) Ionic organic cage-encapsulating phase-transferable metal clusters. Chem Sci 10:1450–1456
Chen W, Ji J, Duan X, Qian G, Li P, Zhou X, Chen D, Yuan W (2014) Unique reactivity in Pt/CNT catalyzed hydrolytic dehydrogenation of ammonia borane. Chem Commun 50:2142–2144
Ye W, Ge Y, Gao Z, Lu R, Zhang S (2017) Enhanced catalytic activity and stability of Pt nanoparticles by surface coating of nanosized graphene oxide for hydrogen production from hydrolysis of ammonia–borane. Sus Energy Fuels 1:2128–2133
Hu Y, Wang Y, Lu ZH, Chen X, Xiong L (2015) Core–shell nanospheres Pt@SiO2 for catalytic hydrogen production. Appl Surf Sci 341:185–189
Dan X, Cui Z, Yang J, Yuan M, Cui X, Zhang X, Dong Z (2017) Pt nanoparticles immobilized in mesoporous silica-coated magnetic nanocapsules: a non-leaching catalyst for hydrogen generation from hydrolysis of ammonia borane. Int J Hydrog Energy 42:27034–27042
Qi X, Li X, Chen B, Lu H, Wang L, He G (2016) Highly active nanoreactors: patchlike or thick Ni coating on Pt nanoparticles based on confined catalysis. Appl Mater Interfaces 8:1922–1928
Yin L, Feng Y, Zhou X, Dai K, Gao X, Zhao Y, Zhang B (2019) Synthesis of Pt nanocatalyst supported on halloysite nanotubes via strong electronic adsorption for hydrolytic dehydrogenation of ammonia borane. Chem Lett 48:1084–1087
Li Z, He T, Matsumura D, Miao S, Wu A, Liu L, Wu G, Chen P (2017) Atomically dispersed Pt on the surface of Ni particles: synthesis and catalytic function in hydrogen generation from aqueous ammonia-borane. ACS Catal 7:6762–6769
Kang N, Djeda R, Wang Q, Fu F, Ruiz J, Pozzo JL, Astruc D (2019) Efficient “Click”-dendrimer-supported synergistic bimetallic nanocatalysis for hydrogen evolution by sodium borohydride hydrolysis. ChemCatChem 11:2341–2349
Hu M, Wang H, Wang Y, Zhang Y, Wu J, Xu B, Gao D, Bi J, Fan G (2017) Ruthenium nanoparticles alumina nanofiber-stabilized ruthenium nanoparticles: highly efficient catalytic materials for hydrogen evolution from ammonia borane hydrolysis. Int J Hydrog Energy 42:24142–24149
Lu R, Xu C, Wang Q, Wang Y, Zhang Y, Gao D, Bi J, Fan G (2018) Ruthenium nanoclusters distributed on phosphorus-doped carbon derived from hyper crosslinked polymer networks for highly efficient hydrolysis of ammonia-borane. Int J Hydrog Energy 43:18253–18260
Zhong F, Wang Q, Xu C, Yang Y, Wang Y, Zhang Y, Gao D, Bi J, Fan G (2018) Ultrafine and highly dispersed Ru nanoparticles supported on nitrogen-doped carbon nanosheets: efficient catalysts for ammonia borane hydrolysis. Appl Surf Sci 45:326–332
Ma Y, Li X, Zhang Y, Chen L, Wu J, Gao D, Bi J, Fan G (2017) Ruthenium nanoparticles supported on TiO2 (B) nanotubes: effective catalysts in hydrogen evolution from the hydrolysis of ammonia borane. J Alloys Cmpds 708:270–277
Li YT, Zhang SH, Zheng GP, Liu P, Peng ZK, Zheng XC (2020) Ultrafine Ru nanoparticles anchored to porous g-C3N4 as efficient catalysts for ammonia borane hydrolysis. Appl Catal A 595:117511
Wu Z, Duan Y, Ge S, Yip ACK, Yang F, Li Y, Dou T (2017) Promoting hydrolysis of ammonia borane over multiwalled carbon nanotube-supported Ru catalysts via hydrogen spillover. Catal Commun 91:10–15
Özgür DO, Şimşek T, Özkan G, Akkuş MS, Özkan G (2018) The Hydroloysis of ammonia borane by using Amberlyst-15 supported catalysts for hydrogen generation. Int J Hydrog Energy 43:10765–10772
Zhao W, Wang R, Wang Y, Feng J, Li C, Chen G (2019) Effect of LDH composition on the catalytic activity of Ru/LDH for the hydrolytic dehydrogenation of ammonia borane. Int J Hydrog Energy 44:14820–14830
Qiu X, Liu J, Huang P, Qiu S, Weng C, Chu H, Zou Y, Xiang C, Xu F, Sun L (2020) Hydrolytic dehydrogenation of NH3BH3 catalyzed by ruthenium nanoparticles supported on magnesium–aluminum layered double-hydroxides. RSC Adv 10:9996–10005
Chu H, Li N, Qiu S, Zou Y, Xiang C, Xu F, Sun L (2019) Ruthenium supported on nitrogen-doped porous carbon for catalytic hydrogen generation from NH3BH3 hydrolysis. Int J Hydrog Energy 44:1774–1781
Wei Z, Liu Y, Peng Z, Song H, Liu Z, Liu B, Li B, Yang B, Lu S (2019) Cobalt-ruthenium nanoalloys parcelled in porous nitrogen-doped graphene as highly efficient difunctional catalysts for hydrogen evolution reaction and hydrolysis of ammonia borane. ACS Sustain Chem Eng 7:7014–7023
Song Q, Wang WD, Hua X, Dong Z (2019) Ru nanoclusters confined in porous organic cages for catalytic hydrolysis of ammonia borane and tandem hydrogenation reaction. Nanoscale 11:21513–21521
Sun Q, Wang N, Bai R, Hui Y, Zhang T, Do DA, Zhang P, Song L, Miao S, Yu J (2019) Synergetic effect of ultrasmall metal clusters and zeolites promoting hydrogen generation. Adv Sci 6:1802350
Yang K, Zhou L, Yu G, Xiong X, Ye M, Li Y, Lu D, Pan Y, Chen M, Zhang L, Gao D, Wang Z, Liu H, Xia Q (2016) Ru nanoparticles supported on MIL-53(Cr, Al) as efficient catalysts for hydrogen generation from hydrolysis of ammonia borane. Int J Hydrog Energy 41:6300–6309
Şen B, Aygün A, Şavk A, Duman S, Calimli MH, Bulut E, Şen F (2019) Polymer-graphene hybrid stabilized ruthenium nanocatalysts for the dimethylamine-borane dehydrogenation at ambient conditions. J Mol Liq 279:578–583
Şen B, Demirkan B, Savk A, Kartop R, Nas MS, Alma MH, Sürdem S, Şen F (2018) High-performance graphite-supported ruthenium nanocatalyst for hydrogen evolution reaction. J Mol Liq 268:807–812
Duman S, Özhava D (2023) Green approaches to dehydrogenation of DMAB catalyzed by starch stabilized Ru(0), Cu(0) and Ni(0) nanoparticles in the absence of a solvent. ChemistrySelect 8:e202204606
Sen B, Kuzu S, Demir E, Okyay TO, Sen F (2017) Hydrogen liberation from the dehydrocoupling of dimethylamine–borane at room temperature by using novel and highly monodispersed RuPtNi nanocatalysts decorated with graphene oxide. Int J Hydrog Energy 42:23299–23306
Cai HK, Jiang ZY, Xu S, Xu Y, Lu P, Dong J (2022) Polymer hydrogel supported Ni/Pd alloys for hydrogen gas production from hydrolysis of dimethylamine borane with a long recyclable lifetime. Polymers 14:4647
Duman S, Özkar S (2013) Oleylamine-stabilized ruthenium(0) nanoparticles catalyst in dehydrogenation of dimethylamine-borane. Int J Hydrog Energy 38:10000–10011
Kang N, Wang Q, Djeda R, Wang W, Fu F, Moro M, MdlA R, Moya S, Coy E, Salmon L, Pozzo JL, Astruc D (2020) Visible-light acceleration of H2 evolution from aqueous solutions of inorganic hydrides catalyzed by gold-transition-metal nanoalloys. ACS Appl Mater Interfaces 12:53816–53826
Wang C, Tuninetti J, Wang Z, Zhang C, Ciganda R, Salmon L, Moya S, Ruiz J, Astruc D (2017) Hydrolysis of ammonia-borane over Ni/ZIF-8 nanocatalyst: high efficiency, mechanism, and controlled hydrogen release. J Am Chem Soc 139:11610–11615
Wang Q, Fu F, Escobar A, Moya S, Ruiz J, Astruc D (2018) “Click” Dendrimer-stabilized nanocatalysts for efficient hydrogen release upon ammonia-borane hydrolysis. ChemCatChem 10:2673–2680
Wang Q, Fu F, Yang S, Moro MM, Ramirez MA, Moya S, Salmon L, Ruiz J, Astruc D (2019) Dramatic synergy in CoPt nanocatalysts stabilized by “Click” dendrimers for evolution of hydrogen from hydrolysis of ammonia borane. ACS Catal 9:1110–1119
Ma H, Na C (2015) Isokinetic temperature and size-controlled activation of ruthenium-catalyzed ammonia borane hydrolysis. ACS Catal 5:1726–1735
Chen W, Li D, Wang Z, Qian G, Sui Z, Duan X, Zhou X, Yeboah I, Chen D (2017) Reaction mechanism and kinetics for hydrolytic dehydrogenation of ammonia borane on a Pt/CNT catalyst. AiChE J 63:60–65
Xu Q, Chandra M (2006) Catalytic activities of non-noble metals for hydrogen generation from aqueous ammonia–borane at room temperature. J Power Sources 163:364–370
Peng CY, Kang L, Cao S, Chen Y, Lin ZS, Fu WF (2015) Nanostructured Ni2P as a robust catalyst for the hydrolytic dehydrogenation of ammonia-borane. Angew Chem Int Ed 54:15725–15729
Kalidindi SB, Sanyal U, Jagirdar BR (2008) Nanostructured Cu and Cu@Cu2O core shell catalysts for hydrogen generation from ammonia–borane. Phys Chem Chem Phys 10:5870–5874
Fu F, Wang C, Wang Q, Martinez-Villacorta AM, Escobar A, Chong H, Wang X, Moya S, Salmon L, Fouquet E, Ruiz J, Astruc D (2018) Highly selective and sharp volcano-type synergistic Ni2Pt@ZIF-8-catalyzed hydrogen evolution from ammonia borane hydrolysis. J Am Chem Soc 140:10034–10042
Li Z, He T, Liu L, Chen W, Zhang M, Wu G, Chen P (2017) Covalent triazine framework supported non-noble metal nanoparticles with superior activity for catalytic hydrolysis of ammonia borane: from mechanistic study to catalyst design. Chem Sci 8:781–788
Ren X, Lv H, Yang S, Wang Y, Li J, Wei R, Xu D, Liu B (2019) Promoting effect of heterostructured NiO/Ni on Pt nanocatalysts toward catalytic hydrolysis of ammonia borane. J Phys Chem Lett 10:7374–7382
Wang C, Ciganda R, Yate L, Tuninetti J, Shalabaeva V, Salmon L, Moya S, Ruiz J (2017) Astru D Redox synthesis and high catalytic efficiency of transition-metal nanoparticle–graphene oxide nanocomposites. J Mater Chem A 5:21947–21954
Guella G, Zanchetta C, Patton B, Miotello A (2006) New insights on the mechanism of palladium-catalyzed hydrolysis of sodium borohydride from 11B NMR measurements. J Phys Chem B 110:17024–17033
Sau SC, Hota PK, Mandal SK, Soleilhavoup M, Bertrand G (2020) Stable abnormal N-heterocyclic carbenes and their applications. Chem Soc Rev 49:1233–1252
Ellul CE, Mahon MF, Saker O, Whittlesey MK (2007) Abnormally bound N-heterocyclic carbene complexes of ruthenium: C-H activation of both c4 and c5 positions in the same ligand. Angew Chem Int Ed 46:6343–6345
Pardatscher L, Bitzer MJ, Jandl C, Kück JW, Reich RM, Kühn FE, Baratta W (2019) Cationic abnormal N-heterocyclic carbene ruthenium complexes as suitable precursors for the synthesis of heterobimetallic compounds. Dalton Trans 48:79–89
Witt J, Pöthig A, Kühn FE, Baratta W (2013) Abnormal N-heterocyclic carbene-phosphine ruthenium(II) complexes as active catalysts for transfer hydrogenation. Organometallics 32:4042–4045
Winterling E, Ivlev S, Meggers E (2021) Chiral-at-ruthenium catalysts with mixed normal and abnormal N-heterocyclic carbene ligands. Organometallics 40:1148–1155
Crabtree RH (2013) Abnormal, mesoionic and remote N-heterocyclic carbene complexes. Coord Chem Rev 257:755–766
Vivancos Á, Segarra C, Albrecht M (2018) Mesoionic and related less heteroatom-stabilized N-heterocyclic carbene complexes: synthesis, catalysis, and other applications. Chem Rev 118:9493–9586
Zhu Y, Ouyang L, Zhong H, Liu J, Wang H, Shao H, Huang Z, Zhu M (2020) Closing the loop for hydrogen storage: facile regeneration of NaBH4 from its hydrolytic product. Angew Chem Int Ed 59:8632–8629
Acknowledgements
AAA thanks Taibah University, Saudi Arabia for a scholarship. We also thank (Dr Tracey Davey) for the SEM images (Faculty of Medical Sciences, Newcastle University) and Zabeada Aslam and the Leeds electron microscopy and spectroscopy centre (LEMAS) at the University of Leeds for TEM analysis. This research was funded through a studentship (Anthony Griffiths) awarded by the Engineering and Physical Sciences Centre for Doctoral Training in Molecules to Product (EP/SO22473/1). The authors greatly acknowledge their support of this work. The X-ray photoelectron (XPS) data collection was performed at the EPSRC National Facility for XPS (“HarwellXPS”) under Contract No. PR16195. This article is dedicated to the memory of Professor Stephen A. Westcott (Canada Research Chair holder in the Department of Chemistry & Biochemistry, Mount Allison University, Canada) who recently passed away; a fantastic and inspired scientist, a great ambassador for chemistry teaching and research in Canada and across the globe, a selfless, generous, and kind human being but most of all a genuine, true and sincere friend who is greatly missed.
Author information
Authors and Affiliations
Contributions
Simon Doherty: Conceptualization, Methodology, Supervision, Writing—original draft, Project Administration, Validation. Adhwa Alharbi: Investigation, Formal Analysis. Corinne Wills: Investigation, Resources, Formal analysis. Casey Dixon: Investigation, Formal Analysis. Elisabetta Arca: Supervision, Writing – Review and Editing, Visualization, Formal Analysis, Validation. Thomas W. Chamberlain: Supervision, Writing – Review and Editing, Visualization, Validation, Resources. Anthony Griffiths: Investigation, Visualization, Formal Analysis. Han Yan: Investigation, Visualization, Formal Analysis. Sean M. Collins: TEM Investigation, Supervision. Kejun Wu: TEM Investigation, Supervision. Richard A. Bourne: Supervision, Writing – Review and Editing. Julian G. Knight: Supervision, Writing—Review and Editing, Validation, Formal Analysis.
Corresponding authors
Ethics declarations
Conflict of Interest
The manuscript was written through contributions from all the authors. All authors have given approval to the final version of the manuscript. There is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is dedicated to the memory of Professor Stephen A. Westcott, a great ambassador for chemistry in Canada and across the globe and the best and most sincere of friends.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alharbi, A.A., Wills, C., Dixon, C. et al. Efficient Hydrogen Evolution from Dimethylamine Borane, Ammonia Borane and Sodium Borohydride Catalyzed by Ruthenium and Platinum Nanoparticles Stabilized by an Amine Modified Polymer Immobilized Ionic Liquid: a Comparative Study. Catal Lett (2024). https://doi.org/10.1007/s10562-024-04725-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10562-024-04725-8