
Abstract
Mesoporous silica materials (MSMs) are widely used materials in many applications due to their diverse pore structures. However, the electrical conductivity of MSMs is poor which limits their use in electrochemical applications. In this study, widely used MSMs of different structural properties such as MCM-41, MCM-48, SBA-15, and SBA-16 were synthesized and reinforced with graphene oxide (GO) to obtain conductive composite supports for enzyme immobilization. MSMs were first synthesized using a hydrothermal method and characterized by Fourier-transform infrared spectroscopy, X-ray crystallography, scanning electron microscopy/energy dispersive X-ray, and MAPPING techniques. Aqueous dispersion of GO:MSM composites were prepared with as-synthesized materials and coated on screen-printed electrodes (SPE). The best composites were chosen based on their electroanalytical performance. Glucose oxidase (GOx) was then immobilized on modified SPEs using a simple drop-casting method to produce enzymatic electrodes. The electroanalytical performance of the enzymatic electrodes was investigated using different glucose concentrations to demonstrate biocatalytic activity. Stability tests were performed using intraday and interday measurements which revealed that SPE/GO:MCM-41/GOx electrode showed a more stable performance (3-folds) than SPE/GO/GOx electrode. This study presents an investigation of MSM mixed with GO in enzymatic electrochemical systems providing insight into the use of such materials to preserve enzyme activity.
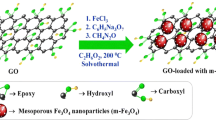
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Mesoporous silica materials (MSMs) have widely been used as adsorbents, thin films, low-k materials, nanowires, and catalysts due to their large specific surface areas and pore sizes ranging from 2 to 50 nm [1]. Some of the most common MSMs known in the market are SBA-15 and M41S-family due to their high surface area, large pore volume, regular pore distribution, and flexible synthesis conditions [2,3,4]. There are different variations of MSMs available with various pore shapes and sizes of both families providing opportunities in catalysis, drug delivery and imaging applications.
MSMs with diverse pore configurations include MCM-41 (hexagonal pores), MCM-48 (cubic pores), and MCM-50 (unstable lamellar pores) [5]. Furthermore, well-known MSMs have space groups of p6mm (MCM-41), Ia3d (MCM-48), p6mm (SBA-15), and Im3m (SBA-16) [6]. Silica materials have a high number of silanol groups on their surface, we can readily adjust the physicochemical nature of the surface through the functionalization process. On the other hand, MSMs have a large surface area and a high pore volume, which means they have a high loading capacity and are unique prospects for various applications [7] such as biomaterials [8], catalyst [9], adsorbent [10], sensor [11], and drug delivery [12].
In the M41S family, the pores of MCM-41 type catalysts are in a two-dimensional hexagonal structure, while the pores of MCM-48 are in a three-dimensional cubic structure [13, 14]. SBA-15, on the other hand, has high thermal stability and a wheat chain structure and SBA-16 is considered the most interesting mesothelium among SBA-type silica materials and is widely used in the field of biomaterials due to its spherical morphology [8, 15]. Different methods are used in the synthesis of MSMs such as impregnation, precipitation/co-precipitation, sol–gel, ion exchange, thermal fusion, solid–liquid leaching, and wet impregnation [16]. However, depending on the thermal strength and the amount of material obtained after synthesis, the hydrothermal synthesis method is widely used in the synthesis of silica-derived mesoporous materials since it can eliminate the use of high-grade materials.
Silica-based materials are known as poor electrical conductors, but they have been utilized in electrochemical systems. There are several examples of the way silica-based materials were employed in these systems such as deposited on conductive electrode surfaces or as thin films, manufactured as metal or carbon composites, and dispersed into conductive composites [17]. Therefore, they can be utilized as an immobilization matrix for selective ligands, electrocatalysts, metal particles, and enzymes [18]. There are also examples of MSMs reinforced with carbon-based materials such as carbon nanomembranes [19] and mesoporous carbon [20] mainly for capacitor applications. This potential of MSMs in electrochemical systems opened up possibilities for their use in immunological sensors, aptasensors, and enzymatic biosensors [21,22,23]. Such systems can offer a variety of opportunities for different applications such as diagnosis of bacterial infections, cancer therapy, cancer detection, detection of heavy metals, contaminated food detection, the detection of viruses, and enzymatic biosensors [22].
The use of MSMs in enzymatic biosensing is a promising application since the critical aspects of enzymatic systems can be supported by MSMs due to their critical properties. MSMs are shown to be effective in preserving the activity of enzymes due to their porous structure providing a protective environment for the enzymes [24, 25]. Furthermore, the high specific pore volume of silica particles can accommodate high enzyme loadings, thus resulting in high current density from enzymatic electrochemical reactions. There are several studies demonstrating the use of MSMs in enzymatic electrochemical systems for the detection of cholesterol [26], catechol [27], ethanol [28], hydrogen peroxide [29], lactic acid [30], uric acid [31], and glucose [28]. Different composites were prepared to increase the conductivity of the MSM-modified electrodes and to retain the enzyme activity for glucose detection such as gold nanoparticles [32, 33], Prussian-blue [34], Nafion [35, 36], and single-walled carbon nanotubes [37].
There are a few studies in the literature demonstrating the use of enzyme-incorporated MSMs for biosensing applications using glucose oxidase (GOx) and laccase. In these studies, Yusan et. al. demonstrated that nanoparticle selenium incorporated MCM-41 could be very effective in retaining enzyme activity [38]. On the other hand, another study by Tvorynska et al. also showed that the MCM-41 incorporated sensor was found to be the most stable sensor configuration for laccase-based biosensors [39]. Most of the studies utilizing MSMs for enzymatic biosensors showed promising performance and good stability, yet there is still a need for comprehensive and systematic studies to demonstrate the effect of the conducting composite materials and the performance of the MSMs on retaining enzyme activity fundamentally.
Herein, four different MSMs from two different types (M41S and SBA) were first synthesized using a hydrothermal method. Then, graphene oxide (GO)-MSM composites were prepared as dispersions of as-synthesized material of optimized amounts into a conductive aqueous GO matrix. Screen-printed electrodes (SPEs) were modified with different material loadings of the prepared composite dispersions and electrochemically characterized using voltammetry and amperometry. The electroanalytical performance of the GOx immobilized electrodes was investigated in terms of the sensitivity and stability of the enzyme. As a result, this study aims to present a comprehensive investigation of the performance of MSMs reinforced with GO in enzymatic electrochemical applications for the first time in the literature. Therefore, it can provide insight into the use of MSMS as additives to carbon-modified electrochemical electrodes (such as graphene, nanotubes, graphite, etc.) to retain enzyme activity.
2 Experimental
2.1 Materials
Pluronic p 123, Pluronic p 127, and the chemicals used in electrochemical studies were obtained at analytical grade from Sigma-Aldrich. Cetyltrimethylammonium bromide (CTMAbr), tetraethyl orthosilicate, (TEOS), and sodium silicate were obtained from Merck.
2.2 Synthesis and Characterization of Mesoporous Silica
The synthesis of MCM-41, SBA-15, MCM-48, and SBA-16 was carried out using the hydrothermal method according to the literature [40,41,42,43]. Briefly, the surfactant was dissolved at different temperatures and durations (MCM-41 and MCM-48; 30 °C, 2–6 h, SBA-15; 40 °C, 2 h, SBA-16; 38 °C, 2 h) using a magnetic stirrer (Elektro-mag, Turkey) following by filtration and drying. The synthesis is then completed with the calcination process at different temperatures to remove impurities in the structure of materials (MCM-41 and MCM-48: 550 °C and 6 h, SBA-15 and SBA-16: 540 °C and 5 h). Although the same hydrothermal synthesis method is used in the synthesis of these mesoporous materials, different chemicals are used as surfactants (CTMAbr, pluronic p 123, and pluronic p 127) and silica sources (TEOS and sodium silicate).
Fourier-transform infrared spectroscopy (FT-IR) analyses of mesoporous materials were performed using the Perkin Elmer IR USA (Attenuated Total Reflectance (ATR) technique) device between 380 and 4000 cm−1. The Panalytical Empryan HT (Netherlands) instrument was used to perform X-ray crystallography (XRD) analyses to determine the structural phases of mesoporous materials using CuK (= 1.540) radiation, 0.066 step size (sensitivity), 30 V (tension), 40 kV (current), and 0° < 2θ < 70° range. The surface morphologies of the samples were analyzed using scanning electron microscopy/energy dispersive X-ray (SEM/EDX, Zeiss SUPRA V40, Germany) analysis. In addition, the MAPPING analysis method was used to determine the distributions of C and Si elements in the structure of the catalyst sample.
2.3 Electrode Preparation and Characterization
All electrochemical experiments were carried out at 23 ± 1 °C using Ivium Potentiostat (Ivium Technologies, Netherlands) and carbon SPE (Model: Dropsens DRP-X1110 with a carbon working electrode surface area of 0.059 cm2, obtained from Metrohm AG, Switzerland). Carbon and silver paste electrodes were used as the counter, and reference electrodes, respectively. SPEs were pre-treated using linear sweep voltammetry (LSV) in a solution containing 0.1 M KCl to remove impurities on the working electrode surface and obtain reproducible results before any experiments [44]. Aqueous dispersions of GO (1 mg/mL, Ultra-pure water, 18.2 MΩ-cm, 4–10% edge oxidized exfoliated graphene nanoplatelets, Sigma-Aldrich) and GO-mesoporous silica mixtures with different mass ratios (1:1, 1:1.5, and 1:2) were prepared and sonicated (Bandelin RK 100 H, Germany) until homogenous mixtures were obtained. Then, the working electrode of the SPEs was drop-coated until a material loading of 0.15 mg/cm2 was achieved [45]. This value was chosen due to physical constraints of the working electrode surface area and above this level, the coating failed. The coated electrodes with different material ratios were then electrochemically characterized using cyclic voltammetry (CV, 50 mV/s) in a solution containing 2 mM K3Fe(CN)6/K4Fe(CN)6 redox couple in 0.1 M KCl. Bare electrode (BE) and GO-only coated SPE were also tested as control experiments. The optimal GO to mesoporous silica amount ratio was chosen based on the anodic and cathodic current changes in CV experiments.
2.4 Enzyme Immobilization and Electrochemical Glucose Oxidation
First, drop-coating 1 µL of ethanoic Nafion solution (0.05% w/w in ethanol) as a supporting layer and dried at room temperature for 15 min. Subsequently, drop-coating 1 µL of GOx [1, 5, and 10 mg/mL in 0.1 M phosphate buffer (PBS), pH 7.4] on Nafion-modified SPEs created an active enzymatic layer for electrochemical glucose oxidation. All prepared electrodes were kept at 4 °C for 24 h and immersed in PBS for 15 min following consecutive washing steps at least 3 times to remove weakly adsorbed species before use. The prepared enzymatic electrodes were denoted as SPE/GO/GOx, SPE/GO:MCM-41/GOx, SPE/GO:MCM-48/GOx, SPE/GO:SBA-15/GOx, and SPE/GO:SBA-16/GOx. The electrochemical glucose oxidation was tested using chronoamperometry (CA) with an applied voltage of 0.14 V for 120 s in 1 mM ferrocene carboxylic acid (FcCOOH, in 0.1 M PBS, pH 7.4) containing 0 and 5 mM glucose. The enzyme concentration for the immobilization was also optimized using LSV (5 mV/s) and used for the electrochemical performance tests. All electrochemical tests were conducted with three independently prepared electrodes unless otherwise stated (N = 3 samples). A schematic representation of electrode preparation steps is shown in Fig. 1.

Schematic representation of electrode preparation steps using MSMs
3 Results and Discussion
3.1 Characterization Studies of the Mesoporous Silica Materials
Characterizations of the synthesized MCM-48, MCM-41, SBA-16, and SBA-15 were first performed using FT-IR, XRD, and SEM. FT-IR analysis results of silica-derived mesoporous materials showed that Si–O–Si bands of the silica structure were seen at wavelengths of 1059 cm−1 [46], 1053 cm−1 [47], 1065 cm−1 [15], and 1064 cm−1 [48], respectively (Fig. 2a). The bands of MCM-41 at 965 and 789 cm−1 correspond to the Si–OH and Si–O structures, respectively (Fig. 2a) [49]. In addition, the peaks at 2927 cm−1 and 2857 cm−1 belong to the expansion of the CH(CH2CH2CH2NH2) structure of MCM-48 (Fig. 2a) [46]. –OH stretch can be seen at the 1635 cm−1 band and the water in the structure shows a wide bandgap of around 3368 cm−1 (especially for MCM-41 and MCM-48; Fig. 2a) [50]. The symmetrical stretching vibration mode of O–H of isolated central silanol (Si–OH) groups are represented by the peaks at 3725 cm−1 (SBA-15; Fig. 2a) and 3749 cm−1 (MCM-48; Fig. 2a) [50, 51]. Although shifts were observed in the Bragg fundamental peaks from the XRD analysis result of MCM-41, the basic Bragg peaks of d(100), d(110), and d(200) reflections were obtained. These baseline Bragg peak values obtained showed that MCM-41 had a regular hexagonal structure (Fig. 2b) [50].

a FT-IR analysis b low-angle XRD patterns and c SEM images of MCM-41, MCM-48, SBA-15, SBA-16, GO and GO:MCM-41
Due to the SiO2 groups in the structures of silica-based materials (MCM-41, MCM-48, SBA-15, and SBA-16), no clear differences may be observed in the FTIR results. In addition, precise determinations may not be possible due to the wide peaks of OH and Si–O–Si structures. After the modification processes, clear differences can be observed in the FT-IR analysis results depending on the functional groups. A more narrow wavelength result for the FT-IR analyses are also given in Figs. S7 and S8 to emphasize some of the peaks in Fig. 2a.
MCM-48’s main Bragg peaks (d(211) and d(220)) were measured at 2Θ:1.33 and 2.4, respectively (Fig. 2b) [42, 46]. Low-angle XRD analysis showed that the main Bragg peak (d110) of the SBA-16 support material was obtained at 2θ:0.84 (Fig. 2b) [15, 41]. XRD analysis for SBA-15 showed that d(100) and d(110) reflections were observed showing the main Bragg peaks of the mesoporous structure (Fig. 2b) [52].
According to the SEM analysis results, the cubic structure of MCM-48 [53], the hexagonal structure of MCM-41 [53], the spherical structure of SBA-16 [15], and the wheat chain structures of SBA-15 [54] were confirmed (Fig. 2c). Moreover, the EDX and MAPPING analysis were used to determine the distributions of C and Si elements in the structure (Figures S1–4). The SEM image of GO shows the randomly aggregated and crumpled sheets align with the previously reported characteristics [55]. Finally, the surface characteristics of the GO:MCM-41 composite were confirmed with SEM images demonstrating the crumbled GO sheets were wrapped around the hexagonal structure of MCM-41. Thereby, the MCM-41 particles are shown to be covered with GO flakes that would provide electrical conductivity for the prepared films (further confirmed by electrochemical measurements).
3.2 Electrochemical Characterization and Optimization of GO-Mesoporous Silica Ratio
To investigate the behaviour of the GO-mesoporous silica-coated SPEs at different material ratios, a series of CV experiments have been conducted in 0.1 M KCl containing 2 mM K3Fe(CN)6/K4Fe(CN)6. Figure 3 shows the voltammograms and respective anodic and cathodic peak current values with changing GO-mesoporous silica ratio.

CVs (50 mV/s) and anodic and cathodic peak current values of a SPE/GO:MCM-41, b SPE/GO:MCM-48, c SPE/GO:SBA-15, d SPE/GO:SBA-16 in 2 mM K3Fe(CN)6/K4Fe(CN)6 redox couple in 0.1 M KCl, (N = 3 samples)
A reversible redox response was observed for BE at ca. 0.18 V and ca. 0.075 V (vs Ag/Ag+) for oxidation and reduction processes, respectively. The voltage separation between anodic and cathodic peaks was ca. 0.1 V (vs Ag/Ag+) and the anodic to cathodic peak current ratio (ipa/ipc) was ca. 1.02 suggesting chemical reversibility and quasi-reversible electron transfer [56]. It can also be seen that the modification of SPEs with GO and GO-MSM at different ratios didn’t cause a significant difference in the electrochemical parameters.. However, different modifications on SPEs made a difference in the catalytic response between electrodes. GO-modified electrode (SPE/GO) showed a significant increase in anodic and cathodic peak current values showing higher peak current values than BE. On the other hand, electrodes modified with the silica materials for all mixing ratios showed lower peak current values than SPE/GO as the integration of electrically insulating silica with GO would be expected the lower the overall conductivity of the composite [57]. However, GO:MCM-41 modified electrode with a 1:1 ratio was the only configuration that resulted in the highest current values. As the pore volume and active surface area of the mesoporous silica increase, the interactions between GO and the silica material would change as well as the electrochemical response due to an increase in total electroactive area. MCM-41 was reported to have a relatively larger surface area and pore-loading capacity than other silica materials used in this study which could be the reason for the better response [40].
3.3 Electrochemical Glucose Oxidation Studies
After the electrochemical investigation of the GO-mesoporous silica materials, enzyme adsorption was performed to evaluate the performance of these compositions in enzymatic glucose oxidation. In this work, the molecular aspects of mesoporous silica materials were not taken into account in the performance of the enzymatic performance, rather it was aimed to compare the performance of the different types of silica materials in the performance of enzymatic glucose oxidation. Therefore a series of CA experiments were performed in 0.1 PBS solution containing 1 mM FcCOOH as an electron transfer mediator. FcCOOH was chosen as a reliable electron transfer mediator in aqueous electrochemistry that was widely used in the literature [58, 59]. The optimization studies for enzyme concentration using LSV revealed that increasing the enzyme concentration for immobilization didn’t cause a significant change in the performance of electrochemical glucose oxidation (Fig. S5). This could be due to the saturation that might be reached in the mesopores of the silica materials, hence further increasing the enzyme concentration wouldn’t make a significant contribution to the catalytic response due to mass transfer limitations. As a result, 1 mg/mL was chosen for the electrochemical experiments. After enzyme loading optimization, CA experiments were performed using modified SPEs with GO, GO:MCM-41, GO:MCM-48, GO:SBA-15, and GO:SBA-16 with a material loading of 0.15 mg/cm2 at a 1:1 ratio to investigate the analytical performance of the prepared electrodes. Figure 4 shows the CA response of SPE/GO/GOx, SPE/GO:MCM-41/GOx, and SPE/GO:MCM-48/GOx with increasing glucose concentration. SPE/GO:SBA-15/GOx and SPE/GO:SBA-16/GOx electrodes were also tested but didn’t show a linear response (Fig. S6). However, MCM-modified electrodes showed a relatively good response to different glucose concentrations up to 7 mM. SPE/GO:MCM-41/GOx showed the best performance among all silica-modified electrodes showing similar performance to GO-only electrodes. In terms of analytical performance the sensitivity of GO, GO:MCM-41, and GO:MCM-48-modified electrodes were calculated as 0.51 µA/mM, 0.39 µA/mM, and 0.17 µA/mM, respectively. These suggest that MCM-modified electrodes showed promising performance compared to other GO-modified electrodes without giving up a significant analytical performance similar to previous investigations without the incorporation of the enzyme. On the other hand, although this study didn’t aim to develop a functional enzymatic biosensor, the following limit of detection (LOD) values were calculated for GO, GO:MCM-41, and GO:MCM-48-modified electrodes as 0.43, 2.62, and 0.80 mM, respectively (LOD: 3.3xSD/Slope) [60].

CA curves (applied voltage: 0.14 V for 120 s) and calibration graphs of (a) and d SPE/GO, b and e SPE/GO:MCM-41, and c and d SPE/GO:MCM-48 electrodes tested in 0.1 M PBS (pH 7.4) containing 1 mM FcCOOH for glucose concentrations between 0 and 7 mM, (N = 3 samples)
Enzyme-modified electrodes usually suffer from low stability in vitro and significant effort has been spent to improve the stability of enzymes immobilized on electrodes [61, 62]. In this study, silica was used to help enhance enzyme stability, therefore, a series of inter-day and intra-day experiments have been performed. The anodic peak current values of the LSV experiments were used to investigate the stability of the enzyme immobilized on different modified electrodes. The tests were conducted with SPE/GO/GOx and SPE/GO:MCM-41/GOx electrodes in 0.1 M PBS containing 1 mM FcCOOH and glucose concentrations of 0 and 5 mM.
Figure 5a shows the intraday experiment results consisting of 6 measurements with 1 h intervals. The electrodes were kept in 0.1 M PBS between experiments at room temperature for intraday and + 4 °C for interday experiments and washed with 0.1 PBS before tests. The intraday experimental results show that both electrodes show similar performance, and the results didn’t show a significant change in the current response regardless of glucose concentration. On the other hand, interday experiments for 14 days (tested on days 0, 7, and 14) revealed that the performance of the SPE/GO:MCM41/GOx electrode showed superior performance over the SPE/GO/GOx electrode (Fig. 5b). Table 1 also summarizes the percentage change for intraday and interday stability measurements. The SPE/GO/GOx electrode showed a 40.57% decrease in current whereas the current change for SPE/GO:MCM-41/GOx was ca. 13.75%. All intraday and interday experiments showed a good degree of repeatability with relative standard deviation (RSD %) values less than 10% except for SPE/GO/GOx is 23.65%. This could be due to the unstable behaviour of the SPE/GO/GOx electrode compared to SPE/GO:MCM-41/GOx supporting that silica-based GO composite might provide a more suitable environment for coating and enzyme stability. The RSD values of the intraday and interday stability experiments are presented in Table S1.

Intraday (a) and interday (b) stability experiments of SPE/GO and SPE/GO:MCM-41 electrodes for 0 and 5 mM glucose concentrations. Electrodes were tested in 0.1 M PBS (pH 7.4) containing 1 mM FcCOOH, (N = 3 samples)
4 Conclusion
In this study, an electrochemical investigation of the performance of different GO composite materials prepared using mesoporous silica materials (MCM-41, MCM-48, SBA-15, and SBA-16) on enzymatic glucose oxidation. Optimization studies were conducted to find the optimal GO-to-mesoporous silica ratio and the effect of enzyme loading on the performance of the prepared electrodes. The results showed that the GO to mesoporous silica ratio was found to be 1:1 and the optimum enzyme working concentration was 1 mg/mL. Among all mesoporous silica materials, MCM:41 showed the most promising performance, therefore it was used for the enzymatic investigations. Enzymatic glucose oxidation experiments showed that GO:MCM-41-modified electrodes showed very promising results in detecting different glucose levels at similar sensitivity values. The sensitivity of enzyme-immobilized GO, GO:MCM-41, and GO:MCM-48-modified electrodes were calculated as 0.51 μA/mM, 0.39 μA/mM, and 0.17 μA/mM, respectively. Furthermore, it showed superior stability compared to GO-only modified enzymatic electrodes. Interday experiments revealed that SPE/GO/GOx electrode showed a 40% decrease in current whereas the current change for SPE/GO:MCM-41/GOx was about 14%. This study shows that an optimized amount of composite materials (1:1 for this study) consisting of GO and mesoporous silica, especially MCM-41 due to its relatively larger surface area and pore-loading capacity, can provide a suitable environment for enzyme retention on the surface. These findings could be important for long-term applications of enzymes such as enzymatic biofuel cells and continuous monitoring of glucose where the stability of the enzymes on silica supports such as MCM-family is one of the key factors for the desired performance.
References
Hoffmann F, Cornelius M, Morell J, Fröba M (2006) Silica-based mesoporous organic–inorganic hybrid materials. Angew Chem Int Ed 45(20):3216–3251
Tang F, Li L, Chen D (2012) Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater 24(12):1504–1534
Etienne M, Zhang L, Vilà N, Walcarius A (2015) Mesoporous materials-based electrochemical enzymatic biosensors. Electroanalysis 27(9):2028–2054
Walcarius A (2018) Silica-based electrochemical sensors and biosensors: recent trends. Curr Opin Electrochem 10:88–97
Øye G, Sjöblom J, Stöcker M (2001) Synthesis, characterization and potential applications of new materials in the mesoporous range. Adv Colloid Interface Sci 89:439–466
Yang Z, Lu Y, Yang Z (2009) Mesoporous materials: tunable structure, morphology and composition. Chem Commun 17:2270–2277
Martínez-Carmona M, Gun’ko YK, Vallet-Regí M (2018) Mesoporous silica materials as drug delivery:“The Nightmare” of bacterial infection. Pharmaceutics 10(4):279
Zhao D, Huo Q, Feng J, Chmelka BF, Stucky GD (1998) Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J Am Chem Soc 120(24):6024–6036
Bachari K, Guerroudj R, Lamouchi M (2017) Catalytic behavior of gallium-containing mesoporous silicas. Arab J Chem 10:S301–S305
Ouargli R, Hamacha R, Benharrats N, Boos A, Bengueddach A (2015) β-diketone functionalized SBA-15 and SBA-16 for rapid liquid–solid extraction of copper. J Porous Mater 22:511–520
Tu J, Wang R, Geng W, Lai X, Zhang T, Li N, Yue N, Li XJS, Chemical AB (2009) Humidity sensitive property of Li-doped 3D periodic mesoporous silica SBA-16. Sens Actuators B Chem 136(2):392–398
Boukoussa B, Hamacha R, Morsli A, Bengueddach A (2017) Adsorption of yellow dye on calcined or uncalcined Al-MCM-41 mesoporous materials. Arab J Chem 10:S2160–S2169
Chen C-Y, Li H-X, Davis ME (1993) Studies on mesoporous materials: I. Synthesis and characterization of MCM-41. Microporous Mater 2(1):17–26
Alfredsson V, Anderson MW (1996) Structure of MCM-48 revealed by transmission electron microscopy. Chem Mater 8(5):1141–1146
Cao Z, Du P, Duan A, Guo R, Zhao Z, Lei Zhang H, Zheng P, Xu C, Chen Z (2016) Synthesis of mesoporous materials SBA-16 with different morphologies and their application in dibenzothiophene hydrodesulfurization. Chem Eng Sci 155:141–152
Campanati M, Fornasari G, Vaccari A (2003) Fundamentals in the preparation of heterogeneous catalysts. Catal Today 77(4):299–314
Walcarius A, Mandler D, Cox JA, Collinson M, Lev O (2005) Exciting new directions in the intersection of functionalized sol–gel materials with electrochemistry. J Mater Chem 15(35–36):3663–3689
Yang X, Qiu P, Yang J, Fan Y, Wang L, Jiang W, Cheng X, Deng Y, Luo W (2021) Mesoporous materials–based electrochemical biosensors from enzymatic to nonenzymatic. Small 17(9):1904022
Zhi J, Wang Y, Deng S, Hu AJRA (2014) Study on the relation between pore size and supercapacitance in mesoporous carbon electrodes with silica-supported carbon nanomembranes. Rsc Adv 4(76):40296–40300
Yang X, Li Z, Zhi J, Ma J, Hu AJL (2010) Synthesis of ultrathin mesoporous carbon through Bergman cyclization of enediyne self-assembled monolayers in SBA-15. Langmuir 26(13):11244–11248
Fang Y, Hu Q, Yu X, Wang L (2018) Ultrasensitive electrochemical immunosensor for procalcitonin with signal enhancement based on zinc nanoparticles functionalized ordered mesoporous carbon-silica nanocomposites. Sens Actuators, B 258:238–245
Kordasht HK, Pazhuhi M, Pashazadeh-Panahi P, Hasanzadeh M, Shadjou N (2020) Multifunctional aptasensors based on mesoporous silica nanoparticles as an efficient platform for bioanalytical applications: recent advances. Trends Anal Chem 124:115778
Zhou G, Fung KK, Wong LW, Chen Y, Renneberg R, Yang S (2011) Immobilization of glucose oxidase on rod-like and vesicle-like mesoporous silica for enhancing current responses of glucose biosensors. Talanta 84(3):659–665
Carlsson N, Gustafsson H, Thörn C, Olsson L, Holmberg K, Åkerman B (2014) Enzymes immobilized in mesoporous silica: a physical–chemical perspective. Adv Colloid Interface Sci 205:339–360
Magner E (2013) Immobilisation of enzymes on mesoporous silicate materials. Chem Soc Rev 42(15):6213–6222
Zhang J, Chen S, Tan X, Zhong X, Yuan D, Cheng Y (2014) Highly sensitive electrochemiluminescence biosensors for cholesterol detection based on mesoporous magnetic core–shell microspheres. Biotechnol Lett 36:1835–1841
Xu X, Lu P, Zhou Y, Zhao Z, Guo M (2009) Laccase immobilized on methylene blue modified mesoporous silica MCM-41/PVA. Mater Sci Eng C 29(7):2160–2164
Zhou M, Shang L, Li B, Huang L, Dong S (2008) Highly ordered mesoporous carbons as electrode material for the construction of electrochemical dehydrogenase-and oxidase-based biosensors. Biosens Bioelectron 24(3):442–447
Fang ZH, Lu LM, Zhang XB, Li HB, Yang B, Shen GL, Yu RQ (2011) A third-generation hydrogen peroxide biosensor based on horseradish peroxidase immobilized in carbon nanotubes/SBA-15 film. Electroanalysis 23(10):2415–2420
Shimomura T, Sumiya T, Ono M, Itoh T, Hanaoka T-a (2012) An electrochemical biosensor for the determination of lactic acid in expiration. Procedia Chem 6:46–51
Mundaca-Uribe R, Bustos-Ramírez F, Zaror-Zaror C, Aranda-Bustos M, Neira-Hinojosa J, Pena-Farfal C (2014) Development of a bienzymatic amperometric biosensor to determine uric acid in human serum, based on mesoporous silica (MCM-41) for enzyme immobilization. Sens Actuators, B 195:58–62
Bai Y, Yang H, Yang W, Li Y, Sun C (2007) Gold nanoparticles-mesoporous silica composite used as an enzyme immobilization matrix for amperometric glucose biosensor construction. Sens Actuators, B 124(1):179–186
Zhang J, Zhu J (2009) A novel amperometric biosensor based on gold nanoparticles-mesoporous silica composite for biosensing glucose. Sci China, Ser B 52(6):815–820
Lai G, Zhang H, Yu A, Ju H (2015) In situ deposition of Prussian blue on mesoporous carbon nanosphere for sensitive electrochemical immunoassay. Biosens Bioelectron 74:660–665
Wang K, Yang H, Zhu L, Liao J, Lu T, Xing W, Xing S, Lv Q (2009) Direct electrochemistry and electrocatalysis of glucose oxidase immobilized on glassy carbon electrode modified by Nafion and ordered mesoporous silica-SBA-15. J Mol Catal B 58(1–4):194–198
Caro-Jara N, Mundaca-Uribe R, Zaror-Zaror C, Carpinelli-Pavisic J, Aranda-Bustos M, Peña-Farfal C (2013) Development of a bienzymatic amperometric glucose biosensor using mesoporous silica (MCM-41) for enzyme immobilization and its application on liquid pharmaceutical formulations. Electroanalysis 25(1):308–315
Boujakhrout A, Sánchez E, Díez P, Sánchez A, Martínez-Ruiz P, Parrado C, Pingarrón JM, Villalonga R (2015) Single-walled carbon nanotubes/Au–mesoporous silica janus nanoparticles as building blocks for the preparation of a bienzyme biosensor. ChemElectroChem 2(11):1735–1741
Yusan S, Rahman MM, Mohamad N, Arrif TM, Latif AZA, MohdAznan MA, Wan WSB (2018) Development of an amperometric glucose biosensor based on the immobilization of glucose oxidase on the Se-MCM-41 mesoporous composite. J Anal Methods Chem. https://doi.org/10.1155/2018/26873412018
Tvorynska S, Barek J, Josypcuk BJB (2022) Influence of different covalent immobilization protocols on electroanalytical performance of laccase-based biosensors. Bioelectrochemistry 148:108223
Şimşek V (2019) Investigation of catalytic sustainability of silica-based mesoporous acidic catalysts and ion-exchange resins in methyl acetate synthesis and characterizations of synthesized catalysts. Arab J Sci Eng 44(6):5301–5310
Veli S, Pinar A (2018) Characterization and catalytic performance of modified sba-16 in liquid phase reaction. Int J Chem React Eng 16(8):20170246
Li H, Wang S, Ling F, Li J (2006) Studies on MCM-48 supported cobalt catalyst for Fischer–Tropsch synthesis. J Mol Catal A 244(1–2):33–40
Şimşek V, Şahin S (2019) Characterization and catalytic performance evaluation of a novel heterogeneous mesoporous catalyst for methanol–acetic acid esterification. J Porous Mater 26:1657–1665
Şahin S, Kaya Ş, Üstündağ Z, Caglayan MO (2022) An electrochemical signal switch–based (on–off) aptasensor for sensitive detection of insulin on gold-deposited screen-printed electrodes. J Solid State Electrochem 26(4):907–915
Şahin S (2020) A simple and sensitive hydrogen peroxide detection with horseradish peroxidase immobilized on pyrene modified acid-treated single-walled carbon nanotubes. J Chem Technol Biotechnol 95(4):1093–1099
Huang HY, Yang RT, Chinn D, Munson CL (2003) Amine-grafted MCM-48 and silica xerogel as superior sorbents for acidic gas removal from natural gas. Ind Eng Chem Res 42(12):2427–2433
Nasiriani T, Nazeri MT, Shaabani A (2023) Cobalt phthalocyanine conjugated SBA-15 mesoporous silica via the Ugi four-component reaction: a potential heterogeneous catalytic nanocomposite for CO2 fixation reaction. Microporous Mesoporous Mater 354:112514
Saikia L, Srinivas D, Ratnasamy P (2007) Comparative catalytic activity of Mn (Salen) complexes grafted on SBA-15 functionalized with amine, thiol and sulfonic acid groups for selective aerial oxidation of limonene. Microporous Mesoporous Mater 104(1–3):225–235
Colilla M, Izquierdo-Barba I, Sánchez-Salcedo S, Fierro JL, Hueso JL, Ma V-R (2010) Synthesis and characterization of zwitterionic SBA-15 nanostructured materials. Chem Mater 22(23):6459–6466
Bhagiyalakshmi M, Yun LJ, Anuradha R, Jang HT (2010) Synthesis of chloropropylamine grafted mesoporous MCM-41, MCM-48 and SBA-15 from rice husk ash: their application to CO2 chemisorption. J Porous Mater 17:475–484
Mirji S, Halligudi S, Mathew N, Jacob NE, Patil K, Gaikwad A (2007) Adsorption of methanol on mesoporous SBA-15. Mater Lett 61(1):88–92
Liu Q-Y, Wu W-L, Wang J, Ren X-Q, Wang Y-R (2004) Characterization of 12-tungstophosphoric acid impregnated on mesoporous silica SBA-15 and its catalytic performance in isopropylation of naphthalene with isopropanol. Microporous Mesoporous Mater 76(1–3):51–60
Schumacher K, Ravikovitch PI, Du Chesne A, Neimark AV, Unger KK (2000) Characterization of MCM-48 materials. Langmuir 16(10):4648–4654
Sayari A, Han B-H, Yang Y (2004) Simple synthesis route to monodispersed SBA-15 silica rods. J Am Chem Soc 126(44):14348–14349
Stankovich S, Dikin DA, Piner RD, Kohlhaas KA, Kleinhammes A, Jia Y, Wu Y, Nguyen ST, Ruoff RS (2007) Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. carbon 45(7):1558–1565
Elgrishi N, Rountree KJ, McCarthy BD, Rountree ES, Eisenhart TT, Dempsey JL (2018) A practical beginner’s guide to cyclic voltammetry. J Chem Educ 95(2):197–206
Shen C, Wang H, Zhang T, Zeng Y (2019) Silica coating onto graphene for improving thermal conductivity and electrical insulation of graphene/polydimethylsiloxane nanocomposites. J Mater Sci Technol 35(1):36–43
Sahin S, Wongnateb T, Chaiyenb P, Yu EH (2014) Glucose oxidation using oxygen resistant pyranose-2-oxidase for biofuel cell applications. Chem Eng Trans. https://doi.org/10.3303/CET1441062
Chaubey A, Malhotra B (2002) Mediated biosensors. Biosens Bioelectron 17(6–7):441–456
Lister AS (2005) Validation of HPLC methods in pharmaceutical analysis. In: Separation Science and Technology, vol 6. Elsevier, pp 191-217
Sassolas A, Blum LJ, Leca-Bouvier BD (2012) Immobilization strategies to develop enzymatic biosensors. Biotechnol Adv 30(3):489–511
Xiao X, Xia H-q, Wu R, Bai L, Yan L, Magner E, Cosnier S, Lojou E, Zhu Z, Liu A (2019) Tackling the challenges of enzymatic (bio) fuel cells. Chem Rev 119(16):9509–9558
Acknowledgements
The authors state that there is no conflict of interest. We would like to thank the Scientific Research Council of Bilecik Seyh Edebali University (BAP), Project no: 2018-02.BŞEÜ.03-09, for financial support. This study also includes a part of Ms. Şevval Kaya's M.Sc. thesis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kaya, Ş., Şimşek, V. & Şahin, S. Investigation of Graphene Oxide/Mesoporous Silica Supports for Enhanced Electrochemical Stability of Enzymatic Electrodes. Catal Lett 154, 2701–2712 (2024). https://doi.org/10.1007/s10562-023-04520-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-023-04520-x