
Abstract
In this study, the promoting effects of ruthenium, palladium, and copper on the performance of a 15%Co-1%K/SiO2 catalyst were evaluated during CO2 hydrogenation in a fixed-bed reactor. Reactions were carried out at atmospheric pressure and 270 °C with H2/CO2 ratio of 3. All catalysts were characterized by X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET) and temperature-programmed reduction (TPR). Ruthenium, palladium and copper facilitated the reduction of cobalt oxides and increased cobalt dispersion. In terms of catalyst’s performance, ruthenium addition led to increased CO2 conversion and methane selectivity with a detrimental effect on C5+ hydrocarbons. Palladium also presented a similar pattern at lower loading but a drop in CO2 conversion and increased reverse water–gas shift activity were observed at 3 wt % Pd loading. Promoting with copper resulted in decreased activity, methane selectivity and C5+ hydrocarbons productivity with a much higher CO selectivity.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The promotional attributes of noble metals on cobalt Fischer–Tropsch catalysts have been widely reported. Depending on parameters such as support type, catalyst preparation method and activation conditions, noble metals are known to induce various modifications in cobalt catalysts properties. These include, among others, hydrogen activation, cobalt dispersion and/or reducibility, cobalt precursors decomposition and the formation of bimetallic compounds. Most of these changes have been linked to alterations in catalyst performance during Fischer–Tropsch synthesis [1].
Within this framework, it appears interesting to investigate these types of catalysts in CO2 hydrogenation. One such investigation has been reported by Zhu et al. [2] where the performance of Co3O4 and (Co0.95Ru0.05)3O4 nanorods catalysts was evaluated. It was demonstrated that the ruthenium doped catalyst possessed a higher activity and methane selectivity than Co3O4. A further comparison with Co/SiO2 and Ru/SiO2 revealed that the bimetallic catalyst was still superior in terms of activity and methane selectivity. It was possible, with the information provided by XPS, to link the observed change in the catalyst performance to the formation of an alloy of cobalt and ruthenium, which segregated to the surface. In contrast, data reported by Owen et al. [3] showed that the addition of 1 wt % Ru to a 20%Co-1%K/SiO2 catalyst led to a drop in CO2 conversion and CO yield while the hydrocarbons yield remained unaffected. Replacing ruthenium with palladium in the same catalyst led to similar observations except for a slight decrease in hydrocarbons yield. In the same study, the promotion of a potassium-free Co/SiO2 catalyst with palladium, however, resulted in a decrease of both CO2 conversion and hydrocarbons yield with a negligible increase in CO yield.
In comparison to ruthenium and palladium, platinum has attracted more attention in CO2 hydrogenation. Boix et al. [4] explored the doping effect of cobalt mordenite catalysts with platinum (0.5, 1 and 5 wt %) prepared by ion exchange of sodium mordenite. The catalyst’s activity was found to increase with the platinum loading, except at 5 wt % where a drop in activity was observed. Nevertheless, all the promoted catalysts possessed a higher activity than the unpromoted one. The increase in activity was ascribed to the synergetic effect of the platinum particle in intimate contact with those of cobalt. This effect was explained in terms of the ability of platinum to dissociate hydrogen and enhance surface oxygen removal due to improved catalyst reducibility as was demonstrated by TPR experiments. The methane selectivity of 0.5 wt % platinum loaded catalyst was found to be higher than that of the unpromoted cobalt. The authors attributed this behaviour to the high dispersion of metallic cobalt particles in the main channels of the mordenite. Conversely at high platinum loadings of 1 and 5 wt % the methane selectivity was suppressed below the level of that of the unpromoted catalyst. At 5 wt % platinum loading, CO was almost exclusively generated. It was argued that the significant drop in methane selectivity could be due to platinum segregation to the surface from Co–Pt bimetallic species. With reference to the work of Bardi et al. [5] on CO chemisorption on CoPt3 alloy, the authors further argued that the probable presence of CoPt3 inside the mordenite main channel was responsible for high water gas shift activity due to changes in the electronic state available for CO–Pt bonding, resulting in lower bond energy. The authors believed that the low CO–Pt bond energy facilitates CO desorption rather than its further dissociation into carbon species and subsequent hydrogenation.
Alayoglu et al. [6] also came to similar conclusions upon investigating cobalt and cobalt–platinum bimetallic alloy (atomic ratio approximately 1:1) nanoparticles grafted on mesoporous silica (MCF-17). While there was little difference in CO2 conversion for both types of catalysts, the methane selectivity dropped to negligible levels for the bimetallic alloy. XPS and TEM of the bimetallic catalyst under a reducing hydrogen atmosphere suggested that surface enrichment of platinum would occur during reaction. Thus, the reactants access to cobalt becomes very limited, resulting almost exclusively in the production of CO. Testing of a similar monometallic platinum catalyst showed similar activity and selectivity to methane under the same conditions, reinforcing the platinum surface enrichment theory.
The role of platinum in Co/SiO2 catalysts has also been studied recently by Beaumont et al. [7]. The particularity of this study lies in the catalyst synthesis step where platinum and cobalt were deposited separately in close proximity (Pt:Co molar ratio of approximately 1:20) rather than as a single bimetallic nanoparticle. Surprisingly, this catalyst showed a methane production rate about 6 times higher compared to monometallic cobalt nanoparticles. Moreover, relatively less CO was produced, in contrast to findings presented beforehand. The evidence of improved cobalt reducibility in the presence of platinum provided by near edge X-ray absorption fine structure (NEXAFS) led the authors to suggest that once dissociated on platinum, the hydrogen atoms may be transferred to cobalt via a spill over mechanism to reduce surface oxide species formed during the reaction, hence generating more active sites.
It was shown in our previous study [8] that the addition of potassium in a Co/SiO2 catalyst negatively affected its reducibility which had an impact on the overall conversion. An economically viable catalyst not only needs to possess a good selectivity towards the component of interest but also an acceptable level of conversion. Noble metals such as the ones mentioned in the preceding lines are known to improve the reducibility of cobalt catalysts [9]. Therefore, including noble metals in the catalyst formulation may result in the improvement of its overall performance. Furthermore, numerous mechanistic investigations have reported CO2 hydrogenation to occur via the reverse water – gas shift reaction to produce CO which then further reacts with hydrogen to form hydrocarbons [3, 10,11,12]. However, cobalt possesses low water – gas shift activity [9]. Thus, promotion with a metal which possess water – gas shift activity like copper may have a significant impact on the catalyst behaviour.
This study aims to investigate the effects of a second promoter, namely ruthenium, palladium and copper on the performance of 15%Co-1%K/SiO2 base catalyst during CO2 hydrogenation.
2 Materials and Methods
2.1 Catalyst Synthesis
The metal precursors used in this study (copper nitrate, ruthenium nitrosyl nitrate and palladium chloride) were all purchased from Sigma-Aldrich. The catalysts were prepared by deposition–precipitation and impregnation depending on whether the metal precursor contained chlorine. Thus, copper and ruthenium were introduced in the catalyst by impregnation while deposition–precipitation was used for palladium. These metals were used to promote a pre-synthesized and calcined 15%Co-1%K/SiO2 catalyst as described in our previous publication [8]. In a typical impregnation, the appropriate amount of precursor was weighed and dissolved or diluted in water and the solution thus obtained added to the pre-synthesized catalyst. As for the deposition–precipitation, the pre-synthesized catalyst was suspended in an aqueous solution of the relevant precursor and an ammonia solution was added dropwise until the pH rose to 8.5. After an hour of ageing, the sample was filtered and washed several times with deionized water to remove chlorine which is considered to be a poison for cobalt FT catalysts [1]. Regardless of the synthesis approach, all the catalysts were then dried overnight at 120 °C and calcined at 400 °C for 6 h.
2.2 Catalyst Characterization
The catalysts investigated in this study were characterized by X-ray diffraction, Brunauer–Emmett–Teller and temperature-programmed reduction. XRD was carried out with a Rigaku Ultima IV instrument equipped with a copper target. The operating current and voltage of the diffractometer were 30 mA and 40 kV respectively. Spectra acquisition was performed at a scanning speed of 1°/min with a step size of 0.01° from 10 to 90°. The BET analysis was performed to evaluate the catalyst porosity using a Micromeritics Tristar 3000 using nitrogen.
An in-house built instrument, equipped with a thermal conductivity detector, was used for temperature-programmed reduction. This analysis was conducted by placing 100 mg of catalyst in a stainless-steel reactor followed by degassing under helium flow (70 NmL/min) at 300 °C for 1 h. The degassing step was necessary for the removal of any ambient contaminants and traces of moisture. After degassing the reactor was cooled to room temperature and helium was replaced with 5% hydrogen in argon flowing at 65 NmL/min. Finally, the temperature was increased to 700 °C at a heating rate of 10 °C/min.
2.3 Catalyst Testing
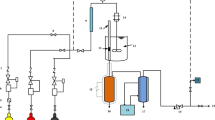
Carbon dioxide hydrogenation was conducted in a fixed-bed reactor system, which was described in our previous publication [8]. In each run, 500 mg of catalyst were used, and activation was carried out at 335 °C for 17 h under 23 NmL/min of pure hydrogen. All hydrogenation reactions were performed at 270 °C, atmospheric pressure, and a space velocity of 0.92 NL/gcath. These conditions were found to be optimum in our previous investigation [8]. The feed gas to the reactor consisted of a mixture containing 21.8% CO2, 68.6% H2 and 9.6% N2. The reduced and passivated catalysts were activated as described in the previous lines. Passivation was done with oxygen diluted in helium (5% O2) flowing at 23 NmL/min for 2 h at room temperature. Gas chromatography was used to analyse the reactor effluent. A Dani Master GC, equipped with a flame ionization detector (FID) and a thermal conductivity detector (TCD), was used for this purpose. The separation of hydrocarbons was done in a capillary column (Supel-Q™ PLOT) while that of H2, N2, CO and CO2 was done in a packed column (60/80 Carboxen 1000).
3 Results and Discussion
3.1 Effect of Ruthenium
3.1.1 X-ray Diffraction
XRD spectra of ruthenium-promoted catalysts after calcination, reduction/passivation, and spent catalysts are illustrated in Fig. 1. Calcined catalysts containing 1 and 3 wt % ruthenium showed similar patterns. Only the reflection planes corresponding to Co3O4 could be identified on both catalysts. Oxides of ruthenium were not detected, indicating a highly dispersed phase. Reduced and passivated catalysts, on the other hand, did not show any reflection line corresponding to Co3O4. After activation, Co3O4 was reduced to CoO and metallic cobalt as indicated by the corresponding spectra. In addition, a ruthenium peak was also detected for both activated catalysts. This peak overlapped with one of metallic cobalt peaks as shown on the diagram. The spent catalysts did not show any diffraction peak indicating that the samples became amorphous after the reaction. Another possibility is that the particle size could be below the detection limit of the instrument. The XRD spectrum of the base catalyst (15%Co-1%K/SiO2) is not included in Fig. 1 as it has been published in our previous work [8]. The calcined base catalyst also presented only diffraction peaks associated with Co3O4.

XRD patterns of a 15%Co-1%K-1%Ru/SiO2 (calcined), b 15%Co-1%K-3%Ru/SiO2 (calcined), c 15%Co-1%K-1%Ru/SiO2 (reduced and passivated), d 15%Co-1%K-3%Ru/SiO2 (reduced and passivated), e 15%Co-1%K-1%Ru/SiO2 (spent), and f 15%Co-1%K-3%Ru/SiO2 (spent)
In order to investigate the effect of ruthenium on particle size, the Scherrer equation was used as previously indicated [8]. Particles size of the base catalyst and ruthenium-promoted catalysts are presented in Table 1. It can be seen that promoting with ruthenium considerably reduces the average particle size of all cobalt species present on the catalyst. The most significant drop in particle size occurred at 1% Ru where a decrease of ca. 20% for Co3O4 and ca. 53% for both CoO and Co was observed. A further increase in the catalyst’s ruthenium content had little effect on the particle size. It can be claimed, within experimental error, that catalysts containing 1 and 3 wt % ruthenium have similar particle size. These findings suggest that ruthenium improved cobalt dispersion. Similar observations have also been made in a number of studies [13, 14]. In fact, Kogelbauer et al. [14] investigated the promoting effect of ruthenium on alumina-supported cobalt catalysts. The average metallic cobalt particle size of all ruthenium-promoted catalysts was found to be roughly half of that of the unpromoted catalyst. It was suggested that ruthenium facilitates the reduction of small cobalt oxide species that strongly interact with the support. This would, in turn, result in additional small particles of metallic cobalt that would eventually decrease the average particle size.
3.1.2 Temperature-Programmed Reduction
TPR profiles of ruthenium-promoted and unpromoted catalysts are illustrated in Fig. 2. The unpromoted catalyst exhibits two maxima at 368 and 425 °C which were previously [8] assigned to the two reduction steps of cobalt species, namely, Co3O4 to CoO and CoO to Co respectively. It is evident that the addition of ruthenium shifted both maxima to lower temperatures. The peak associated with the reduction of Co3O4 to CoO shifted from 368 to 212 °C and that assigned to the reduction of CoO to Co decreased from 425 to 373 °C upon promoting the catalyst with 1 wt % ruthenium. This corresponds to a temperature drop of 156 and 52 °C respectively. Increasing the catalyst’s ruthenium content to 3 wt % further shifted both peak maxima to lower temperatures but to a much smaller extent. This is in good agreement with data reported by Jacobs et al. [15]. Our data indicate that ruthenium improved the catalyst’s reducibility. Xu et al. [16] have shown that RuO2, when supported on TiO2-Al2O3, can be reduced between 146 and 162 °C depending on the calcination temperature. This narrow temperature range falls below the reduction maxima observed in this study for the unpromoted catalyst. It can therefore be argued that RuO2 is first reduced to metallic ruthenium which activates hydrogen and spills it over cobalt oxide species. As a consequence, the reduction of cobalt oxide species occurs at much lower temperatures, leading to increased extent of reduction [13, 14].

TPR profiles for a 15%Co-1%K/SiO2, b 15%Co-1%K-1%Ru/SiO2, c 15%Co-1%K-3%Ru/SiO2
3.1.3 Surface Areas and Textural Data
The BET measurements were performed on the calcined ruthenium-promoted catalysts. The BET surface area, total pore volume, and pore sizes are shown in Table 2.
The BET surface area of the catalysts increased with the addition of ruthenium promoter compared to the catalyst promoted with potassium only. The total pore volume increased when ruthenium was introduced to the catalyst and decreased slightly when the ruthenium content was increased to 3 wt %. The increase in surface area when ruthenium was added could mean that the ruthenium was deposited mostly on the surface of the catalyst and not in the pores. The decrease in the surface area and the total pore volume when ruthenium content was increased could be due to some pores being obstructed due to ruthenium being deposited inside the pores of the catalyst.
3.1.4 Catalyst Testing
The effect of ruthenium incorporation in the base catalyst is shown in Table 3. It can be seen that at a ruthenium loading of 1 wt %, there was a slight improvement in CO2 conversion from 16 to 18.6% and an increase in methane selectivity from 37.6 to 44.4%. This is in line with TPR data which revealed an improved degree of reduction for ruthenium-promoted catalysts. In fact, the high degree of reduction resulted in increased number of active sites available for CO2 hydrogenation. These findings are corroborated by a number of studies [10, 17, 18] which have shown that cobalt catalysts possessing a higher degree of reduction are more selective towards methane and possess a higher CO2 hydrogenating activity. It is understood that this behaviour is dictated by the amount metallic cobalt available on the catalyst. The CO selectivity, on the other hand, was observed to decrease from 31.9 to 26.2% while the C5+ selectivity dropped from 7.8 to 5.9%. The change in C2+ yield was less than a percentage and can be considered negligible within experimental error.
Adding more ruthenium to the catalyst (3 wt %) did not induce significant changes in its performance except increased methane selectivity at the expense of C2+ hydrocarbons. The yield of hydrocarbons other than methane was not affected by the presence of ruthenium, with fluctuations of less than a percentage. This is consistent with both XRD and TPR data which showed similar dispersion and reducibility for all ruthenium-promoted catalysts. It is important, however, to underline that ruthenium has been reported to be active in CO2 hydrogenation and highly selective towards methane [19,20,21]. Since the amounts of ruthenium employed in this study are relatively high, contributions associated with ruthenium in catalyst’s performance cannot be excluded.
3.2 Effect of Palladium
3.2.1 X-ray Diffraction
The diffractograms of palladium-promoted catalysts are shown in Fig. 3. It is evident that, as in the case of ruthenium-promoted catalysts, only reflection planes associated with Co3O4 were visible in the calcined catalysts. Palladium species were not detected in the calcined catalysts, suggesting the presence of amorphous or highly dispersed species. The reduced and passivated catalysts, however, exhibited reflection planes corresponding to metallic palladium, cobalt and CoO as indicated in Fig. 3. As observed on the ruthenium-promoted catalysts, the spent catalysts did not show any diffraction peak indicating that the samples became amorphous after the reaction. Another possibility is that the particle size could be below the detection limit of the instrument. The effect of palladium on cobalt species particle size was also evaluated. The Sherrer equation was used for this purpose and the obtained data are presented in Table 4. It is seen that adding palladium to the base catalyst reduces the particle size of all cobalt species. This behaviour is more pronounced at 1 wt % Pd loading compared to the catalyst promoted with 3 wt % Pd which shows little change in particle size. It is worth noting that this pattern was also observed with ruthenium-promoted catalysts as discussed earlier. Thus, a similar argument can be made with respect to possible reasons behind the decrease in particle size in the presence of palladium.

XRD patterns of a 15%Co-1%K-1%Pd/SiO2 (calcined), b 15%Co-1%K-3%Pd/SiO2 (calcined), c 15%Co-1%K-1%Pd/SiO2 (reduced and passivated), d 15%Co-1%K-3%Pd/SiO2 (reduced and passivated), e 15%Co-1%K-1%Pd/SiO2 (spent), and f 15%Co-1%K-3%Pd/SiO2 (spent)
3.2.2 Temperature-Programmed Reduction
The reduction behaviour of palladium-promoted catalysts along with the base catalyst is shown Fig. 4. Two major hydrogen consumption peaks, assigned to the two-step reduction of cobalt species, can be seen for all catalysts. Palladium appears to act as a reduction catalyst, shifting both peaks to much lower temperatures. Upon promoting with 1 wt % Pd, the peak associated with Co3O4 reduction decreased from 368 to 165 °C, corresponding to a 203 °C shift. The peak associated with the reduction of CoO, on the other hand, dropped from 425 to 406 °C, representing a shift of 19 °C. Increasing palladium content to 3 wt % further lowered the reduction temperatures of cobalt oxides. Peaks associated with palladium oxide which has been reported to undergo reduction at room temperature [22,23,24] were not detected. Thus, palladium oxide reduction probably occurred during signal conditioning of the thermal conductivity detector.

TPR profiles for a 15%Co-1%K/SiO2, b 15%Co-1%K-1%Pd/SiO2, c 15%Co-1%K-3%Pd/SiO2
3.2.3 Surface Aareas and Textural Data
The BET measurements were performed on the calcined palladium-promoted catalysts. The BET surface area, total pore volume, and pore sizes are shown in Table 5.
The BET surface area of the catalysts increased with the addition of palladium promoter compared to the catalyst promoted with potassium only. Contrary to ruthenium, the total pore volume increased when palladium was introduced to the catalyst and increased with its content in the catalyst. There was no explanation we could give for the behavior observed on the BET surface area and the pore volume and size for catalysts containing Pd where the surface area decreased with increasing Pd loading while the pore volume and pore size also increased.
3.2.4 Catalyst Testing
The impact of palladium promotion on the catalyst’s activity and products distribution is given in Table 4. Promoting the catalyst with 1 wt % Pd resulted in increased CO2 conversion from 16 to 19.1%. The methane productivity also increased at the expense of C2+ hydrocarbons as indicated by selectivity and yield data. These findings are consistent with the TPR analysis which demonstrated that palladium-promoted catalysts possess a higher degree of reduction compared to the base catalyst. In fact, previous reports [10, 17, 18] have linked high activity and methane selectivity to metallic cobalt. Since the palladium-promoted catalysts possess a higher degree of reduction, this means more metallic cobalt is available for the reaction. Nevertheless, the CO selectivity remained relatively constant.
Interestingly, increasing the palladium loading to 3 wt % led to a CO2 conversion similar to that obtained with the base catalyst as shown in Table 6. Since TPR and XRD data of the catalysts promoted with 1 and 3 wt % Pd suggested similar degree of reduction and cobalt particle size, both catalysts were expected to perform similarly in CO2 hydrogenation. However, increasing the palladium content from 1 to 3 wt % resulted in a drop in CO2 conversion from 19.1 to 16.6% and in C2+ hydrocarbons productivity. The methane selectivity also decreased from 45.4 to 41.8% while the CO selectivity increased from 31.5 to 40%.
A lower activity and methane selectivity for the catalyst containing 3 wt % Pd suggests that this catalyst possess a lower degree of reduction compared to the one promoted with 1 wt % Pd. However, this argument can be ruled out in the light of TPR data as pointed out earlier. One probable explanation for the observed performance could be that at high palladium loading (3 wt %), some of the metallic cobalt particles become covered by palladium particles and thus inaccessible for CO2 hydrogenation. In addition, CO2 hydrogenation over a series of palladium-supported catalysts has revealed that palladium is active in the reverse water–gas shift reaction [25, 26]. This would justify the significant increase in CO selectivity at high palladium loading. It is worth noting that a similar behaviour has also been reported for a platinum-promoted cobalt catalyst where platinum surface enrichment was found to be responsible for increased reverse water–gas shift activity [6].
3.3 Effect of Copper
3.3.1 X-ray Diffraction
The XRD patterns of potassium – promoted cobalt catalysts containing 1 to 3% copper are illustrated in Fig. 5. Calcined catalysts containing 1 and 3% copper showed identical diffraction lines which have been assigned to Co3O4. The reduced and passivated counterpart, on the other hand, indicated that after reduction, some of the cobalt species remained in oxide form (CoO) along with metallic cobalt. Copper oxide and alloys of copper and cobalt could not be detected in all catalysts. This suggests that the copper phase is highly dispersed given the fact that it only represents a small fraction of the catalyst. The spent catalysts did not show any diffraction peak indicating that the samples became amorphous after the reaction. Another possibility is that the particle size could be below the detection limit of the instrument. The particle size of the different cobalt phases before and after reduction was calculated as earlier using the Scherrer equation and the related data are presented in Table 7. For the freshly calcined catalysts, it is evident that the size of Co3O4 clusters decreased with increased copper loading. However, for the reduced and passivated catalysts, a different behaviour was observed. In fact, there was a negligible increase of less than a nanometer for CoO and Co clusters at 1 wt % Cu loading. A decrease in CoO and Co cluster sizes was only observed at a higher copper loading of 3 wt %. The CoO cluster size decreased from 7.30 to 4.90 nm while the cluster size of metallic cobalt decreased from 7.34 to 5.05 nm. These observations lead to the conclusion that the introduction of copper in the catalyst, especially at a higher loading (3 wt %), improves the dispersion of the different cobalt phases, namely Co3O4, CoO and Co.

XRD patterns of a 15%Co-1%K-1%Cu/SiO2 (calcined), b 15%Co-1%K-3%Cu/SiO2 (calcined), c 15%Co-1%K-1%Cu/SiO2 (reduced and passivated), d 15%Co-1%K-3%Cu/SiO2 (reduced and passivated), e 15%Co-1%K-1%Cu/SiO2 (spent), and f 15%Co-1%K-3%Cu/SiO2 (spent)
3.3.2 Temperature-Programmed Reduction
The TPR profiles of copper-containing catalysts are compared relative to that of the base catalyst (15%Co-1%K/SiO2) in Fig. 6. As discussed earlier, the major peaks for the base catalyst observed at 368 and 425 °C were attributed to the two-step reduction of Co3O4 to CoO and CoO to Co respectively. Upon introduction of 1 wt % copper in the base catalyst, only one hydrogen consumption peak appears at 386 °C. Since XRD revealed the presence of Co3O4 clusters in calcined catalysts, this peak can be attributed to both reduction steps overlapping. Furthermore, because the emergence of the base catalyst peak at 368 °C coincides with that of the promoted catalyst peak at 386 °C, it can be argued that only the second reduction step, namely CoO to Co was promoted by the presence copper. In addition, increasing the amount of copper in the catalyst to 3 wt % revealed no apparent improvement in the catalyst reducibility. In fact, at 3 wt % Cu loading, the TPR profile was identical to the one obtained at 1 wt % Cu. Increased cobalt catalyst reducibility upon copper addition has also been observed by Leite et al. [27].

TPR profiles for a 15%Co-1%K/SiO2, b 15%Co-1%K-1%Cu/SiO2, c 15%Co-1%K-3%Cu/SiO2
3.3.3 Surface Areas and Textural Data
The BET measurements were performed on the calcined copper-promoted catalysts. The BET surface area, total pore volume, and pore sizes are shown in Table 8.
The BET surface area of the catalysts increased with the addition of copper promoter compared to the catalyst promoted with potassium only. The total pore volume increased when copper was introduced to the catalyst and decreased slightly when the copper content was increased to 3 wt %. The increase in surface area when copper was added could mean that the copper was deposited mostly on the surface of the catalyst and not in the pores. The decrease in the surface area and the total pore volume when copper content was increased could be due to some pores being obstructed due to copper being deposited inside the pores of the catalyst.
3.3.4 Catalyst Testing
Copper addition effects in CO2 hydrogenation are presented in Table 9. It is evident that the presence of copper negatively affected the catalyst activity which dropped from 16 to 13% when 1 wt % copper was introduced. However, an increased CO selectivity and a drop in hydrocarbons productivity can be noted. A similar pattern was also observed with the catalyst containing 3 wt % Cu. According to TPR data, copper-containing catalysts possess a higher degree of reduction in comparison to the base catalyst. This should in turn result in more metallic cobalt and therefore in more active catalysts as discussed earlier. The decreased activity has been attributed the poisoning effect of copper on cobalt sites as a consequence of copper surface enrichment [28]. In addition, the suppressed methane selectivity and increased CO selectivity in copper-promoted catalysts indicate a higher rate of the reverse water–gas shift reaction for these catalysts. It is worth noting that there was a drop in cobalt particle size in the catalyst promoted with 3 wt % Cu. This parameter could also be a contributing factor in catalyst deactivation. In fact, Iablokov et al. [29] specifically investigated the influence of cobalt particle size in the hydrogenation of carbon dioxide. The model catalysts contained cobalt nanoparticles with sizes of 3, 7 and 10 nm deposited on mesoporous silica (MCF-17). It was found that CO2 conversion increases with particle size. This is in line with similar studies on the hydrogenation of carbon monoxide, where this behaviour has been attributed to a greater susceptibility to oxidation of smaller cobalt particles. The products distribution was not significantly altered by the variation in particle size. The main products consisted of methane and CO. However, the methane selectivity dependence on cobalt particle size was not established.
4 Conclusion
The aim of the present study was to investigate ruthenium, palladium, and copper as promoters in CO2 hydrogenation. All these promoters were found to improve the catalyst reducibility and dispersion. Ruthenium and palladium promoted both steps of cobalt reduction while copper only promoted the second reduction step. Ruthenium increased CO2 conversion and methane selectivity but negatively affected C5+ hydrocarbons productivity. This behaviour was ascribed to increased active site density for ruthenium containing catalysts. Similar data were also recorded for the catalyst containing 1 wt % palladium. However, a drop in CO2 conversion and increased reverse water–gas shift activity were observed at 3 wt % Pd loading. It was suggested that some of the Co active sites were covered by palladium and hence could not be accessed by the reactants. Promoting with copper resulted in decreased activity, methane selectivity and C5+ hydrocarbons productivity with a much higher CO selectivity. Although copper improved the catalyst reducibility, it also acted as a poison which resulted in poor activity.
References
Diehl F, Khodakov AY (2009) Promotion of cobalt Fischer-Tropsch catalysts with noble metals: a review. Oil & Gas Sci Technol-Revue de l’IFP 64(1):11–24
Zhu Y, Zhang S, Ye Y, Zhang X, Wang L, Zhu W, Cheng F, Tao F (2012) Catalytic conversion of carbon dioxide to methane on ruthenium–cobalt bimetallic nanocatalysts and correlation between surface chemistry of catalysts under reaction conditions and catalytic performances. ACS Catal 2(11):2403–2408
Owen RE, O’Byrne JP, Mattia D, Plucinski P, Pascu SI, Jones MD (2013) Cobalt catalysts for the conversion of CO2 to light hydrocarbons at atmospheric pressure. Chem Commun 49(99):11683–11685
Boix AV, Ulla MA, Petunchi JO (1996) Promoting effect of Pt on Co mordenite upon the reducibility and catalytic behavior of CO2 hydrogenation. J Catal 162(2):239–249
Bardi U, Beard BC, Ross PN (1990) CO chemisorption on the [111] and [100] oriented single crystal surfaces of the alloy CoPt3. J Catal 124(1):22–29
Alayoglu S, Beaumont SK, Zheng F, Pushkarev VV, Zheng H, Iablokov V, Liu Z, Guo J, Kruse N, Somorjai GA (2011) CO2 hydrogenation studies on Co and CoPt bimetallic nanoparticles under reaction conditions using TEM. XPS and NEXAFS Topics Catal 54(13–15):778–785
Beaumont SK, Alayoglu S, Specht C, Michalak WD, Pushkarev VV, Guo J, Kruse N, Somorjai GA (2014) Combining in situ NEXAFS spectroscopy and CO2 methanation kinetics to study Pt and Co nanoparticle catalysts reveals key insights into the role of platinum in promoted cobalt catalysis. J Am Chem Soc 136(28):9898–9901
Iloy RA, Jalama K (2019) Effect of operating temperature, pressure and potassium loading on the performance of silica-supported cobalt catalyst in CO2 hydrogenation to hydrocarbon fuel. Catalysts 9(10):807
Vosoughi V, Dalai AK, Abatzoglou N, Hu Y (2017) Performances of promoted cobalt catalysts supported on mesoporous alumina for Fischer–Tropsch synthesis. Appl Catal A 547:155–163
Gnanamani MK, Jacobs G, Keogh RA, Shafer WD, Sparks DE, Hopps SD, Thomas GA, Davis BH (2015) Fischer–Tropsch synthesis: effect of pretreatment conditions of cobalt on activity and selectivity for hydrogenation of carbon dioxide. Appl Catal A 499:39–46
Russell WW, Miller GH (1950) Catalytic hydrogenation of carbon dioxide to higher hydrocarbons. J Am Chem Soc 72(6):2446–2454
Khangale PR, Meijboom R, Jalama K (2020) CO2 hydrogenation to liquid hydrocarbons via modified Fischer–Tropsch over alumina-supported cobalt catalysts: Effect of operating temperature, pressure and potassium loading. J CO2 Util 41:101268
Parnian MJ, Najafabadi AT, Mortazavi Y, Khodadadi AA, Nazzari I (2014) Ru promoted cobalt catalyst on γ-Al2O3: influence of different catalyst preparation method and Ru loadings on Fischer–Tropsch reaction and kinetics. Appl Surf Sci 313:183–195
Kogelbauer A, Goodwin JG Jr, Oukaci R (1996) Ruthenium promotion of Co/Al2O3 Fischer–Tropsch catalysts. J Catal 160(1):125–133
Jacobs G, Das TK, Zhang Y, Li J, Racoillet G, Davis BH (2002) Fischer-Tropsch synthesis: support, loading, and promoter effects on the reducibility of cobalt catalysts. Appl Catal A 233(1–2):263–281
Xu J, Lin Q, Su X, Duan H, Geng H, Huang Y (2016) CO2 methanation over TiO2–Al2O3 binary oxides supported Ru catalysts. Chin J Chem Eng 24(1):140–145
Melaet G, Ralston WT, Li CS, Alayoglu S, An K, Musselwhite N, Kalkan B, Somorjai GA (2014) Evidence of highly active cobalt oxide catalyst for the Fischer–Tropsch synthesis and CO2 hydrogenation. J Am Chem Soc 136(6):2260–2263
Das T, Deo G (2011) Synthesis, characterization and in situ DRIFTS during the CO2 hydrogenation reaction over supported cobalt catalysts. J Mol Catal A: Chem 350(1):75–82
Weatherbee GD, Bartholomew CH (1984) Hydrogenation of CO2 on group VIII metals: IV. Specific activities and selectivities of silica-supported Co, Fe, and Ru. J Catal 87(2):352–362
Li D, Ichikuni N, Shimazu S, Uematsu T (1998) Catalytic properties of sprayed Ru/Al2O3 and promoter effects of alkali metals in CO2 hydrogenation. Appl Catal A 172(2):351–358
Kwak JH, Kovarik L, Szanyi J (2013) CO2 reduction on supported Ru/Al2O3 catalysts: cluster size dependence of product selectivity. ACS Catal 3(11):2449–2455
Noronha FB, Schmal M, Moraweck B, Delichere P, Brun M, Villain F, Fréty R (2000) Characterization of niobia-supported palladium–cobalt catalysts. J Phys Chem B 104(23):5478–5485
Juszczyk W, Karpinski Z, Lomot D, Pielaszek J, Paal Z, Stakheev AY (1993) The structure and activity of silica-supported palladium–cobalt alloys I: alloy homogeneity, surface composition, and activity for neopentane conversion. J Catal 142(2):617–629
Stonkus V, Edolfa K, Leite L, Sobczak JW, Plyasova L, Petrova P (2009) Palladium-promoted Co–SiO2 catalysts for 1, 4-butanediol cyclization. Appl Catal A 362(1–2):147–154
Erdöhelyi A, Pásztor M, Solymosi F (1986) Catalytic hydrogenation of CO2 over supported palladium. J Catal 98(1):166–177
Díez-Ramírez J, Valverde JL, Sánchez P, Dorado F (2016) CO2 hydrogenation to methanol at atmospheric pressure: influence of the preparation method of Pd/ZnO catalysts. Catal Lett 146(2):373–382
Leite L, Stonkus V, Edolfa K, Ilieva L, Plyasova L, Zaikovskii V (2006) Copper-promoted cobalt catalysts for 2, 3-dihydrofuran synthesis. Appl Catal A 311:86–93
Jacobs G, Ribeiro MC, Ma W, Ji Y, Khalid S, Sumodjo PT, Davis BH (2009) Group 11 (Cu, Ag, Au) promotion of 15% Co/Al2O3 Fischer–Tropsch synthesis catalysts. Appl Catal A 361(1–2):137–151
Iablokov V, Beaumont SK, Alayoglu S, Pushkarev VV, Specht C, Gao J, Alivisatos AP, Kruse N, Somorjai GA (2012) Size-controlled model Co nanoparticle catalysts for CO2 hydrogenation: synthesis, characterization, and catalytic reactions. Nano Lett 12(6):3091–3096
Khangale PR (2021) Hydrogenation of CO2 to hydrocarbons over zirconia-supported cobalt catalyst promoted with potassium. Catal Lett 2021(152):2745–2755
Acknowledgements
The authors wish to acknowledge the National Research Foundation (Grant: UID TTK2203301181) and the University of Johannesburg Global Excellence Stature (GES) program.
Funding
Open access funding provided by University of Johannesburg. This work is supported by the National Research Foundation (Grant No. TTK2203301181).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Iloy, R.A., Jalama, K. & Khangale, P.R. Effect of a Second Promoter on the Performance of a Potassium Doped Silica-Supported Cobalt Catalyst During CO2 Hydrogenation to Hydrocarbons. Catal Lett 154, 2818–2828 (2024). https://doi.org/10.1007/s10562-023-04527-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-023-04527-4