
Abstract
The genetic traits of seagrass populations are a crucial aspect of their ecology and evolution, influencing their adaptability and resilience. Here, we studied the genetic diversity, population structure, and connectivity of eighteen Posidonia oceanica meadows extending in the Eastern Mediterranean Sea (Aegean, Ionian and Cretan Seas, Greece), combining twelve microsatellite markers and Lagrangian particle drift modelling. Our findings revealed a strong genetic differentiation between the Ionian Sea and the Aegean and Cretan Seas, suggesting limited genetic exchange between these two groups. High gene flow was observed within the meadows of the Aegean and Cretan Seas, indicating a well-connected group of populations. Notably, populations of the North Aegean Sea displayed the lowest genetic diversity and the highest clonality compared to the rest of the populations. The lack of substantial oceanographic connectivity between Ionian and Aegean/Cretan Sea populations supported their genetic differentiation. However, the Lagrangian simulations did not fully support gene flow patterns in the Aegean Sea, suggesting that in addition to contemporary processes, historical events may have contributed to the formation of the observed genetic pattern. The genetic information provided here can be incorporated into management strategies aimed at identifying suitable areas as management units in conservation efforts and determining meadows that may serve as donor sites in transplantation initiatives.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Seagrasses rank among the most valuable coastal habitats, providing a wide range of services to the marine environment and humans (Mtwana Nordlund et al. 2016). However, seagrass ecosystems are threatened by the intensification of human activities around the coast, such as dredging, mooring, infrastructure development, fish farming, and climate change (Halpern et al. 2019). Impacts on seagrass ecosystems include reduction in their growth and function (Collier and Waycott 2014; Repolho et al. 2017; Nguyen et al. 2021), decline in extent (Waycott et al. 2009; Dunic et al. 2021) or total habitat loss (Kendrick et al. 2019; Strydom et al. 2020). Amidst these challenges, studying the genetic diversity, structure, and connectivity of seagrass populations offers insights into their health and resilience to a changing environment (Kendrick et al. 2017; Hu et al. 2021). High genetic diversity has been associated with increased abundance and growth (Reynolds et al. 2012; Salo and Gustafsson 2016), as well as higher resilience and recovery from a range of stressors, including warming, shading, and eutrophication (Ehlers et al. 2008; Evans et al. 2017; Plaisted et al. 2020). Its formation depends on a combination of life-history traits (e.g., reproductive strategy) and evolutionary processes (e.g., founder effects, bottlenecks), while its maintenance is highly related to population connectivity via gene flow. Strong connectivity among populations can promote the exchange of genetic variants and the spread of advantageous alleles, contributing to their long-term viability (Foley et al. 2010; Carr et al. 2017). Recognizing the significance of genetic diversity and connectivity is essential, as it facilitates well-connected seascapes that promote genetic exchange (Selkoe et al. 2016; Carr et al. 2017). Consequently, incorporating genetic information into the conservation and restoration planning (Unsworth et al. 2015; Jahnke et al. 2020; Nielsen et al. 2023) emerges as a powerful tool for promoting seagrass persistence and recovery, considering the current necessity for marine protected areas and ecosystem restoration (Pazzaglia et al. 2021; Phair et al. 2021).
The iconic and endemic seagrass of the Mediterranean Sea, Posidonia oceanica (L.) Delile, has undergone a significant decline over the last decades (Blanco-Murillo et al. 2022; Litsi-Mizan et al. 2023), and is projected to show bigger losses during the 21st century (Chefaoui et al. 2018). The fast pace of environmental change in the Mediterranean Sea raises concerns regarding the long-term persistence of the species (Jahnke et al. 2019), especially given its long generation time and small effective population size which in turn leads to slow evolutionary rates (Aires et al. 2011). Moreover, the species’ irregular sexual reproduction (Diaz-Almela et al. 2006) may limit the emergence of new genetic variants that can facilitate the adaptability (Pazzaglia et al. 2021). A meta-analysis of multiple of P. oceanica populations indicated that low genetic diversity could lead to local or regional extinctions (Jahnke et al. 2015), which is alarming considering that low genetic diversity and monoclonal meadows have been reported in several occasions (Procaccini et al. 2001, 2002; Ruggiero et al. 2002; Arnaud-Haond et al. 2012; Jahnke et al. 2017). However, the maintenance of large clones does not necessarily imply decreased resilience (Diaz-Almela et al. 2007; Arnaud-haond et al. 2010), as robust clones can promote phenotypic plasticity (Arnaud-Haond et al. 2012) and acquire genetic diversity through somatic mutations that result in genetic mosaics (Jahnke et al. 2017). Therefore, the balance between two reproductive strategies can be advantageous under different circumstances, and results from a combination of evolutionary processes (Chefaoui et al. 2017), dispersal capacities and population connectivity (Kendrick et al. 2017), but also local environmental conditions (Micheli et al. 2005; Sinclair et al. 2014).
Genetic differentiation between P. oceanica meadows of the Eastern and Western Mediterranean (Arnaud-Haond et al. 2007; Serra et al. 2010) reflects the different evolutionary history and environmental conditions between the two parts of the basin (Serra et al. 2010). The genetic distinctiveness has been attributed to historical events during the Last Glacial Maximum (LGM, ~ 26,000–19,000 BP) which appears to be preserved over time due to geographic barriers, current movements, and the limited lifespan of P. oceanica floating fruits (Serra et al. 2010; Chefaoui et al. 2017). Only one region, the Strait of Sicily in the Central Mediterranean Sea, has been identified as a contact zone between the two parts of the basin, where genetic exchange occurs and the genetic diversity of the species is the highest (Arnaud-Haond et al. 2007; Serra et al. 2010). However, the Eastern Mediterranean genetic group was poorly represented, with only five meadows in the Adriatic and Ionian Seas and one meadow in the Aegean and Levantine Seas, respectively, leaving most of the Eastern part of the basin largely unexplored, despite evidence of unique diversity with private alleles in this part of the basin (Chefaoui et al. 2017). Only one extensive study at the easternmost edge of the species distribution (i.e., along the Turkish coasts) revealed high genetic diversity and population structure between the populations of the Eastern Aegean Sea and Levantine Sea (Tutar et al. 2022). The water bodies surrounding Greece (Ionian, Aegean, and Cretan Seas) offer a complex coastal configuration and a heterogeneous scenery of biotic and abiotic factors (Simboura et al. 2019), which could promote genetic differentiation in macrophytes (Hu et al. 2020). For example, the genetic differentiation found within the Aegean meadows of the Mediterranean native seagrass species, Cymodocea nodosa, was attributed to geographic barriers and local hydrodynamic conditions that impede dispersal, apart from evolutionary processes (Gkafas et al. 2016; Konstantinidis et al. 2022). Although the characteristics of the Greek Seas could promote genetic differentiation within the P. oceanica meadows extending in the region (Panayotidis et al. 2022), only three sites in the Greek Seas have been assessed so far (Diaz-Almela et al. 2007; Jahnke et al. 2017; Serra et al. 2010), leaving a substantial part of the Eastern basin poorly investigated.
Here, we aim to study the genetic pattern of P. oceanica meadows in the Eastern Mediterranean. We do so by estimating the genetic diversity, population structure and connectivity of several meadows situated around Greece using microsatellite markers. In addition, we incorporate Lagrangian particle drift models to examine seagrass propagule dispersal patterns via water currents. Given the current UN Decade of Ecosystem Restoration and the major efforts underway through newly implemented policies, the genetic information provided can be incorporated in the identification of specific management units across our study area and in the selection of appropriate donor sites in transplantation efforts.
Materials and methods
Study area and sampling
A total of 18 monospecific P. oceanica meadows were sampled along the different water bodies of Greece (Ionian, Aegean, and Cretan Seas). In the Aegean Sea, meadows were sampled within the sub-basins of the North Aegean Sea and the South Aegean Sea, where they extended in the insular regions of Cyclades Islands and Dodecanese Islands and towards the mainland, in the Saronikos Gulf. Meadows in the Cretan Sea included those sampled around the island of Crete (Fig. 1, Table S1). The samplings were conducted between 2014 and 2021 with SCUBA diving, targeting meadows situated at depths ranging from 4 to 15 m (Table S1). At each site, 20–30 orthotropic shoots were randomly collected keeping a minimum distance of 2 m. The green leaves per shoot were individually stored at − 80 °C. In the case of sites NIK, SFA and LEN leaves were dried at 60 °C until constant weight.

Map of the sampling sites along the different water bodies of Greece
DNA extraction and genotyping
A total of 20–30 mg of frozen or dry leaf tissue per shoot was powdered using a TissueLyser II (Qiagen). DNA extractions were carried out following a modified Cetyltrimethylammonium bromide (CTAB) chloroform/isoamyl alcohol (24:1) isolation protocol (Doyle and Doyle 1990). The resulting DNA pellet was resuspended in 50 µl of Tris-EDTA (TE) buffer. DNA quality and quantity were checked using a NanoDrop ND 1000 (NanoDrop Technologies, Wimington, DE, USA). All samples were genotyped using a set of 12 microsatellite loci, comprising of 7 neutral and 5 EST-linked loci (Alberto et al. 2003; Arranz et al. 2013); the loci were PCR-amplified in four groups (multiplexes) presented in Table S2. PCR reactions were performed using the Qiagen Multiplex PCR Kit (Qiagen, UK) on a 10 µl reaction, containing 1× Mastermix, 0.2 µM of each primer, and 4 ng of DNA. The PCR conditions consisted of an initial denaturation step at 95 °C for 5 min, followed by 35 cycles at 95 °C for 30 s, 57 °C for 1.30 min and 72 °C for 30s, ending with a final extension step at 68 °C for 30 min. All PCR reactions were conducted using a Bio-Rad T100 Thermal Cycler with a heated lid. Subsequently, the PCR products were diluted (1:200) and genotyped using an Applied Biosystems 3730x automated DNA sequencer.
Data analysis
Clone identification and microsatellite loci polymorphism assessment
All DNA fragments were scored using STRAND (Veterinary Genetics Laboratory, University of California, Davis; http://www.vgl.ucdavis.edu/STRand). To identify potential scoring errors caused by stuttering, all loci were checked with Microchecker (Van Oosterhout et al. 2004) and corrected when necessary. Only samples successfully genotyped in more than eleven loci were considered for subsequent analysis. Since the species exhibits a partially clonal reproductive strategy, clones were identified and removed from the dataset. First, to assess the discriminatory power of the microsatellite set for clone detection, the probability of identity (PI) was calculated using Genalex 6.5 (Peakall and Smouse 2012). Subsequently, multilocus genotypes (MLGs) were determined per site using GeneClone 2.0 (Arnaud-Haond and Belkhir 2007). MLGs with a low probability of originating from distinct sexual events (psexFIS < 0.01) were considered clones and excluded from further analysis. After the MLGs were identified, the twelve loci were tested for putative deviations from neutrality. The frequency of null alleles per locus was estimated using ‘PopGenReport’ (Adamack and Gruber 2014) and the Brookfield 1996 method. Deviations from the Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) at each site and locus were tested using Genepop 4.7 (Raymond and Rousset 1995) with 1000 dememorizations, 1000 batches and 10,000 iterations per batch. The significance of the results was evaluated by applying Bonferroni corrections [alpha (0.05)/number of tests]. Monomorphic loci (> 80%) were identified using ‘hiersftat 0.5–11’ (Goudet 2005). To identify potential outlier loci, the Bayescan software was used (Foll and Gaggiotti 2008), which utilizes a Monte Carlo Markov Chain (MCMC) model to generate posterior probability distribution and test for deviations of loci from neutrality expectations. A locus was considered an outlier when its posterior probability was significant (q value < 0.05).
Genetic diversity, population structure and genetic connectivity
The genotypic richness (R) per site was calculated using the formula R= (MLG-1/N-1) (Arnaud-Haond et al. 2005) where N represents the number of individuals and MLG the number of distinct multilocus genotypes. At each site, the mean number of alleles per locus (NA), the number of private alleles (NPA) and the percentage of polymorphic loci (% p) were estimated using Genalex 6.5 (Peakall and Smouse 2012). Allelic richness (AR) standardized for the minimum number of samples between sites, observed heterozygosity (Ho), unbiased expected heterozygosity (Hs) and inbreeding coefficient (FIS) per site were estimated using ‘adegenet 2.1.10’ (Jombart 2008). To determine whether there was a significant excess or deficit of heterozygotes, confidence intervals were estimated based on 1000 permutations.
The genetic differentiation between sites was estimated using Weir’s and Cockerham’s FST, with confidence intervals estimated through 1000 permutations in ‘hierfstat 0.5–11’ (Goudet 2005). Jost’s D (Dest) genetic distance (Jost 2008) was also calculated between sites using ‘mmod’ (Winter 2012). Spatial genetic differentiation was analysed using STRUCTURE 2.3.4 (Pritchard et al. 2000). Ten independent runs were performed for K values ranging from 1 to 18, with a burn-in period of 100,000 followed by 1,000,000 MCMS steps, assuming admixture and correlated allele frequencies. The optimal number of genetic clusters (K) was determined using the delta-K criterion (Evanno et al. 2005) in STRUCTURE HARVESTER (Earl and vonHoldt 2012) and CLUMPAK (Kopelman et al. 2015), which was also used for the visualization of the results. Additionally, a discriminant analysis of principal components (DAPC; Jombart et al. 2010) was performed selecting the number of principal component (PCs) that achieved the lowest mean standard error.
Patterns of gene flow between sites were inferred using two approaches. First, assignment tests and computation of first-generation migrants were conducted using GENECLASS2 (Piry et al. 2004). The likelihood that an individual belonged to the site from which it was sampled was estimated using the partially Bayesian criterion proposed by Rannala and Mountain (1997). A statistical threshold was determined by comparing the likelihood of excluding an individual from its site of origin to a distribution of likelihoods from 1,000,000 simulated individuals (Paetkau et al. 2004). Individuals were excluded from their site of origin with a 95% probability and assigned to the site with the highest probability (Underwood et al. 2007). Significant first-generation migrants were identified by performing 10,000 MCMC permutations using the Rannala and Mountain (1997) criterion and a probability threshold of 0.01. Secondly, directional migration rates based on pairwise genetic distances (Dest), and 1000 bootstraps were estimated using divMigrate online (https://popgen.shinyapps.io/divMigrate-online/) (Sundqvist et al. 2016). The results were visualized using the R package ‘qgraph’ (Epskamp et al. 2012). All analyses were performed in R version 4.2.2 (R Core Team 2021).
Oceanographic connectivity
We examined the oceanographic (potential) connectivity of P. oceanica floating fruits using a Mediterranean basin-scale hydrodynamic model with a horizontal resolution of approximately 0.05° (approximately 5 km). This model was coupled with a Lagrangian particle drift model (Tsabaris et al. 2021; Tsiaras et al. 2021). A 10-year simulation, spanning from 2010 to 2019, was conducted using the coupled hydrodynamic and particle drift model. To align with the species seedling period (Buia and Mazzella 1991), a batch of 19,000 particles representing P. oceanica fruits was released from each sampling site (Fig. 1). These particles were randomly distributed within a 4 km radius, starting on the 1st of March of each year (2010–2019), and continued for the subsequent 90 days (Buia and Mazzella 1991). The particles were assumed to be passive tracers and confined in the near-surface layer at approximately 5 m depth (Serra et al. 2010). Based on the position of the particle (x) after 28 days of advection from each release site (i), the horizontal probability distribution on the model grid with a 5 km resolution was calculated as:
where Ni(x) represents the number of particles in grid cell x and Ntot the total number of released particles from each site (19,000 particles multiplied by 90 release days). This distribution was then averaged over the different years (2010–2019) of simulations. To determine the potential connectivity C(i,j) from a source site i to a destination site j, the sum of Pi(x) values within a 20 km radius of the destination site was used. Based on the mean connectivity observed during the simulated years, each site was characterized as a sink (where released propagules successfully settled), source (where released propagules reached other sites successfully), or retainer (where propagules tended to remain at the release site) (Jahnke et al. 2017). Further details regarding the hydrodynamic and Lagrangian drift model can be found in Materials and Methods Supplementary material.
Results
Clone identification and microsatellite loci assessment
A total of 442 individuals were successfully genotyped using 12 microsatellite loci. The set of 12 microsatellite loci exhibited a relatively low probability of identity (PI), ranging from 9.8 × 10−6 to 2.4 × 10−3 (Waits et al. 2001), indicating that the microsatellite loci have sufficient power to discriminate between clones. After the removal of individuals identified as clones, 334 unique MLGs were retained. The EST- linked locus Pooc-153 was identified as monomorphic and was therefore excluded from further analysis. In terms of linkage disequilibrium (LD), only 15 out of 555 tests (2.7%) were considered significant after Bonferroni corrections, indicating no major LD. Regarding Hardy–Weinberg equilibrium (HWE), 55 out of 216 tests (25%) per site and locus showed a significant deviation after Bonferroni corrections. Specifically, EST-linked locus Pooc-PC045G11F exhibited deviations from HWE expectations at 12 of 18 sites. Given that this locus was also identified as an outlier based on Bayescan analysis (Fig. S1) and exhibited a high frequency of null alleles (28%, Chapuis and Estoup 2007), it was removed from further analysis. Therefore, the final dataset for population genetics analyses consisted of 10 microsatellite loci.
Genotypic and genetic diversity
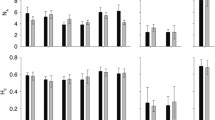
Overall, genotypic richness (R) was high with an average of 0.8 ± 0.2, ranging lower in the meadows of N. Aegean Sea (0.3–0.7) compared to the meadows of the Ionian Sea (0.6–0.7), S. Aegean Sea (0.7–1) and Cretan Sea (0.6–1) (Table 1; Fig. 2a). The highest mean number of alleles per locus (NA) was found in SER (NA = 3.5), which also exhibited 100% polymorphic loci. Similarly, SOU and KEF displayed 100% of polymorphic loci, with relatively high NA (2.8 and 3.0, respectively). On the other hand, LES had the lowest NA (NA = 1.8) and the lowest percentage of polymorphic loci (50%). In terms of allelic richness (Ar10), N. Aegean Sea had the lowest range (1.79–2.46) compared to the Ionian Sea (2.92–2.97), S. Aegean Sea (2.19–2.81) and Cretan Sea (2.06–2.72) (Table 1; Fig. 2b). The same pattern was observed for the standardized expected heterozygosity (Hs), with lower values in the N. Aegean Sea (0.24–0.37) compared to the Ionian Sea (0.44–0.49), S. Aegean Sea (0.33–0.40) and Cretan Sea (0.3–0.41) (Table 1; Fig. 2c). Most of the private alleles were found in meadows located in the regions of Crete (GOU = 4 NPA and NIK = 5 NPA) and Dodecanese Islands (STR = 5 NPA). Most of the sites exhibited negative FIS values, but only 4 out of 18 sites (TRI, SOU, PER, NIK) showed a significant heterozygote excess based on 1,000 permutations (Table 1).

Maps of a genotypic richness (R), b allelic richness (Ar) and, c expected heterozygosity (Hs) of P. oceanica meadows along the Greek Seas
Population structure and genetic (realized) connectivity
Pairwise FST values ranged from 0.04 to 0.50, with the lowest being between PAR and KEF in the Ionian Sea (0.04) and the highest between LES and LIM in N. Aegean Sea (0.50). All pairwise comparisons were significant except for the two sites in the Ionian Sea (PAR and KEF; Fig. S2). Jost’D (Dest) pairwise distances showed similar patterns with FST (Fig. S3). STRUCTURE analysis and delta-K values indicated that K = 2 was the most likely number of population clusters. However, when higher values of K were investigated, high levels of admixture were observed for most populations, while in some cases further substructure became more apparent. In particular, when K = 6 was selected, which had the second highest delta-K value (Fig. S4), putative subpopulations were identified (Fig. 3). According to K = 6, the two sites located in the Ionian Sea (green color; PAR and KEF) form a distinct cluster, with minimal admixture with the other sites. The meadows of the N. Aegean Sea did not form a unique cluster with LIM and TRI (blue color), showing greater differentiation from the other sites. Conversely, AFY and LES displayed higher levels of admixture with sites in the S. Aegean Sea. The S. Aegean and Cretan Sea meadows showed extensive admixture, particularly in the region of Cyclades Islands, where they appeared to be admixed with sites of the Dodecanese Islands and Crete. Only NIK and LEN, located in Crete, appeared to form separate subpopulations. The DAPC analysis yielded similar results to the STRUCTURE analysis (based on K = 6), providing support for the genetic differentiation of the Ionian Sea sites (PAR and KEF) and indicating the distinction of LEN in south Crete (Fig. 4).

Population structure analysis using STRUCTURE (Pritchard et al. 2000) assuming admixture and correlated allelic frequencies. Based on delta-K values, the two most likely K-values are presented (K = 2 and K = 6, see also Fig. S4)

Discriminant principal component analysis (DAPC) retaining 35 PCs axis
Regarding membership probabilities, in total 269 individuals (81%) were correctly assigned to their respective site of origin, and 19% were assigned to other sites (Table 2). The analysis also identified 22 individuals originating from 13 sites as significant first-generation migrants (P < 0.01) (Table 2). Within the Ionian Sea, first-generation migrants remained within the same sea, except for one migrant from KEF (Ionian Sea) found in SOU (S. Aegean Sea, Saronikos Gulf). Similarly, migrants within the Aegean Sea tended to stay within their respective sea of origin (S. Aegean or N. Aegean Sea) or followed a predominant direction from S. Aegean to N. Aegean Sea. Notably, several meadows of Cyclades Islands, primarily SER and KOU but also SAN and IOS, played a significant role as sources of first-generation migrants, contributing a total of 9 migrants to meadows of N. Aegean Sea and Cretan Sea (Table 2). Based on divMigrate analysis, high levels of gene flow were observed between the meadows located in the Ionian Sea (KEF and PAR), whereas lower rates of exchange were observed between the Ionian Sea and the rest of the meadows. High dispersion levels were observed in the S. Aegean Sea, particularly in the sites SER, KOU, and NIS. The meadows of these sites functioned as both sinks and sources for the wider S. Aegean Sea, as well as, to a lesser extent for the N. Aegean, and Cretan Sea. Gene flow in the N. Aegean Sea mainly occurred within the geographical barriers of the region itself or received gene flow from the S. Aegean Sea, primarily originating from Cyclades Islands (Fig. 5a).

a Genetic (realized) connectivity based on divMigrate analysis and Dest genetic distances. Cut-off value was selected based on the average migration rate (0.15) and, b mean oceanographic (potential) connectivity after an advection time of 28 days, based on Lagrangian particle dispersion simulations for the period 2010–2019. The arrow thickness reflects the magnitude of gene flow and oceanographic connectivity, while the arrow edges indicate their respective directions
Oceanographic (potential) connectivity
Potential oceanographic connectivity patterns were derived by analysing the water circulation patterns (Fig. S5) and the dispersal patterns of particles representing P. oceanica fruits. Considering the period of 2010 to 2019 and an advection time of 28 days, it was observed that the particles released from the sites in the Ionian Sea (PAR and KEF) were not dispersed to the Aegean or Cretan Seas. Instead, they remained confined within the Ionian Sea, facilitating the exchange of propagules between these sites (Fig. 5b, Fig. S6, S7). Similarly, the propagules released from the N. Aegean Sea predominantly remained within this area, with occasional dispersal to some sites of the two closest regions of the S. Aegean Sea, namely the Saronikos Gulf or the Cyclades Islands (Fig. 5b, Fig. S6, S7). In contrast, the sites located in the S. Aegean Sea exhibited high oceanographic connectivity, allowing for the exchange of particles among sites. High oceanographic connectivity was also found between S. Aegean Sea and the sites located in the north of the Cretan Sea, while less exchange was noted between the sites located in south Crete and the Aegean Sea. Regarding their retention capacity, the sites located in the Ionian Sea were both weak sources and sinks, as were the sites located in the southern part of the Cretan Sea. On the other hand, the sites of the S. Aegean Sea acted as both sink and sources, having relatively weak retention capacity. Among the strongest retainers were found in the N. Aegean Sea, particularly at AFY, but also at TRI and LES sites (Fig. 6).

Retention, source, and sink strength of each one of the 18 sampling sites based on mean oceanographic (potential) connectivity for the period 2010–2019 and an advection time of 28 days. See also Fig. S6, S7
Discussion
Our data analysis revealed the genetic characteristics of P. oceanica meadows extending along the different water bodies of Greece. Spatial differences were found in genetic and genotypic diversity, which was overall lower in the meadows of N. Aegean Sea. Genetic discontinuity was found between the meadows of the Ionian Sea and the rest of the meadows, indicating limited genetic exchange between them. However, within each sea, substantial connectivity was found between the meadows, highlighting the complex interactions between population dynamics and environmental settings, especially the prevailing hydrodynamic conditions.
Considerable admixture and gene flow among populations was found, despite the evidence of moderate genetic structure, as supported by the significant pairwise FST values and the small number of first-generation migrants. The most distinct genetic separation was observed between the main water bodies of the Greek Seas, the Ionian Sea on one hand, and the Aegean and Cretan Seas on the other hand. This result combined with the previously found genetic separation between the Aegean and Levantine Sea meadows (Tutar et al. 2022), suggests that there is more complexity and genetic differentiation of P. oceanica meadows within the Eastern Mediterranean than the main established separation between Eastern and Western Mediterranean meadows. The genetic separation of the Ionian Sea is consistent with the results obtained from the Lagrangian modelling, which indicated low propagule exchange probability between the Ionian and the rest of the Greek Seas. In contrast, the hydrodynamic conditions that occur in the Aegean and Cretan Seas facilitate the transport of propagules, leading to a significant admixture of their meadows. Indeed, the Aegean and Cretan Sea is characterized by a cyclonic circulation with a strong north-to-south component (Fig. S5; Olson et al. 2007; Politikos et al. 2017). This cyclonic circulation pattern, driven by the Black Sea Water inflow through the Dardanelles and the northward flow of Levantine waters, along the eastern side of the Aegean Sea, combined with the predominant southward winds (Olson et al. 2007), appears to facilitate the admixture of the Aegean and Cretan Sea meadows. In addition to water currents circulation, other factors, such as the high shipping traffic of the Aegean Sea can contribute to the long-distance dispersal of propagules. Nonetheless, potential sub-populations were also identified in the region of Crete, mainly at LEN, located in the southern part of the island. This result could be linked to the weak connectivity of southern Crete with the rest of the Aegean Sea, influenced by the circulation patterns (Fig. S5). On the other hand, SFA, also located in south Crete, did not show evidence of genetic separation, suggesting that, by expanding the sampling effort towards Crete, it is possible to discover intermediate populations that could act as sources of gene flow towards the southern part of the island.
Although the meadows from the Aegean and Cretan Seas were overall well-connected, spatial differences in genetic and genotypic diversity were still found. These differences likely arise from a complex interplay between contemporary processes, as indicated by the patterns of oceanographic (potential) connectivity, and evolutionary processes whose signals are reflected in the pattens of the genetic (realized) connectivity. Indeed, the differences in the retention capacity between meadows, like the strong retention capacity of some meadows in the N. Aegean Sea (e.g., AFY, which exhibits the highest retention capacity among all meadows), their location in enclosed bays (e.g., TRI, LES) or their proximity to cyclonic gyres (e.g., LIM and LES located inside the Limnos–Lesvos plateau) can limit the connectivity among populations and contribute to the maintenance of a small number of successful genotypes with limited exchange capacity with other meadows. Except for the contemporary processes, historical processes could have played a crucial role in shaping the present-day genetic characteristics of P. oceanica meadows of the Greek Seas. The lower genetic diversity and higher clonality found in the N. Aegean meadows likely reflect historical processes related to the paleoclimatic conditions during the Quaternary period, particularly the LGM climatic oscillations (~ 26,000–19,000 years BP). Previous research indicated that the probability of P. oceanica occurrence during the LGM was higher in the southern parts of the Mediterranean Sea, where glacial refugia likely existed, and environmental conditions were more favourable for the species (Chefaoui et al. 2017). Thus, it is likely that P. oceanica meadows had to persist in the southern parts of the specie’s distribution range and the present-day genetic diversity of N. Aegean meadows is likely influenced by founder effects, arising from post-colonization events from southern meadows after the LGM. Especially for meadows located in the eastern N. Aegean (LIM, LES) post-colonization events may have taken place through Asia Minor, as suggested by recent findings regarding N. Aegean meadows of C. nodosa and their post-settlement from Cyprus (Konstantinidis et al. 2022). The persistence of P. oceanica meadows in southern regions during the LGM is further supported by the high number of private alleles found at specific locations of the S. Aegean and Cretan Seas, aligning with previous findings (Chefaoui et al. 2017). The hypothesis of post-colonization of N. Aegean meadows from the South, also provides an explanation for the seemingly inconsistent results between the prevailing direction of gene flow (south to north) and the primary direction of propagule dispersal suggested by the Lagrangian modelling (north to south). Genetic differentiation and lower genetic diversity due to founder effects associated with LGM refugia have been previously reported in seagrasses in the Mediterranean Sea, such as P. oceanica in the Adriatic Sea (Ruggiero et al. 2002), C. nodosa in the Aegean Sea (Konstantinidis et al. 2022), but also for other seagrasses worldwide such as Posidonia australis (Sinclair et al. 2016) and in other taxa, such as algae (Assis et al. 2016) and coral reefs (Ludt and Rocha 2015).
Our study extends the limited knowledge of the genetic characteristics of Eastern Mediterranean P. oceanica meadows. Compared to the Western Mediterranean studies, no strictly monoclonal meadows were found, as in the Adriatic Sea (Ruggiero et al. 2002) or the Balearic Islands (Arnaud-Haond et al. 2012). On the contrary, our study area was characterized by high genotypic richness, coupled with a general absence of significant deviations from HWE, which suggests that no extensive clonal propagation occurs (Arnaud-Haond et al. 2020), while it indicates the occurrence of substantial sexual recruitment (Krueger-Hadfield et al. 2011). The interplay between sexual and asexual reproduction is influenced by multiple factors, including the prevailing environmental conditions, like light availability and temperature (Jahnke et al. 2015; Jarvis and Moore 2015). A recent study suggested that high genotypic richness in the easternmost edge of the specie’s distribution is associated with the high temperatures occurring in the Eastern Mediterranean Sea (Tutar et al. 2022), which can trigger the switch between reproductive strategies through heat-induced flowering (Ruiz et al. 2018). However, P. oceanica flowering records over long time periods are scarce and mainly pertain to Western Mediterranean meadows (Diaz-Almela et al. 2006), making it challenging to draw firm assumptions about the role of sexual reproduction in our study area. Furthermore, the substantial oceanographic connectivity in our study area promotes the dispersal of P. oceanica propagules over long distances and thus increases the chance of the arrival of new genetic variants via sexual reproduction (Bricker et al. 2011). Significant heterozygote excess, which is often associated with asexual reproduction (Arriesgado et al. 2015; Arnaud-Haond et al. 2020) and the accumulation of mutations at different alleles in a locus over time in clonal organisms (known as the “Meselson effect”, Balloux et al. 2003), was observed in only four meadows (TRI, SOU, PER, NIK). However, all four meadows had low clonality, suggesting that heterozygote excess is likely a result of outbreeding, underscoring the need to monitor these sites, which may be suffering from a reduction in fitness and resilience (Williams et al. 2014).
In the context of the whole Mediterranean Sea, the measured genetic diversity indices, namely allelic richness and expected heterozygosity, are within the previously reported range for the Eastern Mediterranean meadows (Ar: 1.92–3.54, Hs: 0.24–0.40), including three meadows previously analysed in Greece (Ar: 2.68–3.54, Hs: 0.37–0.40; Arnaud-Haond et al. 2007; Diaz-Almela et al. 2007; Jahnke et al. 2017). These indices are also consistent with the range reported in previous studies conducted on Western and Central Mediterranean meadows (Ar: 1.77–5.23, Hs: 0.13–0.53; Procaccini et al. 2001; Arnaud-Haond et al. 2007; Serra et al. 2010; Jahnke et al. 2017). In some cases, such as in the meadows of the Ionian Sea, the reported values were close to the ones previously reported in the Strait of Sicily in the Central Mediterranean (average Hs: 0.42; Serra et al. 2010), the most genetically diverse region of the species. These results indicate that no distinct differences in genetic diversity are found at the basin scale, implying that genetic differentiation primarily occurs at a local level. Nonetheless, the presence of private alleles in our study area reinforces the notion that the Eastern Mediterranean P. oceanica meadows possess unique genetic diversity (Chefaoui et al. 2017), which signifies genetic traits exclusive to the region and highlights the importance of targeted preservation of these meadows.
The genetic patterns discussed in the present study result from the application of microsatellites, which play a crucial role as molecular markers in the field of population genetics due to their high polymorphism and versatility which provide valuable insights into the genetic diversity of populations. However, we acknowledge that the use of microsatellites may be gradually replaced by new technologies. Although genome-wide methods, like single nucleotide polymorphisms (SNPs), have become increasingly accessible, their application in non-model species, such as most seagrasses, has been focused on the detection of potential adaptive DNA polymorphisms which provide insights into their resilience and resistance (Phair et al. 2020; Ruocco et al. 2022; Nguyen et al. 2023). In addition, comparative analysis has indicated that microsatellites remain an informative technology capable of yielding results like those obtained with SNPs (Hauser et al. 2021). Therefore, the use of microsatellite markers in the present study not only allowed us to uncover the genetic characteristics of P. oceanica populations but also enabled us to generate data that can be compared with the existing literature, which remains biased towards the use of microsatellites.
Given the relationship between genetic diversity, resilience, and adaptability (DuBois et al. 2021), as well as the numerous threats faced in the Greek Seas, including high sea surface temperatures, marine heat waves of substantial magnitude and intensity (Darmaraki et al. 2019; Androulidakis and Krestenitis 2022), and direct anthropogenic disturbances, the genetic information presented here are critical in the realm of management decisions. The incorporation of genetic information into these decisions plays a crucial role, especially when determining management units and formulating strategies for seagrass restoration. The comprehension of populations’ evolutionary capacity through genetic diversity and connectivity aids in prioritizing conservation endeavours by identifying populations that act as sources of genetic diversity and those that depend on migratory individuals to augment their genetic makeup (Foley et al. 2010; Jackson et al. 2021). Considering our study area, meadows in the Ionian Sea should be treated as a distinct management unit (MU) since the genetic exchange with the rest of the Greek Seas is minimal. On the other hand, the region of Cyclades Islands could also be considered as a separate MU due to the well-connected nature of its meadows which serve as sources of gene flow to various sites, primarily to other regions of the S. Aegean Sea but also in the N. Aegean Sea. Moreover, meadows located in the Cyclades Islands can serve as significant source populations for degraded meadows in transplantation efforts, since selecting diverse sites as donor meadows is essential to mitigate the negative effects associated with low genetic diversity and inbreeding (Jackson et al. 2021). However, to ensure successful and long-term seagrass conservation, it is crucial to adopt a holistic approach that will incorporate the genetic information alongside environmental, ecological, and demographic aspects (Jahnke et al. 2020; Pazzaglia et al. 2021). Ultimately, a collaborative and integrated approach is necessary to ensure the continued provision of the ecological, economic, and social benefits offered by seagrass ecosystems (Mtwana Nordlund et al. 2016) and the enhancement of the resilience of our coasts (Unsworth et al. 2019).
Data availability
All data generated in this study are provided in a separate excel file.
Change history
22 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s10531-024-02805-0
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5:384–387. https://doi.org/10.1111/2041-210X.12158
Aires T, Marbà N, Cunha RL et al (2011) Evolutionary history of the seagrass genus Posidonia. Mar Ecol Prog Ser 421:117–130. https://doi.org/10.3354/meps08879
Alberto F, Correia L, Arnaud-Haond S et al (2003) New microsatellite markers for the endemic Mediterranean seagrass Posidonia oceanica. Mol Ecol Notes 3:253–255. https://doi.org/10.1046/j.1471-8286.2003.00415.x
Androulidakis YS, Krestenitis YN (2022) Sea Surface temperature variability and Marine heat waves over the Aegean, ionian, and Cretan seas from 2008–2021. J Mar Sci Eng 10:42. https://doi.org/10.3390/jmse10010042
Arnaud-Haond S, Belkhir K (2007) GENCLONE: a computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol Ecol Notes 7:15–17. https://doi.org/10.1111/j.1471-8286.2006.01522.x
Arnaud-Haond S, Texeira S, Procaccini G et al (2005) Assessing genetic diversity in clonal organisms: low diversity or low resolution ? Combining power and cost efficiency in selecting markers. J Hered 96:434–440. https://doi.org/10.1093/jhered/esi043
Arnaud-Haond S, Migliaccio M, Diaz-Almela E et al (2007) Vicariance patterns in the Mediterranean Sea: East-West cleavage and low dispersal in the endemic seagrass Posidonia oceanica. J Biogeogr 34:963–976. https://doi.org/10.1111/j.1365-2699.2006.01671.x
Arnaud-haond S, Marbà N, Diaz-almela E et al (2010) Comparative analysis of stability—genetic diversity in Seagrass (Posidonia oceanica) Meadows yields unexpected results. Estuaries Coasts 33:878–889. https://doi.org/10.1007/s12237-009-9238-9
Arnaud-Haond S, Duarte CM, Diaz-Almela E et al (2012) Implications of extreme life span in clonal organisms: millenary clones in Meadows of the threatened Seagrass Posidonia oceanica. PLoS ONE 7:e30454. https://doi.org/10.1371/journal.pone.0030454
Arnaud-Haond S, Stoeckel S, Bailleul D (2020) New insights into the population genetics of partially clonal organisms: when seagrass data meet theoretical expectations. Mol Ecol 29:3248–3260. https://doi.org/10.1111/mec.15532
Arranz SE, Avarre J, Balasundaram C et al (2013) Permanent genetic resources added to molecular ecology resources database 1 December 2012–31 January 2013. Mol Ecol Resour 13:546–549. https://doi.org/10.1111/1755-0998.12095
Arriesgado DM, Kurokochi H, Nakajima Y et al (2015) Genetic diversity and structure of the tropical seagrass Cymodocea serrulata spanning its central diversity hotspot and range edge. Aquat Ecol 49:357–372. https://doi.org/10.1007/s10452-015-9529-0
Assis J, Coelho NC, Lamy T et al (2016) Deep reefs are climatic refugia for genetic diversity of marine forests. J Biogeogr 43:833–844. https://doi.org/10.1111/jbi.12677
Balloux F, Lehmann L, De Meeûs T (2003) The population genetics of clonal and partially clonal diploids. Genetics 164:1635–1644. https://doi.org/10.2135/cropsci1967.0011183X000700040005x
Blanco-Murillo F, Fernández-Torquemada Y, Garrote-Moreno A et al (2022) Posidonia oceanica L. (Delile) meadows regression: long-term affection may be induced by multiple impacts. Mar Environ Res. https://doi.org/10.1016/j.marenvres.2022.105557
Bricker E, Waycott M, Calladine A, Zieman JC (2011) High connectivity across environmental gradients and implications for phenotypic plasticity in a marine plant. Mar Ecol Prog Ser 423:57–67. https://doi.org/10.3354/meps08962
Brookfield JFY (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5:453–455. https://doi.org/10.1046/j.1365-294X.1996.00098.x
Buia MC, Mazzella L (1991) Reproductive phenology of the Mediterranean seagrasses Posidonia oceanica (L.) Delile, Cymodocea nodosa (Ucria) Aschers., and Zostera noltii Hornem. Aquat Bot 40:343–362
Carr MH, Robinson SP, Wahle C et al (2017) The central importance of ecological spatial connectivity to effective coastal marine protected areas and to meeting the challenges of climate change in the marine environment. Aquat Conservation: Mar Freshw Ecosyst 27:6–29. https://doi.org/10.1002/aqc.2800
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Chefaoui RM, Duarte CM, Serrão EA (2017) Palaeoclimatic conditions in the Mediterranean explain genetic diversity of Posidonia oceanica seagrass meadows. Sci Rep 7:1–8. https://doi.org/10.1038/s41598-017-03006-2
Chefaoui RM, Duarte CM, Serrão EA (2018) Dramatic loss of seagrass habitat under projected climate change in the Mediterranean Sea. Glob Change Biol 24:4919–4928. https://doi.org/10.1111/gcb.14401
Collier CJ, Waycott M (2014) Temperature extremes reduce seagrass growth and induce mortality. Mar Pollut Bull 83:483–490. https://doi.org/10.1016/j.marpolbul.2014.03.050
Darmaraki S, Somot S, Sevault F et al (2019) Future evolution of Marine heatwaves in the Mediterranean Sea. Clim Dyn 53:1371–1392. https://doi.org/10.1007/s00382-019-04661-z
Diaz-Almela E, Marbà N, Álvarez E et al (2006) Patterns of seagrass (Posidonia oceanica) flowering in the Western Mediterranean. Mar Biol 148:723–742. https://doi.org/10.1007/s00227-005-0127-x
Diaz-Almela E, Arnaud-Haond S, Vliet MS et al (2007) Feed-backs between genetic structure and perturbation-driven decline in seagrass (Posidonia oceanica) meadows. Conserv Genet 8:1377–1391. https://doi.org/10.1007/s10592-007-9288-0
Doyle J, Doyle J (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
DuBois K, Williams SL, Stachowicz JJ (2021) Experimental warming enhances effects of eelgrass genetic diversity via temperature-induced niche differentiation. Estuaries Coasts 44:545–557. https://doi.org/10.1007/s12237-020-00827-9
Dunic JC, Brown CJ, Connolly RM et al (2021) Long-term declines and recovery of meadow area across the world’s seagrass bioregions. Glob Change Biol 27:4096–4109. https://doi.org/10.1111/gcb.15684
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Ehlers A, Worm B, Reusch TBH (2008) Importance of genetic diversity in eelgrass Zostera marina for its resilience to global warming. Mar Ecol Prog Ser 355:1–7. https://doi.org/10.3354/meps07369
Epskamp S, Cramer AOJ, Waldorp LJ et al (2012) Qgraph: network visualizations of relationships in psychometric data. J Stat Softw. https://doi.org/10.18637/jss.v048.i04
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Evans SM, Vergés A, Poore AGB (2017) Genotypic diversity and short-term response to shading stress in a threatened seagrass: does low diversity mean low resilience? Front Plant Sci. https://doi.org/10.3389/fpls.2017.01417
Foley MM, Halpern BS, Micheli F et al (2010) Guiding ecological principles for marine spatial planning. Mar Policy 34:955–966. https://doi.org/10.1016/j.marpol.2010.02.001
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Gkafas GA, Orfanidis S, Vafidis D et al (2016) Genetic diversity and structure of Cymodocea nodosa meadows in the Aegean Sea, eastern Mediterranean. Appl Ecol Environ Res 14:145–160. https://doi.org/10.15666/aeer/1401_145160
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8278
Halpern BS, Frazier M, Afflerbach J et al (2019) Recent pace of change in human impact on the world’s ocean. Sci Rep 9:11609. https://doi.org/10.1038/s41598-019-47201-9
Hauser SS, Athrey G, Leberg PL (2021) Waste not, want not: microsatellites remain an economical and informative technology for conservation genetics. Ecol Evol 11:15800–15814. https://doi.org/10.1002/ece3.8250
Hu ZM, Zhong KL, Weinberger F et al (2020) Linking ecology to genetics to better understand adaptation and evolution: a review in Marine macrophytes. Front Mar Sci. https://doi.org/10.3389/fmars.2020.545102
Hu ZM, Zhang QS, Zhang J et al (2021) Intraspecific genetic variation matters when predicting seagrass distribution under climate change. Mol Ecol 30:3840–3855. https://doi.org/10.1111/mec.15996
Jackson EL, Smith TM, York PH et al (2021) An assessment of the seascape genetic structure and hydrodynamic connectivity for subtropical seagrass restoration. Restor Ecol. https://doi.org/10.1111/rec.13269
Jahnke M, Olsen JL, Procaccini G (2015) A meta-analysis reveals a positive correlation between genetic diversity metrics and environmental status in the long-lived seagrass Posidonia oceanica. Mol Ecol 24:2336–2348. https://doi.org/10.1111/mec.13174
Jahnke M, Casagrandi R, Melià P et al (2017) Potential and realized connectivity of the seagrass Posidonia oceanica and their implication for conservation. Divers Distrib 23:1423–1434. https://doi.org/10.1111/ddi.12633
Jahnke M, Esposito DD, Orrù L et al (2019) Adaptive responses along a depth and a latitudinal gradient in the endemic seagrass Posidonia oceanica. Heredity 122:233–243. https://doi.org/10.1038/s41437-018-0103-0
Jahnke M, Moksnes PO, Olsen JL et al (2020) Integrating genetics, biophysical, and demographic insights identifies critical sites for seagrass conservation. Ecol Appl. https://doi.org/10.1002/eap.2121
Jarvis JC, Moore KA (2015) Effects of seed source, sediment type, and Burial depth on mixed-annual and perennial Zostera marina L. seed germination and seedling establishment. Estuaries Coasts 38:964–978. https://doi.org/10.1007/s12237-014-9869-3
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. https://doi.org/10.1186/1471-2156-11-94
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026. https://doi.org/10.1111/j.1365-294X.2008.03887.x
Kendrick GA, Orth RJ, Statton J et al (2017) Demographic and genetic connectivity: the role and consequences of reproduction, dispersal and recruitment in seagrasses. Biol Rev 92:921–938. https://doi.org/10.1111/brv.12261
Kendrick GA, Nowicki R, Olsen YS et al (2019) A systematic review of how multiple stressors from an extreme event drove ecosystem-wide loss of resilience in an iconic seagrass community. Front Mar Sci 6:1–15. https://doi.org/10.3389/fmars.2019.00455
Konstantinidis I, Gkafas GA, Papathanasiou V et al (2022) Biogeography pattern of the marine angiosperm Cymodocea nodosa in the eastern Mediterranean Sea related to the quaternary climatic changes. Ecol Evol. https://doi.org/10.1002/ece3.8911
Kopelman NM, Mayzel J, Jakobsson M et al (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Krueger-Hadfield SA, Collén J, Daguin-Thiébaut C, Valero M (2011) Genetic population structure and mating system in Chondrus crispus (Rhodophyta). J Phycol 47:440–450. https://doi.org/10.1111/j.1529-8817.2011.00995.x
Litsi-Mizan V, Efthymiadis PT, Gerakaris V et al (2023) Decline of seagrass (Posidonia oceanica) production over two decades in the face of warming of the Eastern Mediterranean Sea. New Phytol 293:2126–2137. https://doi.org/10.1111/nph.19084
Ludt WB, Rocha LA (2015) Shifting seas: the impacts of Pleistocene sea-level fluctuations on the evolution of tropical marine taxa. J Biogeogr 42:25–38. https://doi.org/10.1111/jbi.12416
Micheli C, Paganin P, Peirano A et al (2005) Genetic variability of Posidonia oceanica (L.) Delile in relation to local factors and biogeographic patterns. Aquat Bot 82:210–221. https://doi.org/10.1016/j.aquabot.2005.03.002
Mtwana Nordlund L, Koch EW, Barbier EB, Creed JC (2016) Seagrass ecosystem services and their variability across genera and geographical regions. PLoS ONE 11:e0163091. https://doi.org/10.1371/journal.pone.0163091
Nguyen HM, Ralph PJ, Zoologica S et al (2021) Seagrasses in an era of ocean warming: a review. Biological reviews 2015:2009–2030. https://doi.org/10.1111/brv.12736
Nguyen HM, Ruocco M, Dattolo E et al (2023) Signs of local adaptation by genetic selection and isolation promoted by extreme temperature and salinity in the Mediterranean seagrass Posidonia oceanica. Mol Ecol 32:4313–4328. https://doi.org/10.1111/mec.17032
Nielsen ES, Hanson JO, Carvalho SB et al (2023) Molecular ecology meets systematic conservation planning. Trends Ecol Evol 38:143–155. https://doi.org/10.1016/j.tree.2022.09.006
Olson DB, Kourafalou VH, Johns WE et al (2007) Aegean surface circulation from a satellite-tracked drifter array. J Phys Oceanogr 37:1898–1917. https://doi.org/10.1175/JPO3028.1
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65. https://doi.org/10.1046/j.1365-294X.2004.02008.x
Panayotidis P, Papathanasiou V, Gerakaris V et al (2022) Seagrass meadows in the Greek seas: presence, abundance and spatial distribution. Bot Mar 65:289–299. https://doi.org/10.1515/bot-2022-0011
Pazzaglia J, Nguyen HM, Santillán-Sarmiento A et al (2021) Review the genetic component of seagrass restoration: what we know and the way forwards. Water. https://doi.org/10.3390/w13060829
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Phair NL, Toonen RJ, Knapp ISS, Von Der Heyden S (2020) Anthropogenic pressures negatively impact genomic diversity of the vulnerable seagrass Zostera capensis. J Environ Manage 255:109831. https://doi.org/10.1016/j.jenvman.2019.109831
Phair NL, Nielsen ES, Von Der Heyden S (2021) Applying genomic data to seagrass conservation. Biodivers Conserv 30:2079–2096. https://doi.org/10.1007/s10531-021-02184-w
Piry S, Alapetite A, Cornuet JM et al (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539. https://doi.org/10.1093/jhered/esh074
Plaisted HK, Novak AB, Weigel S et al (2020) Eelgrass genetic diversity influences resilience to stresses associated with Eutrophication. Estuaries Coasts 43:1425–1438. https://doi.org/10.1007/s12237-019-00669-0/Published
Politikos DV, Ioakeimidis C, Papatheodorou G, Tsiaras K (2017) Modeling the fate and distribution of floating litter particles in the Aegean Sea (E. Mediterranean). Front Mar Sci 4:191. https://doi.org/10.3389/fmars.2017.00191
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Procaccini G, Orsini L, Ruggiero MV, Scardi M (2001) Spatial patterns of genetic diversity in Posidonia oceanica, an endemic Mediterranean seagrass. Mol Ecol 10:1413–1421
Procaccini G, Ruggiero MV, Orsini L (2002) Genetic structure and distribution of microsatellite population genetic diversity in Posidonia oceanica in the Mediterranean basin. Bull Mar Sci 71:1291–1297
Rannala B, Mountain JL (1997) Detecting immigration by using Multilocus genotypes. Proc Natl Acad Sci USA 94:9197–9201
Raymond M, Rousset F (1995) GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J Hered 86:248–249
R Core Team (2021) A Language and environment for statistical computing
Repolho T, Duarte B, Dionísio G et al (2017) Seagrass ecophysiological performance under ocean warming and acidification. Sci Rep 7:1–12. https://doi.org/10.1038/srep41443
Reynolds LK, McGlathery KJ, Waycott M (2012) Genetic diversity enhances restoration success by augmenting ecosystem services. PLoS ONE. https://doi.org/10.1371/journal.pone.0038397. 7:
Ruggiero MV, Turk R, Procaccini G (2002) Genetic identity and homozygosity in North-Adriatic populations of Posidonia oceanica: an ancient, post-glacial clone? Conserv Genet 3:71–74. https://doi.org/10.1023/A:1014207122382
Ruiz JM, Marín-Guirao L, García-Muñoz R et al (2018) Experimental evidence of warming-induced flowering in the Mediterranean seagrass Posidonia oceanica. Mar Pollut Bull 134:49–54. https://doi.org/10.1016/j.marpolbul.2017.10.037
Ruocco M, Jahnke M, Silva J et al (2022) 2b-RAD genotyping of the seagrass cymodocea nodosa along a latitudinal cline identifies candidate genes for environmental adaptation. Front Genet 13:866758. https://doi.org/10.3389/fgene.2022.866758
Salo T, Gustafsson C (2016) The effect of genetic diversity on ecosystem functioning in vegetated coastal ecosystems. Ecosystems 19:1429–1444. https://doi.org/10.1007/s10021-016-0014-y
Selkoe K, D’Aloia C, Crandall E et al (2016) A decade of seascape genetics: contributions to basic and applied marine connectivity. Mar Ecol Prog Ser 554:1–19. https://doi.org/10.3354/meps11792
Serra IA, Innocenti AM, Di Maida G et al (2010) Genetic structure in the Mediterranean seagrass Posidonia oceanica: disentangling past vicariance events from contemporary patterns of gene flow. Mol Ecol 19:557–568. https://doi.org/10.1111/j.1365-294X.2009.04462.x
Simboura N, Maragou P, Paximadis G et al (2019) Greece. World seas an environmental evaluation. Elsevier, Amsterdam, pp 227–260
Sinclair E, Krauss S, Anthony J et al (2014) The interaction of environment and genetic diversity within meadows of the seagrass Posidonia Australis (Posidoniaceae). Mar Ecol Prog Ser 506:87–98. https://doi.org/10.3354/meps10812
Sinclair EA, Anthony JM, Greer D et al (2016) Genetic signatures of bassian glacial refugia and contemporary connectivity in a marine foundation species. J Biogeogr 43:2209–2222. https://doi.org/10.1111/jbi.12822
Strydom S, Murray K, Wilson S et al (2020) Too hot to handle: unprecedented seagrass death driven by marine heatwave in a World Heritage Area. Glob Change Biol 26:3525–3538. https://doi.org/10.1111/gcb.15065
Sundqvist L, Keenan K, Zackrisson M et al (2016) Directional genetic differentiation and relative migration. Ecol Evol 6:3461–3475. https://doi.org/10.1002/ece3.2096
Tsabaris C, Tsiaras K, Eleftheriou G, Triantafyllou G (2021) 137Cs ocean distribution and fate at East Mediterranean Sea in case of a nuclear accident in akkuyu nuclear power plant. Prog Nucl Energy. https://doi.org/10.1016/j.pnucene.2021.103879
Tsiaras K, Hatzonikolakis Y, Kalaroni S et al (2021) Modeling the pathways and accumulation patterns of micro- and macro-plastics in the Mediterranean. Front Mar Sci. https://doi.org/10.3389/fmars.2021.743117
Tutar O, Ruocco M, Dattolo E et al (2022) High levels of genetic diversity and population structure in the Mediterranean seagrass Posidonia oceanica at its easternmost distribution limit. ICES J Mar Sci 79:2286–2297. https://doi.org/10.1093/icesjms/fsac163
Underwood JN, Smith LD, Van Oppen MJH, Gilmour JP (2007) Multiple scales of genetic connectivity in a brooding coral on isolated reefs following catastrophic bleaching. Mol Ecol 16:771–784. https://doi.org/10.1111/j.1365-294X.2006.03187.x
Unsworth RKF, Collier CJ, Waycott M et al (2015) A framework for the resilience of seagrass ecosystems. Mar Pollut Bull 100:34–46. https://doi.org/10.1016/j.marpolbul.2015.08.016
Unsworth RKF, Mckenzie LJ, Collier CJ et al (2019) Global challenges for seagrass conservation. Ambio 48:801–815. https://doi.org/10.1007/s13280-018-1115-y
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Waits LP, Luikart G, Taberlet P (2001) Estimating the probability of identity among genotypes in natural populations: cautions and guidelines. Mol Ecol 10:249–256. https://doi.org/10.1046/j.1365-294X.2001.01185.x
Waycott M, Duarte CM, Carruthers TJB et al (2009) Accelerating loss of seagrasses across the globe threatens coastal ecosystems. Proc Natl Acad Sci 106:12377–12381. https://doi.org/10.1073/pnas.0905620106
Williams AV, Nevill PG, Krauss SL (2014) Next generation restoration genetics: applications and opportunities. Trends Plant Sci 19:529–537. https://doi.org/10.1016/j.tplants.2014.03.011
Winter DJ (2012) Mmod: an R library for the calculation of population differentiation statistics. Mol Ecol Resour 12:1158–1160. https://doi.org/10.1111/j.1755-0998.2012.03174.x
Acknowledgements
We acknowledge lab training received by Ester Serrao during a short stay at the Algarve Centre of Marine Sciences (CCMAR, Portugal) funded by the European Union’s Horizon 2020 research and innovation programme under grant agreement No 730984, ASSEMBLE Plus project. We thank Julius Glampedakis, Thanos Dailianis, Oscar Serrano, Miguel Angel Mateo, Paul Lavery, Karina Inostroza, and Eduard Serrano for participating in the sample collection. We also thank Vaso Terzoglou and Jon Kristoffersen for their help during the lab work.
Funding
Open access funding provided by HEAL-Link Greece. This research was co-financed by DRESSAGE (MIS5045792) (through the Operational Program ‘Competitiveness, Entrepreneurship and Innovation’ (EPAnEK 2014–2020) and the Water Framework Directive (WFD) and Habitat Directive (92/43/EC) implementation within the framework of the National Monitoring Project of Greece.
Author information
Authors and Affiliations
Contributions
ETA and VLM conceived and designed the study. ETA and VG performed the samplings. VLM and CAGE performed the laboratory analysis and analysed the data. VLM drafted the manuscript. KT performed the biophysical modelling. ETA and CST acquired the funding and supervised the study. All co-authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Communicated by Pradeep K. Divakar.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Include article note in XML - The original online version of this article was revised: text was changed
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Litsi-Mizan, V., García-Escudero, C.A., Tsigenopoulos, C.S. et al. Unravelling the genetic pattern of seagrass (Posidonia oceanica) meadows in the Eastern Mediterranean Sea. Biodivers Conserv 33, 257–280 (2024). https://doi.org/10.1007/s10531-023-02746-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10531-023-02746-0