
Abstract
In a pyrolysis reactor, organic polymers from biomass or plastic waste are thermally decomposed into volatile gases, condensable vapours (tar or bio-oil) and solid residues (char). Since these products may serve as building blocks for downstream chemical refinement or form the basis of bio-derived fuels, pyrolysis is thought to be instrumental in our progress towards a circular economy. A pyrolysis reactor constitutes a multiphase reactive system whose operation is influenced by many chemical and physical phenomena that occur at different scales. Because the interactions and potential reinforcements of these processes are difficult to isolate and elucidate experimentally, the development of a predictive modelling tool, for example, based on the CFD-DEM (discrete element method) methodology, is attracting increasing attention, particularly for pyrolysis reactors operated with biomass as feedstock. By contrast, CFD-DEM descriptions of plastic pyrolysis remain a challenge at present, mainly due to an incomplete understanding of their melting behaviour. In this article, we provide a blueprint for describing a pyrolysis process within the scope of CFD-DEM, review modelling choices made in past investigations and detail the underlying assumptions. Furthermore, the influence of operating conditions and feedstock properties on the key metrics of the process, such as feedstock conversion, product composition and residence time, as determined by past CFD-DEM analyses is surveyed and systematised. Open challenges that we identify pertain to the incorporation of particle non-sphericity and polydispersity, the melting of plastics, particle shrinkage, exothermicity on part of the gas-particle chemistry and catalytic effects.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
1 Introduction
Pyrolysis is the decomposition of organic matter, both naturally occurring and synthetic, into a range of light weight compounds through thermal treatment in the absence of oxygen. The liberated compounds can be used as energy sources or building blocks for further chemical processing (Sadaka and Boateng 2009). In addition to being an end-to-end process on its own, pyrolysis is also encountered as a preceding step of other processes, such as combustion and gasification (Grønli and Melaaen 2000). Within the scope of coal combustion, for instance, pyrolysis can be employed as a means of stabilising the flame, as loose volatiles are released before the oxidation commences. This initial devolatilisation has been found to have a considerable influence on the subsequent combustion behaviour of coal particles (Solomon and Fletcher 1994; Zhang et al. 2022b).
While the reactors carrying out pyrolysis may be operated with any organic polymer as feedstock, there is an apparent interest in biomass or plastics for this role. By virtue of its wide availability and carbon-neutrality, biomass has certainly asserted dominance over other renewable energy sources during the past decade. In fact, bio-energy is the largest renewable energy source, accounting for two-thirds of the total renewable energy throughput. Currently, it occupies about 14% of the global energy production (World Bioenergy Association 2020). Pyrolysis, alongside other conversion routes, such as liquefaction, fermentation, and enzymatic conversion, enables the extraction of bio-oil from biomass. Once upgraded, this bio-oil has the potential to serve as an alternative to fossil fuels (Matayeva et al. 2019). Plastics, on the other hand, face several operational challenges during their conventional conversion procedure, i.e., mechanical recycling. These challenges include the intrinsic irreversibility of thermosetting plastics and the need for sorting and separation based on plastic type prior to recycling—a process that is naturally labour-intensive (Eze et al. 2021). Consequently, from 6300 Mt of plastics that had been produced until 2015, only 9% has been recycled, whereas an alarming 79% has ended up as landfill (Geyer et al. 2017). A recycling strategy based on pyrolysis, by contrast, does not suffer from the aforementioned limitations concerning recycling; it provides a strategy to convert thermosetting plastics to reusable substances and can readily be implemented on unsorted feedstocks, e.g., municipal plastic waste. Nowadays, plastics are even allowed in the same pyrolyser as biomass, in pursuit of energy optimisation and better product quality (Wang et al. 2021c).
The extensive list of pyrolysis products can be classified into three main groups: pyrolysis-oil, pyrolysis-gas and char residue. Pyrolysis-oil, which is the desired product of most applications, is recovered by condensing the vapour collected during the process. The non-condensable fraction of this vapour is termed the pyrolysis-gas and can be utilised for heat and power generation or as a feedstock in manufacturing industrial chemicals, such as methanol and ammonia. The solid residue, which remains after the reactions, is rich in carbon and has excellent absorbent qualities. It is important for the metallurgical industry as a reducing agent, or for households as a cooking fuel or as a soil amender with the ability to sequestrate carbon (Grønli and Melaaen 2000). Clearly, the yield and the composition of pyrolysis product groups strongly depend on the composition of the feedstock. For example, a particle of woody biomass mainly encompasses three distinct polymeric components, namely cellulose \((\text {C}_6\text {H}_1\text {O}_{5})_{n}\), hemicellulose, and lignin. As listed in Table 1, this lignocellulosic biomass displays a different composition depending on the wood type from which it is sourced (Gellerstedt et al. 2009). The compositions may also fluctuate depending on the seasons and during the prepossessing of the feedstock (Williams et al. 2016). Due to its comparatively shorter chain lengths, the decomposition of hemicellulose commences at the lowest temperature (200 \(^\circ \)C), followed by cellulose at 240 \(^\circ \)C and lignin at 280 \(^\circ \)C. During the initial decomposition, both hemicellulose and cellulose produce more condensable vapour, while lignin predominantly yields char residue. The extracted bio-oil from hemicellulose mainly contains acids and furfural derivatives. Levoglucosan \((\text {C}_6\text {H}_{10}\text {O}_5)\) is the most abundant species in bio-oil, which is obtained mostly from the conversion of cellulose (Rabinovich et al. 2009). The average molecular weight of the bio-oil is seen to increase with the amount of lignin in the feedstock, since the resulting vapour contains heavier compounds (Fahmi et al. 2008).
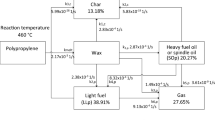
Most of our municipal plastic waste consists of polypropylene (PP). PP along with high and low density polyethylenes (HDPE and LDPE), polyethylene terephthalate (PET), and polystyrene (PS) are the most common plastic types which are being pyrolysed (Vijayakumar and Sebastian 2018). A forecast of plastic pyrolysis products is possible based on the proximate analyses that break-down a plastic into four components: moisture, fixed carbon, volatiles, and ash. Generally, high volatile and low ash contents are known to result in high yields of pyrolysis-oil (Anuar Sharuddin et al. 2016). Pyrolysis of HDPE, LDPE, PP, and PS are characterised by a higher pyrolysis-oil yield since they are rich in volatile matter (Vijayakumar and Sebastian 2018). PET, on the other hand, converts mostly into pyrolysis-gas, while its oil is unfeasibly acidic with benzoic acid (Çepelioǧullar and Pütün (2013)). Although pyrolysis seems to be a convenient pathway to mitigate the issues of plastic recycling, it also possesses limitations, mainly due to the chemical compositions of certain plastics. The appearance of polyvinyl chloride (PVC) in pyrolysis feedstock raises health concerns as it decomposes into toxic hydrochloric fumes when undergoing pyrolysis. Furthermore, the high sulphur contents found in the liquid fuel obtained from pyrolysis of plastics render it unsuitable for deployment in existing combustion engines without any down-stream purification (Eze et al. 2021).
Based on the operating conditions, pyrolysis can be categorised as either slow, fast or flash pyrolysis. Slow pyrolysis is characterised by slow heating rates \(\left( {0.1}{-}{1}\,^\circ \text {C s}^{-1}\right) \) and long particle and vapour residence times \(\left( {450}{-}{550}\text {s} \right) \). These conditions promote the devolitilisation to occur in favour of the char residue. Also, if the residence time is long, then the produced vapour will inevitably partake in secondary decomposition reactions, further reducing the yield of pyrolysis-oil. On the contrary, fast pyrolysis requires much faster heating rates \(\left( {10}{-}{100}\,^\circ \text {C s}^{-1} \right) \), and the residence time \(\left( {0.5}{-}{10}\text {s} \right) \) of particles and the liberated vapour is minimised by employing rapid gas flow rates. In this heating regime, the devolatilisation produces more condensable vapours. Due to their rapid release from the reactor, these vapours are largely prevented from further decomposition. The flash pyrolysis features even faster heating \(\left( > {1000}{\,^\circ }\text {C} \text {s}^{-1} \right) \) and particle and vapour entrainment rates \(\left( < {0.5}\text {s} \right) \). Consequently, the product will largely contain pyrolysis-oil and only a little amount of char residue (Uddin et al. 2018).
A generic pyrolysis reactor epitomises a complex multiphase reactive system, wherein a decomposing particulate phase continuously interacts with a moving gas that, too, contains reactive species. In some applications, the solid phase may be a combination of the feedstock and inert bed material that acts as a heat capacitance and improves mixing and heat transfer inside the reactor. As illustrated in Fig. 1, pyrolysis can be viewed as a multi-scale problem, where attributes at different scales co-dependently co-influence the efficacy and outcome of the overall process, measured in terms of the final conversion, product composition, particle residence time, etc. Apart from the feedstock composition, the existence of catalytic species in the system also tremendously influences the rates and paths of decomposition reactions at the molecular level. For instance, the naturally existing potassium in biomass has proven catalytic effects on thermochemical conversion processes, including pyrolysis (Safar et al. 2019; Fan et al. 2021). At the particle scale, the physiothermal properties of a particle, such as its size, shape, density, heat capacity, and conductivity, govern the intra-particle distributions of temperature and composition and control the shrinkage behaviour. Lastly, at the reactor scale, operating conditions, such as temperature and inert flow rate, largely influence the interphase heat, mass, and momentum interactions. Moreover, these scale-attributed phenomena are inherently linked to functions on other scales as well. For example, the temperature and the velocity of the carrier gas contribute to the intra-particle temperature distribution, which in turn affects its local decomposition rates.

Chemical and physical processes and interactions on different scales in a pyrolyser. The pyrolysis system is affected by the chemical composition at the molecular scale, by particle properties at the particle scale, and by operating conditions at the reactor scale
The most common choices for pyrolysis reactor systems include fluidised bed reactors, fixed bed reactors, auger reactors, and entrained flow reactors (Uddin et al. 2018). Moreover, pyrolysis may be operated as an auto-thermal process using a circulating fluidised bed setup, where the energy demand for thermal decomposition in the primary bed is supplied by the heat extracted from the combustion of char residue inside a secondary bed through a circulating heat carrier (Liu et al. 2017b). Recently, novel technologies, such as microwave pyrolysis, grinding pyrolysis, and solar pyrolysis have also been researched in this field (Chen et al. 2019). Due to their large heat and mass transfer rates and mixing degrees, fluidised bed reactors are often preferred to carry out pyrolysis (Ku et al. 2019). However, fluidised beds, too, are not free of drawbacks. The backmixing effect and premature exits of particles, which are characteristic of fluidised beds, are absolutely undesired in pyrolysis (Wang et al. 2021b). Furthermore, a fluidised bed may not be a viable option for the conversion of plastics, as they melt rather easily causing adhesion in particles, resulting in de-fluidisation (Zhang et al. 2022a). Many experimental studies are available on pyrolysis of various feedstocks (biomass (Guizani et al. 2017; Septien et al. 2012; Shen et al. 2009), plastics (Mastral et al. 2002; Chaudhary et al. 2023; Stelmachowski 2010; Lin and Yen 2005)), whose main focus has been analysing the effect of operating conditions. Despite being essential in the process of developing and optimising real-life reactors, they offer the barest insight into the physiochemical interactions taking place inside the system. Additionally, building an experimental setup is expensive and time-consuming. Its operation is also fairly challenging because of the accumulation of tar, that ends up causing blocks or even erosion of equipment (Chen et al. 2020). Consequently, researchers have shifted to employing numerical simulations, or what is known as “virtual experiments”, employing computational fluid dynamics (CFD) to investigate the behaviour of pyrolysis reactors across scales. In particular, CFD-based tools have permitted the analysis of many-particle systems without sacrificing intricate features thanks to the exponential growth in computing power over the past few decades.
In the past, pyrolysis reactors have been modelled using two main approaches, the two-fluid model (TFM) and the CFD framework combined with the discrete element method (DEM), on which we focus here. The two approaches differ in terms of the physical description of the particulate feedstock and possess distinct advantages and challenges. In the TFM, the particulate feedstock and the carrier gas phase are considered as two interpenetrating and coexisting continua that may exchange mass, momentum and heat. While these exchanges are driven by physical and chemical processes that live on the single particle level, the particulate phase is conceptually described on the level of an entire particle population in terms of the particle number or mass density, the mean particle velocity (or momentum density) and an enthalpy density. Strictly, these densities represent means that are defined or computed as averages over all particles in a control volume about the present spatial location and can be related to statistics of the so-called particle property distribution. The property distribution quantifies how the particles within a control volume differ in terms of property values, for example, the particle size, velocity or temperature. The evolution in space and time of the property distribution can be described by the population balance equation (PBE) (Rigopoulos 2010), but its solution is practically challenging owing to its high dimensionality. For the particulate phase, the two-fluid equations follow from the PBE as the equations for the zeroth and first order moments of the property distribution and include two distinct types of closures that require modelling. The first type encompasses all assumptions that have led to the formulation of the PBE, for example, a continuity hypothesis on the distribution of the particles in physical and property space (Hulburt and Katz 1964) or assumptions on two-particle statistics influencing collisions (Marchisio and Fox 2013, Chapter 6). The second type originates from the moment reduction of the PBE and requires assumptions on how the particle-averaged behaviour of the particulate phase is modified by property-polydispersity. In this regard, the closure of collisional interparticle momentum transfer, for instance, is achieved with the aid of a pseudo-stress model (Lun et al. 1984). Despite these closure challenges, the TFM has been very popular for the investigation of pyrolysis reactors, as it has many practical advantages. First, since the TFM is a fully Eulerian description, it supports solution through standard CFD tools. Second, the number of fields describing the particulate phase is independent of the number of physical particles, rendering the model particularly economical for applications with a large number of physical particles. Third, particle-particle interactions are accounted for statistically and, therefore, do not require the computationally expensive contact search and tracking algorithms of CFD-DEM.
In the context of pyrolysis, most of the past contributions to the modelling of gas-particle flows have been based on the TFM, analysing the flow in fluidised beds (Lathouwers and Bellan 2001), spouted beds (Hooshdaran et al. 2021), entrained-flow reactors (Xiu et al. 2008), and auger reactors (Aramideh et al. 2015). The investigation of the effects of operating conditions on biomass pyrolysis in fluidised beds by Lathouwers and Bellan (2001) is considered as one of the seminal works. They concluded that the temperature of the fluidising gas is the most influential parameter for the yield of bio-oil. Ranganathan and Gu (2016) compared the product yields predicted by global, semi-global, and detailed chemical kinetic models of biomass pyrolysis using TFM and demonstrated that the detailed scheme produces the most accurate results. Xiong et al. (2016) coupled TFM with a distributed activation energy model (DAEM) to account for the varying bond strengths across molecules of the same component. Considering a multi-fluid formulation, Liu et al. (2017a) and Xue and Fox (2015) were able to compute particle size distributions and the density of pyrolysis feedstock by adopting the direct quadrature method of moments (DQMOM) to solve the PBE.
Within the scope of the CFD-DEM approach, on the other hand, the dispersed particles are considered as separate individual entities, possessing distinct sets of degrees of freedom. As mass, momentum, and heat are exchanged between a particle and the carrier medium, the particle’s degrees of freedom evolve in time. Both the number of degrees of freedom per particle and their physical meaning depend on the fidelity of the particle-level model. For example, a single particle temperature is sufficient if the heat conduction within the particle is much faster than the rate at which heat is supplied to the particle, while several temperature degrees of freedom are needed to parameterise the intra-particle temperature profile of thermally thick particles. For very small particles, temperature could even be entirely removed from the list of degrees of freedom if the particle and surrounding gas equilibrate (nearly) instantaneously to a common temperature. From the perspective of the continuous carrier phase, the DEM particles constitute point sources of mass, momentum, and enthalpy and are, therefore, often referred to as point-particles. In particular, the flow around the particles is not spatially resolved; this is in line with population balance models and the TFM and causes CFD-DEM to realise a meso-scale description. The dynamics of DEM point-particles follow from single-particle mass, momentum, and enthalpy balances, including all particle-particle interactions. By design, the CFD-DEM framework can accommodate multi-particle interactions and particle-particle polydispersity without top-level approximations, and amendments of the point-particle model are readily incorporated. Furthermore, the CFD-DEM approach enables the prediction of residence times of individual particles, a feature that is absent in TFM (Yang et al. 2021). This information is instrumental during the design and optimisation cycle of a continuous pyrolysis system to prevent both premature exits and accumulation of fully converted feedstock particles.
While less common than TFMs, the CFD-DEM methodology has been applied to many pyrolysis systems over the course of the past decade, including fluidised beds (Bruchmüller et al. 2012; Wang et al. 2021b), packed beds (Mahmoudi et al. 2016b), entrained flow reactors (Gao et al. 2021a), auger reactors (Qi and Wright 2020), batch reactors (Cordiner et al. 2018), and rotary drums (Wang et al. 2022). Papadikis et al. (2008, 2009b, 2009c, 2009a) pursued a hybrid TFM-DEM modelling approach to model the pyrolysis of biomass in fluidised beds with inert bed material. Here, the inert particles were modelled as a continuous phase using a TFM description, while the feedstock was represented by DEM point-particles. Beyond pyrolysis, the applications of CFD-DEM contain many other multi-phase reactive systems such as gasifiers (Ku et al. 2015a), furnaces (Wei et al. 2019) and shaft kilns (Krause et al. 2015, 2017).
Pyrolysis is a maturing technology with the potential to have a positive impact on society and the environment, as it is a building block for a circular economy based on the recycling and sustainable upgrading of spent resources. In line with our objective to realise these benefits optimally at an industrial scale, CFD-DEM as a computer-based design and analysis tool offers valuable and explanatory insights into the pyrolysis processes that are difficult (or impossible) to gain through experimental investigations. Although many successful attempts at studying pyrolysis of biomass with CFD-DEM are reported in the literature, to the best of our knowledge, it is yet to be applied to the pyrolysis of plastics. We believe that a succinct review of the CFD-DEM framework with regard to pyrolysis systems, and a reevaluation of the insights provided by past studies will motivate researchers to not only analyse more systems in depth in the future, but also to develop effective tools to enhance the usability of CFD-DEM in this context. Our work contrasts with many reviews done by researchers on related topics, which include chemical kinetics of plastic pyrolysis (Armenise et al. 2021) and biomass pyrolysis (Papari and Hawboldt 2015), application of DAEM in biomass pyrolysis (Cai et al. 2014), co-pyrolysis of biomass (Abnisa and Wan Daud 2014), particle-scale models for biomass pyrolysis (Luo et al. 2022), biomass pyrolysis with CFD (Xiong et al. 2017), and MFiX-based CFD simulations of biomass pyrolysis (Lu et al. 2021a). There are several conversion methods, such as torrefaction and hydrothermal carbonisation, which are closely related to pyrolysis but which we do not discuss here because they have hardly been investigated using CFD-DEM tools.
This review article is structured as follows. In Sect. 2, we summarise the fundamental principles of the CFD-DEM framework while presenting the governing equations of both gas and solid phases encountered in a typical pyrolyser. Several potential simplifications for the descriptions of each phase as well as for interphase interactions are also detailed based on the characteristics of the targeted systems. Section 3 reviews the numerous physical mechanisms occurring in a pyrolyser, their influence on the overall process, and possible modelling techniques that lie within the scope CFD-DEM. Section 4 is reserved for a discussion of the impact of operational parameters on the results of pyrolysis, as obtained from past CFD-DEM studies on the subject. In Sect. 5, we identify open challenges with regard to modelling of pyrolysis using CFD-DEM, before conclusions are drawn in Sect. 6.
2 Core Model for Describing the Continuous and Particulate Phases
2.1 Carrier Gas
In the CFD-DEM framework, the continuous gas phase is described in a Eulerian fashion based on field variables that vary both along the spatial coordinates \(\textbf{x}\) and in time t. The mechanical field variables encompass the gas velocity \(\textbf{u}_\textrm{g}\) and the mechanical pressure p and are, strictly, obtained by applying a spatial averaging or filtering operator to the true, spatially completely resolved fields. For conciseness, the averaging operation is not indicated notationally here, but we emphasise that its application is implied throughout. In practice, the averaging length scale is often associated with the grid on which the governing gas phase equations are solved using a direct discretisation scheme. Alternatively, the averaging length scale may be chosen independent of a discretisation in such a way that the gas volume fraction \(\varepsilon _\textrm{g}\) obtained upon filtering is insensitive (or hardly sensitive) to the length scale. Conceptually, the averaging results in a removal of small-scale gradients from the describing fully resolved fields, resulting in averaged fields that vary smoothly on the filter scale. Within the scope of a CFD-DEM description, these averaged fields are the principal descriptors of the continuous phase. Particularly in the presence of turbulence, the influence of the residual scales that were eliminated by the filtering on the behaviour of the averaged fields may be accounted for through an amendment of the single phase constitutive laws that govern, for example, viscous stresses or heat conduction. In this review, however, we exclude both turbulence and the explicit modelling of residual scales other than through gas-particle mass, momentum or heat exchanges.
The filtering approach mentioned above was pioneered by Anderson and Jackson (1967) and leads, upon adaptation to a DEM description of the particulate phase, to the following mass and momentum balance equations governing the gas velocity \(\textbf{u}_\textrm{g}\) and the mechanical pressure p,
The right hand side of Eq. (1) includes the mass \(S^\textrm{mass}_j\) that is released to the gas by particle j per unit of time. The Dirac \(\delta \)-distribution \(\delta (\textbf{x}- \textbf{x}_j(t))\) indicates that the mass is released at the present location \(\textbf{x}_j(t)\) of particle j and that the associated mass density is, therefore, concentrated entirely at \(\textbf{x}_j\). Similarly, the momentum source terms on the right hand side of Eq. (2) encompass the momenta \(\textbf{S}^\textrm{mom}_j\) imparted to the carrier flow by the dispersed particles \(j = 1,\ldots ,N_\textrm{p}\). Lastly, \(\textbf{g}\) (in [N/kg\(_\textrm{g}\)]) indicates a volume force that may be instrumented to accommodate gravity, for instance, while \(\overline{\overline{\varvec{\tau }}}\) denotes the viscous stress tensor. Frequently, the gas phase is assumed to behave like a Newtonian fluid such that \(\overline{\overline{\varvec{\tau }}}\) is given by
where \(\mu _\textrm{g}\) is the dynamic viscosity and \(\overline{\overline{\mathbb {I}}}\) denotes the identity tensor.
The gas density \(\rho _\textrm{g}\) that appears in Eqs. (1) and (2) is a constitutive property that defines the material make-up of the gas and is linked to the gas temperature \(T_\textrm{g}\) and the thermodynamic pressure P through an equation of state. In the modelling of pyrolysis reactors, the gas phase is often considered to behave like an ideal gas, whence the three thermal state variables obey
where R is the universal gas constant and W denotes the mean molecular weight of the gas molecules. If the gas velocity is much smaller than the speed of sound, then acoustic pressure perturbations propagate almost instantaneously through the reactor, causing the thermodynamic pressure P in Eq. (4) to be nearly homogeneous (but possibly time-dependent), \(P = P(t)\). In this case, the mechanical pressure p in Eq. (2) remains distinct from P and takes on the role of a kinematic variable akin to a multiplier that enforces the continuity equation (Eq. (1)). In reactors that are open to the environment or that are operated at constant pressure by means of a movable piston, P is prescribed as the ambient pressure. If a reactor is operated at a prescribed volume V(t), by contrast, P can be computed in terms of the total reactor volume from Eq. (4). Physically, the assumption that the thermodynamic pressure P is homogeneous and independent of p is associated with a low Mach number, \(\textrm{Ma} \lesssim 0.3\). In practice, the low Mach number assumption is very advantageous because it permits the thermal state \((\rho _\textrm{g}, T_\textrm{g}, P)\) of the gas to be defined independent of the gas motion. For example, in a constant pressure reactor and for given W, the gas density (and, hence, the entire thermal state) is only controlled by \(T_\textrm{g}\).
At larger Mach numbers, the propagation of acoustic pressure perturbations is tied in a much stronger fashion to the gas flow field and the almost instantaneous, omnidirectional equidistribution of pressure that is so characteristic of small Mach numbers ceases. The thermodynamic pressure P then coincides with the mechanical pressure p, \(P = p(\textbf{x}, t)\), which acquires the meaning of a kinetic field variable. In the model formulations for pyrolysis reactors we review in this article, the low Mach number assumption is invoked throughout and we, therefore, maintain the simplifications it entails below.
In Eqs. (1) and (2), the constriction of the flow domain due to the presence of pyrolysing particles is accounted for through the gas volume fraction \(\varepsilon _\textrm{g}\). Since \(\varepsilon _\textrm{g}\) is the complement of the particle volume fraction \(\varepsilon _\textrm{p}\), it can be linked to the volumes \(V_j(t)\) presently occupied by all particles \(j = 1,\ldots ,N_\textrm{p}\) according to
If the gas temperature \(T_\textrm{g}\) and the mean molecular weight W are specified and if we leave aside the particle phase variables for a moment, then Eqs. (1) through (3) are completely closed in terms of the main gas phase fields \(\textbf{u}_\textrm{g}\), p and \(\rho _\textrm{g}\). In pyrolysis reactors, however, external heating, conductive heat losses to the particles, radiation, and gas phase chemical reactions induce temperature heterogeneities that evolve through conductive and convective heat transfer. Jointly, these effects enter an evolution equation for a calorific variable that quantifies both the sensible heat and the energy stored chemically within the gas. In open or constant pressure systems, this calorific variable is commonly chosen as the gas enthalpy \(h_\textrm{g}\) (in [J/kg\(_\textrm{g}\)]),
where \(h_i\) denotes the specific enthalpy of gaseous species i, \(\varDelta h_{f,i}^0\) is its enthalpy of formation at the reference temperature \(T_\textrm{ref}\) and \(C_{p,i}\) represents its specific heat capacity at constant pressure. In Eq. (6), the chemical composition of the gas is represented in terms of the mass fractions \(Y_{\text {g}, i}\) of all gaseous species \(i = 1,\ldots ,N_\textrm{g}\). Note that, by definition, the species mass fractions sum to unity.
The evolution equations for the species mass fractions follow from a material balance of the species-specific masses upon localisation and read
where \(S^\mathrm {mass-reac}_{\textrm{g},i}\) is the rate at which species i is produced (\(S^\mathrm {mass-reac}_{\textrm{g},i} > 0\)) or consumed (\(S^\mathrm {mass-reac}_{\textrm{g},i} < 0\)) due to gas phase chemical reactions. Similarly, \(S^\textrm{mass}_{j,i}\) represents the mass of species i released to the gas phase by particle j; jointly, the source terms \(S^\textrm{mass}_{j,i}\) associated with all species yield the gas-particle mass exchange term \(S^\textrm{mass}_{j}\) in Eq. (1),
We emphasise that the works reviewed in this article involve reactor setups that are operated exclusively in pyrolysis conditions, where the injected gas flows exclusively comprise inert species. Therefore, the term \(S^\mathrm {mass-reac}_{\textrm{g},i}\) in Eq. (7) is a consequence only of the secondary thermal decomposition of gas species produced and released by the decomposing feedstock and does not account for combustion or partial oxidation reactions that are possible in actual pyrolysis reactors, which may contain oxidising species in minute quantities.
Strictly, the species-specific diffusion velocities \(\textbf{U}_i\) with
follow from the binary diffusion coefficients through the Maxwell-Stefan equations. In computational practice, however, the solution of these equations for \({\textbf{U}}_i\) is not only expensive, but also difficult to take advantage of because \({\textbf{U}}_i\) drives an advective term whose discretisation may elicit unphysical artificial diffusion unless a costly Riemann solver is invoked. Poinsot and Veynante (2005) summarise two common approximations with different physical degrees of fidelity that result in a conversion of the advective \({\textbf{U}}_i\)-term into a smoothing diffusion term with far less stringent resolution requirements. The first approximation is based on the assumption that all binary diffusion coefficients are identical and equal to \(D_\textrm{g}\), yielding
In a more general approach, Hirschfelder et al. (1964) assumed a single binary diffusion coefficient per species, yet, irrespective of the second species it diffuses into. This admits a species-unique coefficient of diffusion, given by \(D_{\textrm{g},i}\), and results in the diffusion velocity approximation
with (Wilke 1950)
where \(D_{ki}\) is the binary mass diffusion coefficient pertinent to diffusion of species k into species i, while \(X_{\textrm{g},i}\) denotes the mole fraction of species i. Unlike Fick’s law in Eq. (10), the Hirschfelder and Curtiss approximation violates Eq. (9), causing Eq. (7) to be inconsistent with Eq. (1). This mass error can be eliminated by amending the right-hand side of Eq. (11) by a correction velocity \({\textbf {u}}_\text {g}^\text {c}\),
where \(W_i\) is the molecular weight of species i. In an alternative method, Eq. (7) is solved only for \(N_\textrm{g}-1\) species, and the mass fraction of the last species is computed from the condition that all mass fractions sum to unity. In contrast to the approach based on \({\textbf {u}}_\text {g}^\text {c}\), this workaround results in an aggregation of the entire mass conservation error on the last species and should only be used if the last species is abundant (Poinsot and Veynante 2005).
If the gas enthalpy \(h_\textrm{g}\) of Eq. (6) is employed to measure the chemical and sensible energy storage of the carrier gas, then Eq. (7) is complemented by the balance law
where \(S^\textrm{heat}_j\) denotes the heat released by particle j. The flux vector \(\textbf{q}\) that appears in Eq. (14) accounts for the transport of heat across the bounding surface of a material gas volume; it is traditionally modelled as
and encompasses two contributions. The first term in Eq. (15) corresponds to Fourier’s law and represents the conductive heat transfer through the bulk gas that is, physically, mediated by molecular collisions and molecular diffusion. The intensity of this molecular heat migration is measured by the heat conductivity \(\lambda _\textrm{g}\). The second contribution serves to amend the heat flux by the net species-bound enthalpy transport.
The third term on the right-hand side of Eq. (14) represents the heating of the gas by viscous tractions. In low Mach number flows, however, the viscous heating term can be neglected and the mechanical pressure in Eq. (14) may be replaced by a distinct thermodynamic pressure \(P = P(t)\), as in the equation of state above (Eq. (4)) (Poinsot and Veynante 2005, Section 1.2.1). Introducing these simplifications along with Eqs. (1) and (6) into Eq. (14), the enthalpy balance can be recast in terms of the gas temperature, yielding
where \(C_{p,\text {g}}\) denotes the gas’ heat capacity at constant pressure,
2.2 Particulate Phase
In the applications we target in this review, the carrier gas is charged with dispersed particles that represent the pyrolysis feedstock and, possibly, inert bed material. Contrary to the gas, the particulate phase is observed from a Lagrangian perspective (DEM) and modelled as a collection of evolving point-particles. Specifically, the motion of particle j, translating and rotating with velocities \(\textbf{v}_j\) and \(\varvec{\upomega }_j\), obeys Newton’s second law (Fitzpatrick 2021),
where \(\textbf{x}_j\), \(m_j\) and \(\overline{\overline{\textbf{I}}}_j\) are the position, mass and the inertia tensor of particle j. The symbols \(\textbf{F}^\textrm{c}_j\), \(\textbf{F}^\textrm{d}_j\), and \(\textbf{F}^\textrm{b}_j\), moreover, represent the forces experienced by the particle due to particle-particle or particle-wall contacts, drag, and buoyancy, respectively. Commonly, the torque \(\varvec{\uptau }_j\) is computed solely based on particle-particle contact interactions, disregarding the effects of forces exerted by the gas phase. This is tantamount to assuming negligible variations in the flow properties, particularly density and velocity, over the length scale of a particle. Note that in Eq. (18c) the shape of the particle is assumed to be spherical. In the case of non-sphericity, the term \( - \varvec{\upomega }_j \times \overline{\overline{\textbf{I}}}_j \varvec{\upomega }_j \) needs to be added on the right-hand side to account for the particle’s directional variation of inertia and the orientation of the particle must be tracked, for example, through a set of quaternions as additional degrees of freedom (Diebel 2006; Zhao and van Wachem 2013).
In pyrolysis, the change of particle mass is controlled by two phenomena, drying and devolatilisation. Drying describes the evaporation of contained liquid water and requires the supply of heat through exothermal intra-particle reactions or by external heat transfer from the ambience. Devolatilisaion, by contrast, is the process of liberation of gaseous species from particles, produced as a result of the thermal decomposition of the feedstock. In the following, we denote the rates of drying and devolatilisation by \(\dot{m}_j^\textrm{evap}\) and \(\dot{m}_j^\textrm{devol}\), respectively,
These aspects are revisited along with their corresponding modelling approaches in subsequent sections of this article, specifically, drying in Sect. 3.3 and devolatilisation as part of chemical reactions in Sect. 3.4.
Besides the mass \(m_j\), the thermochemical state of a particle is characterised in terms of the particle temperature \(T_j\) and a chemical composition \(Y_j = \left\{ Y_{j,s} \, | \, s \in \{1,\ldots ,N_\textrm{s} \} \right\} \). Here, \(Y_{j,s}\) is the mass fraction of the sth particulate species, which represents either a solid phase educt or product or inherent moisture. From the definition \(Y_{j, s} = m_{j,s} / m_j\), the evolution of a species mass fraction in a particle is obtained upon time differentiation as
where \(m_{j,s}\) is the mass of species s contained in particle j and \(\textrm{d}m_{j,s}/\textrm{d}t\) describes the rate-of-change of \(m_{j,s}\) due to drying or intra-particle chemical reactions.
In a lumped description based on \(T_j\), a particle is assumed to possess a homogeneous internal temperature distribution. By an enthalpy balance, \(T_j\) changes in time according to
where
denotes the specific heat capacity of the particle and \(\dot{Q}_j\) accounts for the radiative and conductive heat transfer to the particle. The modelling approaches that relate \(\dot{Q}_j\) to the particle’s thermochemical state and the ambient conditions are reviewed in Sect. 3.1 below.
In the investigation of pyrolysers, the mass transfer from particles to the continuous phase is commonly assumed to be rate-limited by chemical kinetics, that is, the rate at which gaseous species are liberated from the free surface of the particles, rather than by mass transfer. In view of the generally small size of pyrolysis feedstock and the formation of cracks in the fuel particles undergoing pyrolysis, Rabinovich et al. (2009) emphasised that this assumption is well suited. By Spalding’s mass transfer theory (Spalding 1953), the mass flux emitted from a particle in the continuum regime decreases inversely proportionally to the particle diameter, whence mass transfer limitations apply to large particles. Additionally, the formation of cracks and the small particle size promote a low intra-particle mass transfer resistance, facilitating the transport of gaseous species within the particle. Accordingly, all formed gaseous species are instantaneously released into the carrier gas and the interphase mass transfer term can be formulated without reference to a mass transfer coefficient,
where \(\dot{m}^\textrm{devol}_{j,i}\) is the production rate of gas species \(i \ne \left\{ \text {H}_2\text {O} \right\} \) in particle j following its devolatilisation. Implicit in Eq. (23) is the assumption that water vapour is not produced by chemical reactions. If this assumption is invalid, then \(\dot{m}^\textrm{evap}_j\) needs to be complemented by a term \(\dot{m}^\textrm{devol}_{j,\text {H}_2\text {O}}\) that accounts for the chemical production of water vapour.
2.3 Discretisation and Gas-Particle Coupling
To simulate a flow laden with particles while accommodating the physical and chemical interaction mechanisms, a discretisation strategy for the partial differential equations and a strategy for coupling the Eulerian fluid fields and the states of the Lagrangian particles must be chosen.
An important aspect of the simulation framework for predicting particle-laden flows is how the spacing of the grid points on which the discretisation of the fluid phase equations is based compares to the size of the particles. This aspect is commonly quantified in terms of the ratio of particle size to fluid mesh spacing. Currently, there exists a substantial body of research focusing on fully resolved simulations of particle-laden flows. In these frameworks, the particles are still described in a Lagrangian framework, but spatially resolved. Similarly, the fluid flow and the fluid-particle boundary layers are completely resolved using Direct Numerical Simulation (DNS). This approach is frequently referred to as particle-resolved (PR) DNS or PR-DNS (Tenneti and Subramaniam 2014). The advantage of employing such an approach is that it minimises the reliance on empirical parameters since all significant length and time scales are resolved through the methodology. On the downside, a considerable computational cost is incurred, even when dealing with a small number of particles in the flow, rendering the method only viable for low to moderate Reynolds number flows. As a result, the PR-DNS framework is not suitable for predicting the behavior of most realistic pyrolysis processes. Nonetheless, it can still be employed to investigate highly specific information within very small, typically academic, cases of the pyrolysis of a few particles.
In most particle-laden flow frameworks, the particle surfaces are not fully resolved by the Eulerian mesh, and the interaction between the fluid and the surfaces of the individual particles must be modelled. Here, the Eulerian mesh cells are larger than the individual particles. A commonly used framework (Eaton 2009; Kuerten 2016) is the particle-source-in-cell Eulerian-Lagrangian (PSIC-EL) framework (Crowe et al. 1977), where the particles exchange information only with the cell in which they are present. The common assumption is made, that the Eulerian mesh cells are at least 10 times larger than the particle diameter, although even then errors of up to 10% can occur (Evrard et al. 2021). While various filtering techniques have been developed recently for a more accurate coupling between the Eulerian mesh and the Lagrangian particles (Capecelatro and Desjardins 2013; Evrard et al. 2019), these have not been used in the context of modelling pyrolysis processes with CFD-DEM.
3 Physical and Chemical Mechanisms
The CFD-DEM framework is kinetically driven by different chemical and physical processes that control not only the gas-particle mass, momentum, and heat exchanges, but also cause intra-particle alterations or changes on part of the carrier gas. At the end of this section, in Table 6, we summarise past CFD-DEM investigations of pyrolysis, with an emphasis on the modelling choices made by the investigators. The models referenced in Table 6 are discussed in detail in this section.
3.1 Heat Transfer
Accurate modelling of heat interactions, within and between the phases, is invaluable for an accurate prediction of the overall process for several reasons. The accuracy of many physical phenomena, such as the drying and chemical kinetic rates, strongly depends on the accuracy of the heat transfer rates and the resulting temperature profiles. Also, the enthalpy budget of a reactor is strongly tied to the system’s energy consumption, which is, in turn, mediated by the heat transfer in the system. Fundamentally, three modes of heat transfer can be distinguished in the system: conductive, convective, and radiative heat transfer. Following this distinction, the external heat transfer to a particle, which appears on the right hand side of Eq. (21), is decomposed into
with the terms \(\dot{Q}^\textrm{cond}_j, \, \dot{Q}^\textrm{conv}_j\), and \(\dot{Q}^\textrm{rad}_j\) denoting the conductive, convective, and radiative contributions, respectively.
Conduction describes the heat transfer through direct contact from an element of high temperature to another at a lower temperature, without an apparent motion of matter. In fluids, this is facilitated by collisions and diffusion at the molecular level, whereas in solids, it proceeds through lattice vibrations and the movement of free electrons. The conduction in the gas phase is accounted for by the third term on the right hand side of Eq. (16), and needs no further detailing. On the other hand, the interparticle conductive term \(\dot{Q}^\textrm{cond}_j \) encompasses two distinct mechanisms: the direct heat transfer in colliding particles, and the indirect heat transfer through the gas layer between non-colliding particles. The rates of these transfers, with reference to a particle j, are denoted by \(\dot{Q}^\mathrm {cond-pp}_j \) and \(\dot{Q}^\mathrm {cond-pgp}_j\), respectively, in the following,
Considering the heat transfer from particle \(j_2\) to another particle \(j_1\), the direct component is commonly computed with the model of Batchelor and O’Brien (1977),
while the indirect or particle-gas-particle conduction is modelled according to the scheme introduced by Rong and Horio (1999) as follows,
Here, \(r^\textrm{c}\) is the contact radius and \(\lambda _j, \, T_j\) are the thermal conductivity and the temperature of particle \(j \in \{ j_1,j_2 \}\), respectively. Additionally, \(r^\textrm{in}\) and \(r^\textrm{out}\) define the radial bounds of the corresponding heat transfer region and \(l \! \left( r \right) \) is the thickness of the gas layer separating the particles at the radial distance r, while \(l_\textrm{min}\) is a minimum (threshold) conduction distance. The threshold term in the denominator of Eq. (27) guards against a singularity of the heat transfer rate as \(l \! \left( r \right) \) approaches zero and the particles touch. Schematics of both direct and indirect heat transfer mechanisms are shown in Fig. 2. The conduction term is sometimes neglected in the particle enthalpy balance equation, stating its inferiority compared to the convective term (Bruchmüller et al. 2012; Ku et al. 2019; Li et al. 2017). In this regard, the CFD-DEM study by Hu et al. (2019) reveals that the interphase convective heat transfer is approximately twice more effective than the interparticle conductive heat transfer. Lu et al. (2020) pointed out that artificially low spring constants are generally implemented in DEM to ensure numerical stability. Consequently, the overlaps between colliding particles are severely overestimated and, therefore, also the direct heat transfer rates in Eq. (26). In order to avoid this error, a correction may be imposed on the overlap prior to heat transfer calculations. Studying the impact of shrinkage patterns and operating parameters on biomass pyrolysis, Hu et al. (2019) implemented such a correction based on the scheme proposed by Zhou et al. (2010).

Schematics of a Direct and b Indirect conductive heat transfer between two particles, namely \(j_1\) and \(j_2\). Here, \(r^\textrm{c}, \, l(r), \, l_\textrm{min}, r^\textrm{in}, r^\textrm{out}\) are the contact radius, normal gap, minimum conduction distance, and lower and upper radial bounds of heat transfer regions, respectively. Further, \(r_j\) is the radius of particle \(j \in \left\{ j_1,j_2 \right\} \), and \(\theta \) is the thickness of the thermal boundary layer of the comparatively bigger particle
The convective heat transfer, in contrast, is assisted by a bulk flow of matter that transports heat from a point of high temperature to a point of low temperature. Convective heat transfer can occur in two ways, either through free convection, where fluid flow is caused by density variations in a stagnant medium, or through forced convection, where fluid flow is controlled and directed. The terms \(\dot{Q}^\textrm{conv}_j\) in Eq. (24) and the interphase heat transfer term \(S^\textrm{heat}_j\) in Eq. (16) originate from this same principle, however, with a change in direction by definition. Adopting the definition of an interphase heat transfer coefficient, denoted by \(\alpha _j\) for particle j, the heat transfer rates are given by
where \(A_j\) is the surface area of particle j. Here, \(\alpha _j\) is an indirect measurement of the thickness of the thermal boundary layer between the particle and the surrounding gas and may be related to the dimensionless Nusselt number \(\textrm{Nu}_j\) of particle j according to
with \(L_j\) representing the characteristic length of the particle. Although Eq. (28) is very often applied in the context of pyrolysing DEM particles, it is, strictly, only valid if there is no mass exchange between the particle and the surrounding gas (\(S_{j,i}^{\textrm{mass}} = 0\)). Evaporation or devolatilisation, by contrast, induce a net gas-particle mass transfer that is accompanied by an outward-pointing Stefan flow, affecting both the species and temperature profiles across the gas-particle boundary layer. For spherical particles, the heat transfer rate in the presence of gas-particle mass transfer is obtained as (Turns 2012, Chapter 10)
where \(\bar{T}\) indicates an estimated mean film temperature and may, for example, be computed using the 1/3-rule as \(\bar{T} = T_j + (T_\textrm{g}(\textbf{x}_j) - T_j) / 3\) (Abramzon and Sirignano 1989).
Many empirical correlations exist and are referred in this context for the definition of the Nusselt number, as listed in Table 2. Despite not having a volume fraction correction, the Ranz-Marshall model (Ranz and Marshall 1952) is seemingly the preferred choice of most researchers (Papadikis et al. 2009c; Mahmoudi et al. 2014; Cordiner et al. 2018; Ku et al. 2019; Lu et al. 2020) in studying the pyrolysis of spherical particles. On the other hand, the Tavassoli model (Tavassoli et al. 2015), which is a modified version of the Gunn model (Gunn 1978), is primarily employed for systems of non-spherical particles (Gao et al. 2021b; Lu et al. 2021d). In this case, the characteristic length is replaced by the diameter of a volume-equivalent sphere. Papadikis and Gu (2009) adopted different approaches for evaluating the heat transfer coefficient depending on the position of the particle inside the fluidised bed. The Ranz-Marshall correlation was applied for a particle elutriated by the fluidising gas, while the heat transfer coefficient of a particle in the dense bed was found by applying the penetration theory proposed by Kuipers et al. (1992). Simulating pyrolysis of cylindrical biomass particles using a glued-sphere method, Lu et al. (2021c) found an insignificant effect of the choice of Nusselt number correlation on the particle temperatures and the product compositions. Gao et al. (2021b) also reported a similar behaviour, having studied slow pyrolysis of cubic particles, and further argued that the system had been operated in a chemical kinetics dominated regime.
Radiative heat transfer enables a transfer of heat between elements that are not in direct contact, as it is mediated through electromagnetic waves. Both particles and gas molecules constantly emit and absorb radiation of heat energy to and from the system. The extent of radiative emission is regulated by their absolute temperatures and emissivities, whereas the absorption is controlled by the absorptivities and irradiation. A radiative heat transfer occurs if there is a disparity between the extents of these transactions; the temperature of an element increases following a surplus in absorption, and decreases if emission dominates. Often in fluidised beds, the gas phase is characterised as an optically thin medium, meaning that it transmits radiation of all wavelengths without any absorption. This idealisation makes it immune to heat transfer by radiation. The Stefan–Boltzmann law states that the radiative heat flux is proportional to the fourth power of a body’s absolute temperature. According to this relation, radiative heat has an elevated effect in high-temperature applications, but is usually negligible if the system is operated at a moderate temperature. Stressing this fact, many researchers, also within the context of pyrolysis (Wang et al. 2021a; Hu et al. 2019; Lu et al. 2021d; Gao et al. 2021b), refrain from accounting for the effect of heat transfer by radiation, not only on the gas phase but also on part of the particles. In the following, we discuss how radiative heat transfer to a particle can be formulated and approximated with suitable models.
If \(G_j\) represents the incident radiation at the location of particle j, then the rate of radiative heat transfer to the particle reads
where \(\sigma _\textrm{s}\) and \(a_j\) are the Stefan–Boltzmann constant and the particle absorptivity, respectively. The exact magnitude of \(G_j\) is derived from the radiative intensity field obtained as solution of the radiative transfer equation (RTE) (Modest and Mazumder 2021, Section 10.4). Conditional on effects of emission, absorption, and scattering in the medium, the RTE describes the propagation of radiative intensity through space. From a computational standpoint, solving the complete RTE is far too tedious and also expensive, as it involves the direction and wavelength as additional independent coordinates. To convert the RTE to a more practicable form, one can introduce a simplification to the description of the radiative intensity field, as in the methods of spherical harmonics (\(P_N\)-approximation) (Jeans 1917) or discrete ordinates (\(S_N\)-approximation) (Chandrasekhar 1960). A preferred practice, even in the context of pyrolysis modelling (Ku et al. 2019; Chen et al. 2020; Cordiner et al. 2018), is to simplify the radiative intensity term in the RTE with first order spherical harmonics (Modest and Mazumder 2021, Section 16.5). This is the so-called \(P_1\) model, the simplest of its kind, that yields the incident radiation as solution of the following transport equation
with
where \(a_\textrm{g}, \, \sigma _\textrm{g}\), and n are the absorption and scattering coefficients, and the refraction index of the gas, respectively. The coefficient C accounts for the anisotropic scattering effects. The term \(S^G\) on the right hand side of Eq. (35) can be instrumented to include the influence of radiation sources in the system. Following a low emissivity assumption for the gas phase, Bruchmüller et al. (2012, 2013) took a different approach to determine the incident radiation \(G_j\) in Eq. (34). Their approximation reads
with \(\bar{T}\) being the arithmetic mean temperature of the particles found in near proximity of particle j. The eligible region when considering a particle for the calculation of \(\bar{T}\) was composed of the same and adjacent CFD cells as the cell that contained particle j at the time of measurement.
3.2 Momentum Transfer
The exchange of momentum, both between particles and between the particles and the gas phase, plays a significant role in the desired operation of pyrolysis reactors. Fluidised bed reactors frequently contain inert particulate material to improve the intensity and uniformity of momentum transfer from the carrier gas to the feedstock particles. This supports the suspension of the feedstock particles, allowing for an enhanced heat and mass transfer and promoting thermal decomposition while ensuring a uniform conversion across the pyrolysis reactor. Furthermore, a well-regulated momentum transfer is vital to maintain the balance between conversion and removal rates, in order to achieve a proper and efficient utilisation of the feedstock.
Both the linear and angular momentum of a particle are affected when it collides with other particles in the system. Physically, a collision lasts for a small but measurable time span, during which repulsive and frictional forces act on the colliding particles, inhibiting their compression in the normal direction and the relative motion in the tangential direction, respectively. Correspondingly, the contact force \(\textbf{F}^\textrm{c}_j\) is typically decomposed into a normal component \(\textbf{F}^\textrm{n}_j\) and a tangential component \(\textbf{F}^\textrm{t}_j\) and evaluated separately based on the physics underlying each. Note that the term \(\textbf{F}^\textrm{c}_j\) in particle j’s linear momentum balance equation (Eq. (18b)) represents the net collision force of all the pair-wise computed collisions in which the particle is involved at the current point in time. Similarly, \(\varvec{\uptau }_j\) is the sum of the collision-driven torques pertinent to particle j. For a contact between particles \(j_1\) and \(j_2\), the torque exerted by particle \(j_2\) on particle \(j_1\) reads
where \(\textbf{F}^\textrm{c}_{j_1 \leftarrow j_2}\) is the contact force exerted by particle \(j_2\) on particle \(j_1\) and \(\textbf{x}^\textrm{c}_{j_1j_2}\) represents the point of contact. In DEM, the particle-particle contact force is typically modelled using a soft-sphere approach, whereby the points of first contact of both particles are interconnected by two spring-dashpot pairs, one along the normal direction and one in the tangential direction. The tangential force is additionally limited in magnitude by the Coloumb friction condition that specifies a maximum value in terms of the instantaneous normal force and the friction coefficient \(\mu _\textrm{f}\). Accordingly, the normal and tangential forces \(\textbf{F}^\textrm{n}_{j_1 \leftarrow j_2}\) and \(\textbf{F}^\textrm{t}_{j_1 \leftarrow j_2}\) on particle \(j_1\) due to its collision with particle \(j_2\) are given by
and
respectively. Here, \(k_{j_1j_2}\), \(v_{j_1j_2}\), \(\delta _{j_1j_2}\) and \(\dot{\delta }_{j_1j_2}\) represent the spring constants, damping coefficients, overlaps, and collision speeds, respectively, and the superscripts ‘\(\textrm{n}\)’ and ‘\(\textrm{t}\)’ indicate the normal and tangential contact directions. The normal unit vector \(\hat{\textbf{n}}\) points from the centre of particle \(j_1\) to that of particle \(j_2\). The tangential unit vector \(\hat{\textbf{t}}\), on the other hand, points along the direction of the initial tangential velocity of contact (Cundall and Strack 1979).
As mentioned in Sect. 2.2 above, the flow of gas is commonly assumed to leave the angular momentum of a particle unaffected. However, the gas flow influences the linear momentum of a particle via two distinct forces, the buoyancy force and drag. The buoyancy force \(\textbf{F}^\textrm{b}_j\) results from the spatial change in the mechanical pressure across the particle’s extent and can be computed according to
Conversely, the drag force results from tangential traction or viscous shear exerted by the gas on the particle surface, and is driven by the relative motion between a particle and the carrier gas. The main constitutive parameters affecting the shear-based momentum transfer are the local gas density and its dynamic viscosity. On part of the gas phase, drag gives rise to the momentum source \(\textbf{S}_j^\textrm{mom}\) in Eq. (2) as the momentum transferred from a particle to the gas per unit of time. In practice, the drag force is commonly related to an interphase momentum transfer coefficient \(\beta \) (Anderson and Jackson 1967), that may either be calibrated experimentally in terms of the particle shape and Reynolds number or determined from spatially fully resolved calculations of the momentum boundary layer about the particle,
Table 3 shows a summary of the drag correlations that have been used in the context of pyrolysis in the past. All of these drag models are particular to spherical particles and are based on the assumption that the DEM point-particles are locally homogeneously distributed, that is, \(\nabla \varepsilon _g \approx \textbf{0}\) near particle j. In reality, the feedstock particles of biomass are non-spherical, in general. To address this non-sphericity, Lu et al. (2021d, 2021c) coupled the drag correction of Ganser (1993), featuring a measure of particle sphericity, with the drag model of Gidaspow et al. (1991). In recent years, researchers have also implemented hybrid drag models that acknowledge the contrasting fluidisation behaviours of different particles as stipulated by their properties. According to the Geldart particle classification (Geldart 1973), biomass is regarded as type A and inert sand as type B by many research groups for the applications in fluidised beds. In the works of Lu et al. (2020) and Houston et al. (2022), the drag on Geldart B sand particles was computed using the Syamlal-O’Brien model (Syamlal et al. 1989), while the filtered drag model of Gao et al. (2018) was used to describe the drag on biomass particles. The drag model of Gao et al. (2018) pertains to the class of heterogeneous drag models and incorporates the effects of unresolved velocity scales on the drag experienced by a particle. In the context of CFD-DEM of pyrolysis reactors, heterogeneous drag laws were also employed by Wang et al. (2021a) and Gao et al. (2021b), who adopted, respectively, the energy-minimisation multiscale (EMMS) model of Yang et al. (2003) and the Di Felice-Hölzer/Sommerfeld hybrid drag model (Di Felice 1994; Hölzer and Sommerfeld 2008).
3.3 Drying
Moisture in the pyrolysis feedstock and its subsequent evaporation inside the reactor has a variety of effects on the pyrolysis process. First, from a product quality point of view, any moisture level is highly undesired as it ultimately contaminates the pyrolysis products (Mahmoudi et al. 2014). The emergence of water vapour increases the gas flow through the reactor, decreasing the ambient temperature and thereby curtailing both heat transfer and reaction rates. Further, a particle’s fluidisation behaviour is also affected by its evaporation, following the change in mass. The moisture content of biomass feedstocks, widely used in pyrolysis and gasification processes, ranges between \(10\!-\!20 \%\) by mass (Basu and Kaushal 2009). On the other extreme, many plastics, according to their proximate analyses, show moisture levels of below 1% (Abnisa and Wan Daud 2014). By consequence, the effect of drying on the pyrolysis of plastics is not as strong as in the pyrolysis of biomass, even to the extent that it can be excluded from the overall physical description.
In a particle of biomass, water exists in one of two forms. Free water resides in and flows through the pores of a particle, while bound water is held in the cell walls by hydrogen bonds formed with the chemical constituents in wood. There is a thermodynamic phase equilibrium between the water vapour at the particle surface and the free liquid water inside. As a result, the surface vapour pressure of water coincides with its saturation vapour pressure at the current particle temperature (Machmudah et al. 2020). Evaporation occurs if the free-stream vapour pressure is lower than the saturation vapour pressure at the particle surface, and water vapour diffuses from the surface into the ambient gas. The migration of water vapour is accompanied by a diffusive flow of other species towards the particle that is balanced, at the semi-permeable gas-solid interface, by an outward-pointing Stefan flow (Akkus 2020). Here, the evaporation is driven both by diffusion and convection and, hence, mass transfer limited. A kinetic limitation, on the other hand, prevails if the vapour flow causes the vapour concentration at the surface to decrease faster than liquid water vaporises and phase equilibrium can be re-established. Physically, the main cause for a kinetic limitation of vaporisation may be an insufficient supply of heat to the particle, for example, due to slow conductive or radiative heating or insufficient exothermicity on the part of intra-particle chemical reactions. Conversely, if the ambient vapour pressure is larger than the saturation vapour pressure at the particle surface, water condenses onto the particle surface, releasing the heat of vaporisation. The released heat may either be transferred to the gas or drive endothermal chemical reactions within the particle. A larger gas temperature is accompanied by a larger heat supply to the particle, potentially mitigating any kinetic limitation and causing the particle temperature to rise. In turn, a larger particle temperature promotes evaporation because the saturation vapour pressure increases with temperature (Bellais 2007).
The influence of drying is introduced to the rest of the system through a drying rate, which quantifies the rate at which water is removed from a particle and added to the flow domain. Typically, it is computed by the heat sink model (Bruchmüller et al. 2013; Mahmoudi et al. 2016a; Ku et al. 2019), the chemical kinetic model (Gao et al. 2021b; Lu et al. 2021b), or the equilibrium model (Mahmoudi et al. 2016b). In the heat sink model, also referred to as the constant evaporation model, drying is assumed to be controlled by the heat transfer from the local surrounding to the particle, and any potential limitation by mass transfer is omitted. Thus, this approach is particularly suited for fast heating conditions where the diffusive evaporation below the boiling temperature is short-lived. The rate of evaporation of particle j, which takes effect at the boiling temperature denoted by \(T^\textrm{evap}\), is derived by the thermodynamic balance between the transferred heat and the enthalpy of the produced water vapour as (Bruchmüller et al. 2012)
where \(Y_{j,\text {H}_2\text {O}}\) represents the current mass fraction of liquid water, while \(H^\textrm{evap}\) and \(\delta t\) are the heat of vaporisation of water and the computational time step, respectively. One disadvantage of the formulation in Eq. (47) is that the evaporation rate is not differentiable in the particle properties, both at \(T^\textrm{evap}\) and when the \(\min \) function switches from returning the first argument to the second one. It also enforces a stagnant effect on the particle phase properties, which can be observed in the works of Ku et al. (2019) and Mahmoudi et al. (2014), in terms of momentary plateaus in both particle temperature and conversion profiles, in regions corresponding to drying in particles. Besides, unlike the models to follow, Eq. (47) does not account for drying below the boiling temperature of water (Bellais 2007).
In the chemical kinetic model, the conversion rate of liquid water to water vapour is assumed to obey a first order Arrhenius relation. In particular, the drying rate can be expressed as
where \(A^\textrm{evap}\) and \(E^\textrm{evap}\) are the pre-exponential factor and the activation energy of the phase change reaction \(\text {H}_2\text {O}(l) \longrightarrow \text {H}_2\text {O}\)(g), respectively. Herein, the Arrhenius coefficients are deliberately set to generate a steep increase in the evaporation rate as \(T_j\) approaches \(T^\textrm{evap}\) (Mehrabian et al. 2012). Compared to the heat sink model above, this model is both numerically well-behaved and easy to implement. In fact, it can be readily appended to the particle phase list of reactions in the chemical reaction model, along with the corresponding reaction heat, i.e., the heat of vaporisation of water (Bellais 2007).
The phase equilibrium between the liquid water (free and bound) and the water vapour residing in the pores of the particle underlies the formulation of the equilibrium model. The evaporation rate in this case is driven by the difference between the equilibrium and the present concentration of water vapour available at the particle surface (Wurzenberger et al. 2002).
Here, \(k^\textrm{evap}\) is a modelling constant, whereas \(P^*_{\text {H}_2\text {O}}\) and \(W_{\text {H}_2\text {O}}\) denote the temperature dependent saturation vapour pressure and the molecular weight of water, respectively. Mahmoudi et al. (2016b) adopted the equilibrium model in their numerical investigation of biomass pyrolysis and noticed that drying levels as high as 90% of the total moisture can be achieved at a temperature of 330 K. As opposed to the previous methods, this approach can capture the possible condensation of water vapour inside particles or parts of particles (if particles feature internal temperature distributions, see Sect. 3.6) with sufficiently low temperature and moisture content (Bellais 2007).
3.4 Chemical Reactions
Chemical reactions lie at the core of pyrolysis as they mediate the conversion of a used or little-valued feedstock into (re)usable and value-enhanced products. At incipient pyrolysis, the polymer chains within the feedstock are cut and reduced in length following the absorption of heat imparted by the ambience. This initial step is referred to as devolatilisation or primary decomposition. As a result, an array of organic vapours and a solid residue is produced, whose specific compositions depend on the chemical composition of the feedstock and the positions of the bond cleavages. The residual solid phase or char is largely non-reactive. The produced vapours may either be instantaneously liberated from a feedstock particle or spread through its pores and be gradually released over time. The fraction of the vapour which is condensable at room temperature is termed pyrolysis-oil or tar. Depending on the conditions to which the feedstock is exposed, particularly the heating rate and vapour residence time inside the reactor, a further disintegration of tar, or what is known as secondary decomposition, is also possible during pyrolysis. Lastly, the non-condensable fraction of vapour constitutes the pyrolysis-gas. Generally, pyrolysis is considered to be an endothermal process overall. Yet, many experimental studies have hinted at the existence of exothermal reactions that may become significant in certain circumstances. In particular, this exothermicity has been observed in the form of central temperature overshoots in large feedstock particles on occasions where the reactors were operated at high pressure or temperature or with a high feedstock loading (Park et al. 2010; Basile et al. 2014; Di Blasi et al. 2014). One of the reasons for this effect is the exothermal nature of the decomposition of some components in the feedstock, for example, lignin in biomass (Di Blasi et al. 1999). On the other hand, this exothermicity has also been ascribed to a set of heterogeneous reactions of tar inside the particle’s pores or at the particle surface that produce a secondary char (Anca-Couce and Scharler 2017; Di Blasi et al. 2017). A supporting evidence is the study of cellulose pyrolysis by Mok and Antal (1983) who observed a reduction of the energy required for pyrolysis with increasing production of char. Furthermore, the effect was found to be more pronounced in conditions that promote gas phase chemical reactions, such as high-pressure and long vapour residence times.
An evaluation of possible reaction mechanisms and their respective kinetic parameters is imperative for a quantitative description of pyrolysis. Additionally, since pyrolysis leads to many different product species (product groups), a plausible description of chemical reactions is required when optimising the process using simulations to maximise the yield of the most desired product. However, a precise determination of all possible reactions is not only difficult experimentally, but also to benefit from during modelling if the proposed mechanism is overly extensive. The inefficiency caused by the comprehensiveness is rather amplified in CFD-DEM, where particles are assessed individually and both temperature and composition may vary across particles. A general chemical reaction of pyrolysis is

where a single educt is converted into either one or several products through an irreversible reaction. In Eq. (50), \(\nu _k\) with \(k = 1,2,\ldots \) represent the stoichiometric coefficients. Also, the chemical reactions are assumed to proceed at rates that follow first-order kinetics. In particular, the rate \(\mathcal {R}_n\) (in [mol m\(^{-3}\) s\(^{-1}\)]) of reaction n is proportional to the current concentration of the educt species whose decomposition is described by the respective reaction. Accordingly, the rate of a particle phase reaction (primary decomposition) is given by
with
where \(Y_{j,s}\) is the mass fraction of species s in particle j, whose decomposition is described by reaction n, and \(k_{n,j}\) represents the rate constant of reaction n in particle j. Similarly, the rate of a gas phase reaction (secondary decomposition) is described by
with
where \(X_{\textrm{g},i}\) is the mole fraction of gas species i participating in reaction s as the educt.
With the aid of the reaction rates in Eqs. (51) and (53), the rate of change of mass of species s in particle j is expressed as
and the rate of formation of gas species i as
where \(\nu _{s,n}\) and \(\nu _{i,n}\) are the stoichiometric coefficients of species s and i of the considered reaction n, while \(N_\textrm{pd}\) denotes the number of primary reactions considered in the model. Furthermore, the species mass source/sink term \(S^\mathrm {mass-reac}_{\textrm{g},i}\) in Eq. (7) is given by
where \(N_\textrm{sd}\) gives the number of secondary decomposition reactions according to the employed chemical reaction model. In the subsequent parts of this section, we will focus on pyrolysis reaction models that have been utilised in CFD-DEM frameworks.
In the simplest approaches, the overall transformation of feedstock is described by a global chemical reaction. The feedstock is described in terms of a single species and the products are also lumped into groups on the basis of their final aggregate states as follows (Mahmoudi et al. 2014; Ku et al. 2019).

A slightly advanced scheme, such as the one proposed by Thurner and Mann (1981), includes either two or three competing reactions to describe the primary decomposition.


An underlying assumption in the mechanisms of Eqs. (58) and (59) is that the characteristic time scales of gas phase reactions are much larger than the vapour residence time in the reactor, such that the released gas appears to be unreactive. However, a competing scheme still improves upon the global scheme in Eq. (58) as it permits capturing the shifts in yields of different product groups when the operating conditions change. In some competing reaction schemes, the primary decomposition of the feedstock is assumed to be preceded by an activation step that does not involve any change in composition or temperature (Beetham and Capecelatro 2019; Cordiner et al. 2018). Also incorporating the secondary decomposition of tar, such a pyrolysis reaction mechanism with lumped species reads


Since the feedstock is represented irrespective of its precise chemical composition in the mechanisms in Eqs. (58) to (60b), they are only applicable to the specific feedstock for which the kinetic parameters have been calibrated. While the chemical make-up of plastics may be inferred from their types (Kayacan and Doğan 2008; Martín-Gullón et al. 2001), it is not that straightforward with biomass, where the composition varies significantly across its sources. Bradbury et al. (1979) first developed a kinetic model for the pyrolysis of cellulose, the most abundant component in woody biomass, including feedstock activation and its primary decomposition, given by two competitive reactions (char and gas are produced simultaneously). Since their model is based on a reduction of the earlier formulation of Broido (1976), it is now known as the Broido-Shafizadeh model. It has been incorporated into CFD-DEM models by Papadikis et al. (2009a) who investigated the pyrolysis of cellulose and by Hu et al. (2019) who, for simplicity, considered the biomass feedstock as cellulose. Later, Di Blasi (1994) extended the Broido-Shafizadeh model by the secondary decomposition of tar. Miller and Bellan (1997) were the first to propose a constituent-based chemical reaction scheme for biomass pyrolysis by superposing the decomposition kinetics of its three major components cellulose, hemicellulose, and lignin, thereby extending the model by Di Blasi (1994). The kinetic parameters of the Miller–Bellan reaction mechanism are summarised in Table 4, with reference to the mechanistic model shown in Eq. (60a). Herein, the reaction heats of hemicellulose and lignin are assumed to be identical to those of cellulose. Furthermore, the char production is exothermal so as to reflect the experimentally observed increase in exothermicity with char yield. The Miller–Bellan reaction scheme has been employed in many CFD-DEM investigations of biomass pyrolysis (Bruchmüller et al. 2012; Rabinovich et al. 2009; Beetham and Capecelatro 2019; Wang et al. 2021a) due to its capability to describe the thermochemical conversion of any biomass feedstock with a lignocellulosic characterisation. Bruchmüller et al. (2012) simulated the pyrolysis of biomass sourced from red oak (cellulose—46.9%, hemicellulose—31.8%, and lignin—21.3%), using a Miller–Bellan scheme without secondary tar tracking. However, the yields of product groups were reasonably well predicted in three separate test cases that differed in terms of the operating temperatures (699 and 758 K) and particle moisture contents (7.0% and 16.1%). On all three occasions, tar and char showed a higher and a lower yield, respectively, compared to the experimental results. The deviation of the gas yield from the experimental measurements changed sign when the temperature of the fluidising inert gas was decreased from 758 to 699 K, while all other conditions remained fixed.
In recent years, more detailed chemical reaction models have been integrated into the CFD-DEM framework. Particularly the mechanistic models developed by the Chemical Reaction Engineering and Chemical Kinetics (CRECK) modelling group in Milan have been applied. An example is the reaction mechanism developed by Debiagi et al. (2018) which describes the primary decomposition in terms of 32 first-order chemical reactions involving 29 solid and 29 gaseous species. Here, the biomass feedstock is taken to consist of cellulose, three types of hemicellulose, three types of lignin, and two extractives. Product groups of tar and gas are composed of 22 and six species, respectively. In deriving the kinetic parameters, the authors examined 80 different biomass samples using thermogravimetric analyses (TGA) and fluidised bed experiments, subjecting the feedstock to different heating rates. Several CFD-DEM investigations of pyrolysis have incorporated this scheme to describe the thermochemical decomposition of biomass (Gao et al. 2021a; Lu et al. 2021c, b). Lu et al. (2021b) compared the yields of pyrolysis products predicted by the detailed kinetic scheme of Debiagi et al. (2018) for Loblolly pine and wood waste as feedstock with experimental values. Based on the percent composition of each main group (tar, char, and gas), the simulation results were comparable with those of the experiments. Particularly, the product yields of waste wood were nearly identical to the measured values at four different operating temperatures (400, 450, 500, and 550 \(^\circ \)C). Gao et al. (2021a) used the same chemical reaction mechanism to simulate the pyrolysis of a single cylindrical particle (8 mm diameter, 19 mm length) as well as a pilot-scale entrained flow reactor with polydispersed cylindrical particles (length-to-diameter ratio 5, diameters in the range 140 to 1791 \(\upmu \)m), comparing the predictive fidelities of isothermal and non-isothermal particle descriptions. For the single particle, the non-isothermal model produced more accurate results, while in the case of the pilot-scale reactor the isothermal characterisation was better suited. Since, by design, the reaction model incorporated intra-particle transport, Gao et al. (2021a) surmised that coupling a particle-scale model in the second instance may have caused intra-particle effects to be accounted for twice. For us, however, this justification is hard to accept because it would then apply to the single particle test case as well. Recently, Houston et al. (2022) combined the primary decomposition mechanism of Debiagi et al. (2018) with the full secondary decomposition scheme developed by the CRECK group for gas phase kinetics (Ranzi et al. 2014; Debiagi et al. 2016; Pelucchi et al. 2019). At its furthest level of detail, the secondary decomposition model contains 509 species and 20,240 reactions. In order to reduce the associated expenses in the context of CFD-DEM, Houston et al. (2022) eliminated reactions that were either three-body or fall-off reactions or involved radicals or reactants that are not present in their application. The reduced mechanism included 66 secondary reactions and 59 gaseous species.
In addition to the chemical kinetic models discussed above, there are many other models available in the literature that describe the devolatilisation process of biomass, individual plastics (Al-Salem and Lettieri 2010; Faravelli et al. 2001) and binary (Jing et al. 2013) and tertiary (Costa et al. 2010) plastic mixtures. However, these models have not yet been incorporated into CFD-DEM descriptions of pyrolysis reactors. The distributed activation model (DAEM) is an interesting modelling approach that has been employed in the context of biomass pyrolysis (Cai et al. 2014), aiming to address the observation that chemical bonds may vary in strength among molecules of the same species. This is done by assuming that the activation energy pertaining to a reaction is no longer constant, but normally distributed.
Particularly for biomass, there exists another branch of chemical kinetic models, called network models, that incorporates the chemical structure of the polymeric species in the determination of kinetic parameters and reaction stoichiometry. There are three main instances of such models, namely the Chemical Percolation Devolatilisation (CPD) model (Fletcher et al. 2012), the Flash-Distillation Chain-Statistics (FLASHCHAIN®) model (Niksa 2000), and the Functional Group-Depolymerisation, Vaporisation and Cross linking (FG-DVC) model (Solomon et al. 1988). Incidentally, all aforementioned models were invented in the context of coal pyrolysis and have then been extended to describe the pyrolysis behaviour of lignocellulosic biomass with suitable modifications. In the CPD model, the polymeric feedstock is assumed to comprises clusters of carbon rings adjoined through a labile bridge which acts as the active site during the decomposition. Within the scope of the CPD model of biomass (bio-CPD), three kinetic schemes have been derived for each representative species, cellulose, hemicellulose, and lignin, separately according to the reaction scheme

by assuming their decomposition behaviours to be independent. These are then superposed to arrive at an overall model which accounts for the variable compositions across feedstocks. The kinetic parameters relevant to the model in Eq. (61) are determined based on five parameters that describe the structure of the feedstock in addition to experimental measurements of pyrolysis products. Similar to the assumption on the structure of coal particles in the original work (Grant et al. 1989), the chemical structure of lignin is also represented by a Bethe lattice and the corresponding lattice statistics have been used in the model development. For cellulose and hemicellulose, the anomeric carbon, which connects to two oxygen atoms, is considered as the active site in the absence of aromatic rings. Furthermore, hemicellulose is modelled as an average between its major components, xylan and glucomannan. All the reactions in Eq. (61) follow first-order kinetics and involve Arrhenius-type rate constants which are evaluated with normally distributed activation energies (Fletcher et al. 2012).
In the FLASHCHAIN model for biomass (bio-FC), biomass is represented by a chain copolymer of cellulose and lignin-like compounds, while hemicellulose is neglected. The solid reactant matrix is assumed to compris units of this copolymer with different chain lengths, ranging from monomers to infinite chains. The reactions proceed via a depolymerisation of the copolymers into smaller fragments, which are categorised based on size either as reactant, intermediate or metaplast. The reactant group is susceptible to further scission, producing either intermediates or metaplasts in the process, or may decompose to char by releasing light gases. The intermediates may similarly break down into metaplasts or condense into char links. The phase equilibrium between the tar species in the metaplast and the interacting gas phase drives the production of tar components, which are thought to be removed by a convective flow emanating from the solid matrix. Additionally, the metaplast groups may partake in recombination reactions and regress to intermediate fragments. All reactions in the proposed model are characterised by distributed activation energies and the reaction parameters are determined by fitting experimental measurements of cellulose and lignin pyrolysis. Furthermore, the stoichiometric coefficients are adjusted to the composition of lignin-based components in biomass (Niksa 2000).
The FG-DVC model comprises two sub-models; the FG model describes the release of gas as various functional groups in the polymeric feedstock decompose, while the DVC model governs the formation of tar and char as a consequence of depolymerisation, internal and external mass transfer, and cross-linking induced by heat treatment. The approximation of the chemical structure of the polymer involves 14 parameters (Solomon et al. 1988) and their calibration requires measurements from experimental techniques such as nuclear magnetic resonance (NMR) spectroscopy and field ionisation mass spectroscopy (FIMS). In its extension to describe the devolatilisation of biomass, an average macromolecule is characterised based on a van Krevlen diagram (plot of H/C versus O/C atomic ratios) to match the atomic make-up of the feedstock (Chen et al. 1998). This differs from the approach adopted in bio-CPD where the overall devolatilisation kinetics are a result of superposing the kinetics of individual representative components. However, similar to previous network models, the reaction rates are formulated based on normally distributed activation energies.
3.5 Shrinkage
As feedstock particles dry or decompose chemically and emit volatiles, their volumes may decrease by up to 70% of the original volumes (Davidsson and Pettersson 2002). Many experimental analyses of pyrolysis have affirmed that this volume reduction is influenced by several factors such as the type, size, and structure of the feedstock as well as the heating rate which may produce uneven rates of contraction along different directions of a particle (Davidsson and Pettersson 2002; Kumar et al. 2006). Correspondingly, an accurate description of how the geometry of a pyrolysing particle changes in time is extremely difficult. Further complications arise from other intriguing observations, for example, delayed shrinking or initial swelling of the feedstock particles (Zhou et al. 2013; Paulauskas et al. 2015; Caposciutti et al. 2019). Irrespective of the precise nature of volume changes, any change in the size or shape of a particle affects its fluidisation behaviour as well as heating and reaction rates (Wang et al. 2021b). Intra-particle processes will also be influenced by changes in the particle size or shape, through density and porosity (Papadikis and Gu 2009). In the context of DEM, these dependencies may be accounted for through a sub-model that describes the shrinkage behaviour of a pyrolysing particle.
The existing CFD-DEM models for pyrolysis that we are aware of disregard the influence of particle drying on their shrinkage behaviour, and, further, the possibilities of swelling or delayed shrinking. Most model formulations employ either a constant size model (Papadikis et al. 2009b; Rabinovich et al. 2010) or a constant density model for this purpose. The constant size model appears to be the simplest and least realistic, as it features no apparent change in particle size. Considering a spherical particle with fixed diameter \(d_j\), the particle density \(\rho _j\) adjusts to the current particle mass according to
Conversely, the density is held constant in the constant density model and the diameter of a spherical particle undergoing pyrolysis is computed from the current mass \(m_j\) (Cordiner et al. 2018; Chen et al. 2020),
Ku et al. (2015b) pointed out that neither Eq. (62) nor Eq. (63) are physical as both the diameter and density of a particle simultaneously change during its decomposition. Bruchmüller et al. (2012, 2013) attempted to address this through a mass-proportional shrinkage model, where the particle diameter was calculated by Eq. (63), using, however, a particle density that varies linearly with the mass fraction \(Y_{j,\textrm{B}}\) of virgin biomass in the particle j,
Here, \(\rho _B\) and \(\rho _C\) are the bulk densities of the biomass feedstock and char, respectively.
Di Blasi (1996) proposed a more flexible approach for modelling shrinkage during the pyrolysis of biomass and argued that the volume \(V_j\) of any particle j consists of a solid volume \(V_j^\textrm{solid}\) and a pore volume \(V_j^\textrm{pore}\),
The solid phase is made up of virgin biomass as well as char that has been produced during the decomposition of the virgin biomass. The volume of the solid phase is assumed to vary linearly with the conversion of the virgin biomass according to
where \(f_1\) is a kinetic parameter, \(m_\textrm{B}\) and \(m_\textrm{C}\) are the current masses of virgin biomass and char, respectively, and \(m_{j,0}\) and \(V^\textrm{solid}_{j,0}\) denote the initial mass and the solid volume of the feedstock particle. The pore volume of the particle, on the other hand, is controlled by two effects. First, it decreases as the feedstock is consumed, corresponding to the overall shrinkage of the particle. In particular, the pore volume is assumed to change linearly with the conversion of the particle, that is, the reduction of \(m_\textrm{B}\), from the initial pore volume \(V_{j,0}^\textrm{pore}\), to the final volume \(f_3 V_{j,0}^\textrm{pore}\) at complete conversion (\(m_\textrm{B}(t) = 0\)). Second, the pore volume increases as the solid phase disappears and the volume it occupied is filled with gases. Combining both effects, the pore volume of the particle is computed as
where \(f_2\) represents the fraction of volume recovered by particle internal pore space following the consumption of the solid phase. Like \(f_1\) and \(f_3\), \(f_2\) lies within the range from zero (total disappearance of particle as \(m_\textrm{B} \rightarrow 0\)) to unity (no shrinkage). In order to complete the specification of Di Blasi’s model, the parameters \(f_1\), \(f_2\), and \(f_3\) as well as the initial pore volume \(V_{j,0}^\textrm{pore}\) must be calibrated based on the observed shrinkage behaviour of the biomass under consideration. Note that \(V_{j,0}^\textrm{pore}\) may be deduced from the particle’s initial porosity. Table 5 shows that all shrinkage schemes discussed above can be formulated as special cases of the Di Blasi shrinking model. Additionally, the Di Blasi model parameters determined by several researchers for the feedstocks they considered are listed in Table 5.
Several CFD-DEM studies have been conducted, specifically in fluidised beds, with the objective of assessing the effect of the applied shrinkage model on pyrolysis behaviour. The single particle simulations of Papadikis and Gu (2009) indicated that a particle attains a higher temperature, both at its surface and the center, if less severe shrinking conditions are imposed via a Di Blasi scheme. This rather counter-intuitive observation may be explained by the larger surface areas of more slowly shrinking particles. Consequently, for a given time course of particle mass and composition changes, a slower size reduction results in larger heat transfer rates and a larger particle temperature. Based on simulations of the pyrolysis of thermally thin biomass particles in a fluidised bed, Ku et al. (2015b) observed that constant size shrinking leads to faster devolatilisation rates and greater residence times of particles compared to constant density shrinking. However, physically, their observation of the influence of the shrinkage model on the particle residence time is difficult to explain. Since the mass and heat transfer rates as well as the momentum transfer rate due to Stokes drag scale linearly with the particle size, we would expect a particle with a given mass to devolatilise quicker and be more susceptible to entrainment and removal if its size remained constant than if the density were unchanged. In a more recent work, Hu et al. (2019) investigated the pyrolysis of biomass with four different shrinking patterns and concluded that the residence time, in fact, increases with the severity of imposed shrinkage. Studies of both Papadikis and Gu (2009) and Hu et al. (2019) demonstrated only a minor effect by the employed shrinkage pattern on the product yields of biomass pyrolysis. By manually calibrating the Di Blasi parameters, Li et al. (2017) were able to achieve an acceptable agreement with the experimental particle size evolution, following the configuration \(f_1=0.3, \, f_2=0, \, f_3=0.3\). Upon further investigation, the product yields were also found to be reasonably well predicted. However, we emphasise that this parameter set is not universal and pertinent only to a specific feedstock as well as, possibly, to a specific set of operating conditions.
3.6 Intra-particle Behaviour
When describing the evolution of a particle’s degrees of freedom, specifically its composition and temperature in Eqs. (20) and (21), we considered the particle to be a homogeneous entity. This characterisation is corroborated by the assumption that a particle features a negligible resistance to intra-particle heat conduction and mass diffusion. Here, any amount of heat transferred to the particle from the outside is transported across its volume within an infinitesimal time span. As a result, the particle does not exhibit any internal temperature gradients and its decomposition will follow an identical route at a similar rate everywhere inside the particle, as shown in Fig. 3a). However, if the external rate of heat transfer exceeds the conductive heat transfer rate within the particle, the near-isothermality ceases and thermal gradients emerge in the outward direction of the particle. The spatially inhomogeneous temperature distribution may also induce intra-particle inhomogeneity on part of the chemical composition through drying and chemical reactions. On the other hand, compositional intra-particle heterogeneity may also result if the intra-particle species diffusion is slower than the species removal or absorption at the surface, a case that has not been considered within the scope of pyrolysis to our awareness. For heat transfer, the amenability to intra-particle temperature gradients is commonly assessed by analysis of the particle’s Biot number
a dimensionless quantity that corresponds to the ratio of the particle’s internal thermal resistance to that at its surface. According to Bryden and Hagge (2003), a particle is characterised as thermally thick or non-isothermal if \(0.2< \textrm{Bi}_j < 10\). In this regime, the isothermal assumption leads to an overestimation of the particle’s overall temperature and conversion, as revealed by many numerical investigations on pyrolysis (Bellais 2007; Luo et al. 2020; Ku et al. 2019) also with detailed chemical kinetic schemes (Gao et al. 2021b; Lu et al. 2021d; Gao et al. 2021a). In order to account for potential intra-particle inhomogeneities, the description of the point-particles can be amended by an intra-particle model. Based on the way of spatial discretisation, these models may be categorised as either 1D models (layer discretisation) or 3D models (control volume discretisation).
The 1D models are limited to regular particle shapes and rely on the assumption that the particle’s heat conductivity is isotropic. Two variants can be encountered in the context of pyrolysis modelling: the continuous model and the interfacial model. The continuous models are based on the one-dimensional heat conduction equation governing the temperature profile \(T_j = T_j\left( r,t \right) \)
in a long plate (\(n = 0\)), a long cylinder (\(n = 1\)), or a ball (\(n = 2\)), respectively. Here, \(S^\mathrm {heat-reac}_{j,r}\) describes the heat source/sink term due to chemical reactions and r corresponds to the thickness coordinate of the plate, the radial coordinate of the cylinder, or the radius of a sphere. Equation (67) is supplemented by boundary conditions that account for the symmetry at the particle centre and the external heat transfer rates at its surface,
An example for the intra-particle distributions of temperature and feedstock mass fraction for a biomass particle is shown in Fig. 3b). Mahmoudi et al. (2014) employed the 1D continuous model to simulate the pyrolysis of biomass particles in a packed bed. The shapes of the particles were simplified as spheres having identical volumes, and, yet, a good agreement with the experiments was achieved, based on the system’s mean temperature and mass loss, for a range of inlet temperatures (\({573}-{803} \text {K}\)). The single particle simulations conducted by Gao et al. (2021b) with spherical and cylindrical particles, produced nearly identical results to that of the experiments, including the centre temperature overshoots. They further investigated the pyrolysis of cubic particles with \(\textrm{Bi} = 1.4\) in a packed bed using both isothermal and 1D continuous intra-particle models. Intriguingly, the results suggested that the isothermal model required a considerably longer time for the complete conversion of the entire batch of particles, causing the total runtime to exceed the one required by the 1D models.
In the 1D interfacial model, by contrast, the volume of a particle is split into three main layers. Within the scope of biomass pyrolysis, these layers are referred to as the wet wood layer, the dry wood layer, and the char layer, in the order of appearance from the centre to the surface of a particle. The boundary between the wet and dry wood layers is called the drying front, while the devolatilisation front separates the dry wood and char layers. Each layer can be further discretised into sub-layers or grid points that may be used to resolve intra-layer temperature gradients. Formally, the main differences between the interfacial model and the continuous model lie with the distinct boundary conditions that apply to each of the main layers. For additional information on the assumptions and the governing equations of the 1D interfacial model, we refer to the pioneering work of Ström and Thunman (2013). Figure 3c) illustrates the partitioning of a particle into layers and indicates the intra-particle profiles of the temperature and feedstock mass fraction for an interfacial model.
Wang et al. (2021b) adopted the interfacial method to simulate the pyrolysis of 0.75 mm poplar sawdust particles inside a three-chambered fluidised bed. Although the experimental feedstock contained \(\sim {7}{\%}\) moisture, the particles in the model were considered to be completely dry and the interfacial model could, therefore, be reduced to having only two layers: an unreacted core and a reacted shell. Using a global kinetic model, Ku et al. (2019) analysed the effect of isothermal and 1D interfacial models on the pyrolysis behaviour of a single biomass particle (\(\textrm{Bi}=0.67\)) suspended in a fluidised bed of inert particles. The results indicated that the devolatisation commences earlier and lasts longer in the interfacial model compared to the isothermal approximation. The authors also observed that the trajectories of the particles are strongly affected by the choice of the intra-particle model, and that the interfacial model results in a longer residence time.

Sample intra-particle distributions of temperature (T) and feedstock mass fraction (Y) predicted by a an isothermal model, b a 1D continuous model, and c a 1D interfacial model for a spherical biomass particle undergoing pyrolysis. Here, \(T_0\) and \(Y_0\) are the initial temperature and feedstock mass fraction of the particle, respectively
Three-dimensional models, on the other hand, permit a more comprehensive description of intra-particle variability, including anisotropy, and allow for irregular particle shapes. In mesh-based approaches, the heat conduction or species transport equations on the volume occupied by a particle are spatially discretised using a direct discretisation technique. An alternative is the glued-sphere approach that can be initialised by replacing the cells in the mesh-based model with equal-volume-spheres. The evolution of the temperature of a composite sphere based on its position of appearance in a particle j is given by
where \(\dot{Q}^\mathrm {cond-intra}_j\) is the intra-particle conduction term evaluated by summing the heat transferred to the current composite sphere from all other spheres inside particle j with which it is in contact.
Lu et al. (2021d) implemented the glued-sphere representation first on spherical and cylindrical particles to validate the model and, later, to investigate the pyrolysis behaviour of irregularly shaped biomass particles subjected to different operating conditions. Since the extent of heat transfer depends on the surface area perpendicular to the heat flow direction, they explicitly recorded the surface areas of the mesh cells composing the non-spherical particles during the initialisation of the glued spheres. Instead of the areas rendered by the generated composite spheres, the recorded areas were considered in heat transfer calculations. As expected, the difference between the surface and the centre temperatures increased with the size of a particle. Comparing with the results from an isothermal approach, it was found that the intra-particle variations are negligible below \(\textrm{Bi} = 0.41\), where the isothermal assumption would become valid.
As pointed out by Papadikis et al. (2009b, 2009c), the intra-particle models have a high computational demand and produce large output files in comparison to an isothermal model. A workaround to alleviate this loss of efficiency in the thermally-thick regime is by incorporating a corrected-isothermal model. Through a process of calibration, a corrected-isothermal model lumps the effect of intra-particle transport onto the external heat transfer and reaction rates. Then these modified rate expressions are used to compute a homogeneous temperature and composition for a physically thermally-thick particle. Luo et al. (2020) devised a corrected-uniform model with the aid of volume-averaged temperature and reaction rates predicted for a particle by a 1D continuous model. The derived model predicted the temperature and conversion of a 9.5 mm particle more accurately than a usual isothermal model, and its results were comparable with the 1D continuous model. Lu et al. (2020) calibrated the Nusselt number and reaction rates of a reactor-scale model by minimising the deviations of predictions from the results of a benchmark particle-resolved simulation.
4 The Effect of Operating Conditions on the Pyrolysis Efficacy and Outcome
In the past, the CFD-DEM framework has been instrumented by many investigators to assess the influence of operating parameters, such as the gas temperature, fluidisation velocity (Houston et al. 2022), reactor type (Lu et al. 2021d), or shape (Wang et al. 2021a), and the feedstock properties, for example, the particle size (Ku et al. 2019), shape (Lu et al. 2021d), and moisture content (Bruchmüller et al. 2012), on the overall pyrolysis behaviour or to optimise reactors with respect to product yields. In this section, we summarise key observations that may guide the design or operation of pyrolysis reactors.
Operating Temperature: Rabinovich et al. (2009) analysed the effects of many operating parameters, including temperature, in the process of optimising the yield of bio-oil attained from pyrolysing biomass in an inert bed. After fixing the velocity of inert gas through the reactor at 1.1 times the hovering velocity of char particles produced during the process, they achieved a maximum yield of bio-oil of 72% for a temperature, particle size, and size of bed particles of 776 K, 440 \(\upmu \)m, and 940 \(\upmu \)m, respectively. They further conducted a sensitivity analysis of these optimum conditions to realise that the yield of bio-oil is more sensitive to an increase in the temperature than to a decrease, as such conditions accelerate the breakdown of heavier components in bio-oil. Having comprehensively validated their CFD-DEM model against results from two rather contrasting experiments, Ku et al. (2019) studied the impact of inert gas temperature on biomass pyrolysis in fluidised beds. Individual simulations were conducted with temperatures of 908, 1098, and 1198 K, while all other parameters influencing the system were fixed. They reported a rapid particle entrainment following faster heating and decomposition of particles, as the temperature was increased.
Similarly, Houston et al. (2022) also observed a reduction of the particle residence time when the inert gas temperature increased from 500 to 600 \(^\circ \)C in a fluidised bed operated at three times the minimum fluidisation velocity. They emphasised that this behaviour was prompted by the increase in the devolatilisation rates of the particles whose peak rate doubled. An analysis of the product composition variation was also reported in their work. Accordingly, a monotonic decrease in char from 25.1 to 21.8% by mass in the final product was recorded as the temperature increased from 500 to 600 \(^\circ \)C. This indicates that the temperature increase has intervened in the progression of decomposition reactions through the route of char production. The bio-oil, too, decreased by about 3%, while the gas yield increased by 13% following the temperature change. The observations by Houston et al. (2022) show that an increase in the operating temperature may cause the overall oil production to decrease, as the increase in the oil yield from the primary decomposition is outweighed by its secondary decomposition.
Similarly, the simulations of Chen et al. (2020), too, revealed an increase in gas and a decrease in char yields, each by 8% by mass, when the operating temperature in the fluidised bed was raised from 500 to 700 \(^\circ \)C. Interestingly, the mass fraction of oil in the final product remained unchanged, implying that the contrasting effects of temperature on the severity of primary and secondary decompositions may have been balanced. The detailed chemical reaction mechanism employed by the authors further permitted an investigation into compositional changes of the bio-oil with operating temperature. Specifically, a temperature increase caused the low-molecular-weight species to be more abundant in the oil phase and the portion of high-molecular-weight species to be smaller. Although elevating the operating temperature accelerates the pyrolysis process through faster decomposition and quicker particle entrainment, Lu et al. (2021d) noticed that the heating of particles across the reactor would become non-uniform if only the temperature is increased. As a result, the routes of decomposition may differ from particle to particle, and, further, lead to undesired retention trends of particles in the reactor. This could be in the form of either prolonged retention of fully converted particles or premature exits of under-utilised feedstock.
Particle size: In order to ascertain the impact of the size of feedstock particles on the bio-oil yield, Rabinovich et al. (2009) carried out another sensitivity analysis of the established optimum operating conditions, varying the particle size from 400 to 1000 \(\upmu \)m. While the change in the bio-oil yield was only minor, corresponding to about 1-2%, the pyrolysis time was found to increase notably. Employing a 1D interfacial model for intra-particle transport, Ku et al. (2019) compared the pyrolysis behaviour of biomass particles with four different sizes, 0.5, 1, 2, and 3mm. Prior to the study, their CFD-DEM model was fully validated against data from two experiments of rather large-sized particles of beech (20 mm) and poplar (9.5 mm) wood. Naturally, with the increase in size, the difference between the core and surface temperatures of the particle followed a rising trend. Furthermore, the time until a particle’s core attains its surface temperature increased exponentially, with each duration reading approximately 0.4, 1.1, 3.2, and 9.0 s, respectively. Aware of how local temperature impacts the decomposition paths in pyrolysis, they highlighted the fact that an increase in particle sizes may promote intra-particle heterogeneity and render the pyrolysis rates more difficult to control.
Following an extensive validation, both against single particle and reactor-scale packed bed experiments, Mahmoudi et al. (2016b) conducted CFD-DEM simulations to investigate the influence of the size of beech wood particles on their pyrolysis behaviour. Three identical packed bed reactor setups that differed only in terms of particle sizes were compared. Setups 1 and 2 contained mono-dispersed particles of sizes 10 and 2.5 mm, respectively, while setup 3 encompassed a combination of particles with these sizes in layers. Due to the comparatively large surface area to volume ratio of small particles, the layer of particles that first contacts the flow of hot gas in setup 2 displayed a faster heating, contrary to that in setup 1. The small particles also have a higher surface area in total and larger heat transfer coefficients, compared to the large particles. Hence, the gas flow in setup 2 lost most of its thermal energy rather quickly, leading to a much narrower hot zone within the particle bed. Mahmoudi et al. (2016b) further examined the pyrolysis behaviour of a 2.5 mm particle in comparison with a 10 mm particle, drawn from a location downstream of the former from setup 3. Despite being downstream of the bigger particle, both drying and devolatilisation in the smaller particle started relatively earlier. Furthermore, the compositions of products from setups 1 and 2 affirmed that bigger particles produce more char compared to smaller particles.
Chen et al. (2020) also observed a slight increase in the char yield as the size of biomass particles was increased in the range of 450-850 \(\upmu \)m, and attributed it to the low heating rates of larger particles. Although the heating and mass loss rates were notably higher with small particles, the ultimate conversion of feedstock remained nearly constant, around 84%, unaffected by the change in particle sizes. In a coarse-grained CFD-DEM study of biomass pyrolysis, Lu et al. (2020) analysed the residence time and conversion distributions of three mono-dispersed particulate systems, of sizes 0.278, 0.344, and 0.426 mm. Unsurprisingly, the average residence time increased monotonically, from 4.07 to 5.62 s, with the size of the particles. In the condition they tested, the time-averaged conversion of particles also exhibited a growing trend with the particle size. The distributions of conversions of particles leaving the reactor uncovered an important concern pertaining to the choice of particle size in pyrolysis. In particular, about 15% of the smallest particles in the reactor leave the fluidised bed with a conversion below 80%. On the contrary, the fractions corresponding to this threshold in the other two systems are relatively insignificant. This behaviour evidences an improper utilisation of feedstock as its size is reduced. However, their study lacks a discussion on the variation of product compositions consequent to changes in feedstock size. In determining an optimum size for pyrolysis feedstock, such information must also be factored in.
Particle shape: Using CFD-DEM, Lu et al. (2021d) simulated the pyrolysis of biomass particles of several shapes, including spheres, cylinders, and one irregular shape. In order to resolve the shapes and accommodate intra-particle distributions of temperature and composition, a glued-sphere approach was implemented. The irregular shape contained a smooth tip at its top, resembling a locality of high surface area to volume ratio compared to the rest of the particle. In comparison to a cylindrical particle of identical volume and surface area, the irregularly shaped particle involved a wider intra-particle temperature profile along with a larger peak temperature owing to the appearance of a hot zone at its tip. With view towards a simplified geometrical representation of irregularly shaped particles, they advocated cylinders that possess the same volumes and surface areas as the irregular target particles instead of volume-equivalent spheres which are most commonly used in practice. Apart from the work of Lu et al. (2021d), Gao et al. (2021b) and Lu et al. (2021c) investigated the pyrolysis of non-spherical particles adopting either cubic shapes, formulated as superquadrics, or an overlapping glued-sphere approach, respectively. Unfortunately, the authors did not report any analysis on the influence of the particle shape on the pyrolysis behaviour.
Inert flow rate: Based on the three packed bed setups briefly described above, Mahmoudi et al. (2016b) also inspected the impact of the gas flow rate on the pyrolysis of beech wood particles. Upon increasing the flow rate from 2.5\(\times 10^{-3}\) to 5.0\(\times 10^{-3}\) m\(^{3}\), a 9% reduction of char yield was observed in the first and third setups, whereas in setup 2, this reduction was more compelling at 24%. From these observations, they concluded that the char yield becomes more sensitive to the changes in the flow rate if the feedstock comprises smaller particles. In a later investigation, Hu et al. (2019) monitored the changes in the composition of products formed during the pyrolysis of biomass as the gas velocity was adjusted to 2.2, 2.6, and 3.0 times the minimum fluidisation velocity. Here, the oil yield increased and the gas yield decreased, while the fraction of char in the pyrolysis product remained unaffected by the flow rate. By reviewing the progression of the multistep chemical reaction model that the authors had adopted, they concluded that the secondary decomposition of oils decreased drastically because of fast removal of produced vapour from the reactor at high flow rates.
For a constant inlet temperature of 550 \(^\circ \)C, Houston et al. (2022) varied the inlet velocity of their fluidised bed simulations between two and six times the minimum fluidisation velocity. As the flow rate increased, the gas yield strictly decreased by 2.8%, while the char yield was found to increase by 4.1%. Oil, on the other hand, showed a weak dependency on the flow rate, featuring changes of only about 1%. Moreover, the distributions of the residence times also became more uniform as the inert flow velocity increased. In a similar vain, Lu et al. (2021d) experienced a reduction of the standard deviation of particle temperature in their pyrolysis system when the inlet flow rate of the fluidised bed reactor was increased. This effect was brought about by the enhanced heat transfer to feedstock particles at high ambient flow speeds.
Reactor type and configuration: Lu et al. (2021d) analysed the heating and mass loss rates of irregularly shaped particles that were subjected to pyrolysis in three different reactor systems, a fluidised bed, a spouted bed, and a packed bed. The three reactors were operated at the same average gas velocity of 5 ms\(^{-1}\) and the same temperature of 783 K. The fluidised bed delivered a rather homogeneous heating rate across all particles in the system as reflected by the low standard deviations in their temperature and density profiles. The fixed bed, on the other hand, yielded a higher mean temperature and a lower mean density for a considerable time span compared to the other two reactors. Concomitantly, the standard deviations of temperature and density were larger, suggesting a higher degree of non-uniformity in both the particle heating and reaction rates. The spouted bed offered the lowest particle heating rate, but had an appreciable uniformity, almost comparable to that of the fluidised bed.
Wang et al. (2021a) employed a CFD-DEM framework to investigate how the fluidisation behaviour of pyrolysing biomass particles in fluidised beds changes with the shape of the reactor cross-section. To this end, they designed three pyrolysis reactors with similar inlet areas, but circular, square, and rectangular cross-sections. All the reactors were injected with nitrogen gas at 773 K and 0.36 ms\(^{-1}\). The reactor with the circular cross-section produced the largest mean bed height, while the lowest one occurred with the rectangular cross-section. They also evaluated a bubble equivalent diameter at various heights for each reactor, accounting for the volumes of neighbouring CFD cells with a void fraction of at least 0.8. Nearly up to the top outlet, the circular bed possessed the largest equivalent diameter, while the rectangular reactor had the smallest at all measured heights. Both the average feedstock temperature and the resulting mass loss were slightly higher in the reactor with the circular cross-section compared to the other reactors. The yield of char appeared to be largely unaffected by the choice of the cross-sectional shape. Furthermore, to compare the erosion in differently shaped beds, they evaluated the cumulative erosion volume per unit area in each, adhering to the model proposed by Finnie (1960). After 10 s of operating time, the circular bed had endured the most wear and the square bed the least. Wang et al. (2021a) further analysed the fluidisation behaviour for different gas inlet areas of 25, 50, and 100% in separate circular-bottomed fluidised beds. The bed heights did not show a great dependency on the inlet area, while the frequency at which the heights fluctuated increased as the area of the inlet was reduced. The bed with the largest inlet area produced the lowest mass loss rate as well as the smallest bubble equivalent diameter. The severest degree of erosion was experienced in the reactor with 50% inlet area.
5 Open Challenges
During the last few years, CFD-DEM has become increasingly popular to predict the behaviour of pyrolysis processes. Different reactor setups have been studied using CFD-DEM, and attempts have been made to improve the level of detail with which pyrolysis systems are described in the scope of CFD-DEM. In terms of particle-side physics, researchers have begun to employ more pragmatic Di Blasi shrinkage schemes instead of the unfounded constant density or constant size shrinkage models. The incorporation of intra-particle models and internal thermal resistance has been demonstrated to impact the pyrolysis outcomes, particularly at high Biot numbers. Recently, a few authors have also examined the pyrolysis of non-spherical particles within the scope of CFD-DEM. Apart from a solitary attempt by Mahmoudi et al. (2016a) to spatially semi-resolve the gas phase in the pyrolysis reactor, the gas phase description remains largely unchanged over all works we reviewed. The interphase interactions have been approximated with many correlations available for the evaluation of the Nusselt number and the drag coefficient. A recent interest has been the use of distinct drag coefficients based on the characteristics of different particles that appear in the system (feedstock or inert bed particles), to closely represent differences in their fluidisation behaviours. The studies which feature analyses of the Nusselt number correlations on the outcome of pyrolysis have shown only a minor influence and have always highlighted the need for more accurate chemical kinetic models for a better prediction. Duly, the chemical kinetic models employed in CFD-DEM models of pyrolysis have become increasingly comprehensive by now. While early models were based on a single global reaction describing the overall transformation, the chemical kinetics now involve up to 32 and 60 reactions representing the primary and secondary decomposition of biomass, respectively.
Although a general framework exists and many chemical kinetic models have been developed, a complete CFD-DEM model describing the pyrolysis of plastics is yet to be formulated and validated. A particular challenge which this task entails is the modelling of the melting of plastic particles and its ensuing effects during the thermal treatment. For a molten feedstock, there are several efforts to model the pyrolysis of plastics (Flor-Barriga and Rodríguez-Zúñiga 2022; Yin et al. 2014) in which the volume-of-fluid method is coupled with CFD to spatially resolve the gas-liquid interface. Zhang et al. (2022a) implemented a CFD-DEM model to ascertain the influence of heat carrier loading and the rolling speed of a rotary furnace pyrolyser on the dynamics and heat transfer rates of polyethylene particles. However, this work hardly counts as an example of plastic pyrolysis modelling with CFD-DEM for two reasons. First, a chemical kinetic model was not included and, second, the melting of particles was omitted, although the system was operated at temperatures as high as 773 K.
In our opinion, one modelling aspect which needs to be improved is the representation of the feedstock particles. This includes not only their shape and size, but also their initial composition which may vary across the feedstock. The shapes of the particles of biomass feedstock often used in conversion processes, such as pyrolysis, differ drastically from spheres and are often closer to cylinders (Chaloupková et al. 2019). As highlighted by Lu et al. (2021d), a shape representation in terms of cylinders that possess the same volumes and surface areas as real particles could lead to improved CFD-DEM predictions. However, for particles of general shape, the geometrical representation becomes more difficult (Lu et al. 2015). Furthermore, the requirement of special collision models and models that approximate the interphase exchange, where the orientation of the particles is also of significant importance, are major challenges. These have been studied in recent contributions on the fundamentals of CFD-DEM for non-spherical particles (e.g., Wachs 2019; Zastawny et al. 2012; Cao and Tafti 2020) that mostly focus on ‘regularly’ shaped axisymmetric geometries, such as cylinders and ellipsoids. Although the CFD-DEM framework allows for the variation of individual particle properties, the interactions among particles as well as between the particles and the gas are complicated to model, and general, shape-independent interaction models do not exist at present. Moreover, the feedstock used for pyrolysis may contain particles of varying sizes and compositions, which are frequently represented by a mean value to simplify the simulations. Initialising the particles’ properties based on experimentally measured size and composition distributions over the feedstock is a challenging but important step to allow for an accurate replication of the process.
The geometrical changes that a particle undergoes during its devolatilisation and the driving forces are yet to be fully understood, let alone to be incorporated into the CFD-DEM framework. In particular, there are experimental observations on the macroscopic behaviour of particle shrinkage, which are not yet reflected in CFD-DEM frameworks applied to pyrolysis reactors. For example, presently, a particle is thought to only shrink and the shrinkage commences at the same time as the mass consumption. Additionally, the shrinkage is also considered to be isotropic, regardless of the directional variations of heat transfer and reaction rates a feedstock particle may exhibit (Gentile et al. 2017). An easy but effective solution could be the calibration of the imposed shrinkage pattern based on a Di Blasi model (Di Blasi 1996), to match the experimental size evolution on the single particle level. This will not only improve the accuracy of the prediction of the particle size, but also all chemical and physical gas-particle interactions that are size-dependent. For a specific feedstock, one may even train the Di Blasi parameters by defining them as functions of variables of direct influence, such as feedstock conversion and heating and reaction rates, for a rather holistic approach. The anisotropy of shrinkage may be accounted for by integrating a 3D intra-particle model, and becomes more relevant if the particles are largely irregularly shaped or exposed to uneven heating rates from different directions. As particles collide, moreover, they may break up or form agglomerates. Although pyrolysis is a particle-centered process, particle breakage and agglomeration seem to have been overlooked and are not included in current CFD-DEM descriptions of pyrolysis reactors. In applications of fluidised beds, such phenomena may directly influence the fluidisation behaviour, in addition to modifying the heating, reaction, and exit rates of particles. In order to capture the effects of particle break-up (Bruchmüller et al. 2011; van Wachem et al. 2020) and agglomeration (Blum 2006), it is possible to enhance the existing CFD-DEM framework by corresponding sub-models.
Some topics that the prevailing chemical reaction models do not specifically address are the existence of heterogeneous tar reactions and the effect of catalytic species. These unaccounted reactions of tar, which are known to be exothermal, have an elevated importance in a predominately endothermal process such as pyrolysis. The heat release from exothermal reactions can promote endothermal reactions, particularly during primary decomposition, while lowering the overall energy demand of the process. Anca-Couce and Scharler (2017) extended the primary decomposition scheme of Debiagi et al. (2018) to account for this potential exothermicity; however, the extended chemical kinetics have not been deployed in a CFD-DEM model of pyrolysis yet. Although pyrolysis systems are operated in the presence of industrial catalysts for the purposes of enhancing the selectivity of desired species and minimising the energy demand, some species and products, such as ash, also possess natural catalytic effects which have been experimentally investigated (Yildiz et al. 2015; Di Blasi et al. 2018). In the absence of reliable chemical kinetic models, a study of a catalytic pyrolysis reactor remains an open challenge to date.
6 Conclusions
The computer-based modelling of pyrolysis systems permits an evaluation of effects and interactions in and across different scales that are difficult to isolate or observe experimentally. Although CFD-DEM models are computationally more expensive than fully Eulerian modelling approaches, they offer a direct resolution of particle-level phenomena. Within the scope of CFD-DEM, the carrier gas phase is described in a Eulerian fashion, while the dispersed feedstock and inert bed particles are tracked in a Lagrangian framework. A CFD-DEM description encompasses submodels that detail the rates at which mass, momentum, and heat are exchanged between the carrier gas and the particles, characterise the particles’ shrinkage and drying behaviour, or treat particle-particle interactions. In the first part of this article, we summarise the existing closure models to describe these phenomena, and discuss their underlying physical assumptions or simplifications.
Subsequently, the article provides an overview of the influence of operating conditions on the pyrolysis behaviour, as reported in works that sought to optimise the process with the aid of CFD-DEM models. The optimisation of a pyrolyser is an attempt at maximising the degree of feedstock conversion and the yield of pyrolysis-oil, while minimising the residence time. In addition to accelerating the process, a high temperature is usually preferred for the operation of a pyrolyser, as it increases the selectivity of the pyrolysis-oil during the primary decomposition. On the downside, the secondary decomposition of the produced oils is promoted. Further, an incomplete utilisation of feedstock may also result through the inhomogeneous distribution of temperature across feedstock particles. On the other hand, increasing the gas flow rate through the pyrolysis reactor has been seen to homogenise both interparticle temperature distributions and particle residence times. Yet, a high flow rate can adversely affect the feedstock conversion as the particles may leave the reactor prematurely, particularly for excessively small sizes. Conversely, large-sized particles are not favoured in pyrolysis because they may feature inhomogeneous intra-particle temperature and composition distributions and lead to a larger production of char.
In the present article, we also discuss open challenges in the modelling of pyrolysis with CFD-DEM. Firstly, the extension of the CFD-DEM methodology to arbitrarily-shaped particles and size or shape distributions is a topic of fundamental CFD-DEM research and recent developments here are only beginning to be applied to pyrolysis processes. Secondly, almost all CFD-DEM studies in pyrolysis have been performed with biomass as feedstock and there are currently no CFD-DEM descriptions for the pyrolysis of plastic feedstock, although this is an increasingly important process. Thirdly, the feedstock particles change as they undergo pyrolysis; they typically shrink, but can also expand, break, melt or fuse. Although ‘simple’ shrinkage models do exist in the framework of CFD-DEM, such models are currently elementary, and do not treat the other changes in particle shape and size as the particles undergo pyrolysis. Finally, the complexity of the chemical reaction models currently applied in CFD-DEM descriptions of pyrolysis processes is limited, and further developments are required to include exothermicity and catalytic effects.
Data Availability
N/A.
Abbreviations
- \(\alpha \) :
-
Interphase heat transfer coefficient (W m\(^{-2}\) K\(^{-1}\))
- \(\beta \) :
-
Interphase momentum transfer coefficient (kg m\(^{-3}\) s\(^{-1}\))
- \(\varvec{\upomega }\) :
-
Rotational velocity vector (rad s\(^{-1}\))
- \(\varvec{\uptau }\) :
-
Torque (N m)
- \(\delta (\textbf{x}- \textbf{x}_j)\) :
-
Dirac \(\delta \)-distribution (m\(^{-3}\))
- \(\delta ^\textrm{n}\) :
-
Normal overlap during a collision (m)
- \(\delta ^\textrm{t}\) :
-
Tangential overlap during a collision (m)
- \(\lambda \) :
-
Thermal conductivity (W m\(^{-1}\) K\(^{-1}\))
- \({\mu }_\textrm{g}\) :
-
Gas phase dynamic viscosity (Pa s)
- \(\mu _\textrm{f}\) :
-
Coefficient of friction \((-)\)
- \(\nu \) :
-
Stoichiometric coefficient \((-)\)
- \(\overline{\overline{\varvec{\tau }}}\) :
-
Viscous stress tensor (Pa)
- \(\rho \) :
-
Density (kg m\(^{-3}\))
- \(\sigma \) :
-
Scattering coefficient (m\(^{-1}\))
- \(\sigma _\textrm{s}\) :
-
Stefan–Boltzmann constant = 5.67 \(\times 10^{-8}\) W m\(^{-2}\) K\(^{-4}\)
- \(\theta \) :
-
Thickness of thermal boundary layer (m)
- \(\varepsilon \) :
-
Volume fraction \((-)\)
- \(\delta t\) :
-
Computational time step (s)
- \(\dot{m}^\textrm{devol}\) :
-
Rate of devolatilisation (kg s\(^{-1}\))
- \(\dot{m}^\textrm{evap}\) :
-
Rate of evaporation, i.e., drying rate (kg s\(^{-1}\))
- \(\dot{Q}\) :
-
Rate of total heat transfer (W)
- \(\dot{Q}^\mathrm {cond-intra}\) :
-
Rate of intra-particle conductive heat transfer (W)
- \(\dot{Q}^\mathrm {cond-pgp}\) :
-
Rate of indirect conductive heat transfer (W)
- \(\dot{Q}^\mathrm {cond-pp}\) :
-
Rate of direct conductive heat transfer (W)
- \(\dot{Q}^\textrm{cond}\) :
-
Rate of conductive heat transfer (W)
- \(\dot{Q}^\textrm{conv}\) :
-
Rate of convective heat transfer (W)
- \(\dot{Q}^\textrm{rad}\) :
-
Rate of radiative heat transfer (W)
- \(\hat{\textbf{n}}\) :
-
Contact normal unit vector \((-)\)
- \(\hat{\textbf{t}}\) :
-
Contact tangential unit vector \((-)\)
- \(\textbf{F}^\textrm{b}\) :
-
Buoyancy force (N)
- \(\textbf{F}^\textrm{c}\) :
-
Contact force (N)
- \(\textbf{F}^\textrm{d}\) :
-
Drag force (N)
- \(\textbf{g}\) :
-
Volume force (m s\(^{-2}\))
- \(\textbf{S}^\textrm{mom}\) :
-
Interphase momentum transfer rate (N)
- \(\textbf{U}\) :
-
Diffusion velocity vector (m s\(^{-1}\))
- \(\textbf{u}\) :
-
Translational velocity vector (m s\(^{-1}\))
- \({u}^{\textrm{c}}\) :
-
Correction velocity (m s\(^{-1}\))
- \(\textbf{x}\) :
-
Position vector (m)
- \(\textbf{x}^\textrm{c}\) :
-
Collision point (m)
- \(\mathcal {R}\) :
-
Rate of reaction (mol m\(^{-3}\) s\(^{-1}\))
- \(\textrm{Ma}\) :
-
Mach number \((-)\)
- \(\textrm{Nu}\) :
-
Nusselt number \((-)\)
- \(\textrm{Pr}\) :
-
Prandtl number \((-)\)
- \(\textrm{Re}\) :
-
Reynolds number \((-)\)
- \(\overline{\overline{\mathbb {I}}}\) :
-
Identity tensor \((-)\)
- \(\overline{\overline{\textbf{I}}}\) :
-
Inertia tensor (kg m\(^{2}\))
- A :
-
Surface area (m\(^{2}\))
- a :
-
Absorption coefficient (m\(^{-1}\))
- \(A^\textrm{evap}\) :
-
Pre-exponential factor of phase change reaction \(\text {H}_2\text {O}\)(l)\(\longrightarrow \) \(\text {H}_2\text {O}\) (s\(^{-1}\))
- \(C_p\) :
-
Specific heat capacity (J kg\(^{-1}\) K\(^{-1}\))
- D :
-
Diffusion coefficient \((\text {m}^{2} \text {s}^{-1}\))
- d :
-
Diameter of a spherical particle (m)
- \(E^\textrm{evap}\) :
-
Activation energy of phase change reaction \(\text {H}_2\text {O}\)(l)\(\longrightarrow \) \(\text {H}_2\text {O}\)(g) (J mol\(^{-1}\))
- \(f_1, f_2, f_3\) :
-
Modelling parameters of Di Blasi’s shrinkage model \((-)\)
- G :
-
Incident radiation (W m\(^{-2}\))
- \(h^0_f\) :
-
Enthalpy of formation (J kg\(^{-1}\))
- \(H^\textrm{evap}\) :
-
Heat of vaporisation of water (J kg\(^{-1}\))
- \(h_i\) :
-
Specific enthalpy of species i (J kg\(^{-1}\))
- \(k^\textrm{evap}\) :
-
Modelling constant in equilibrium model for drying \((\mathrm{kg m^{3} mol^{-1} s^{-1}})\)
- L :
-
Characteristic length of a particle (m)
- l(r):
-
Distance between two particles at radial distance r (m)
- \(l_\textrm{min}\) :
-
Minimum conduction distance (m)
- n :
-
Refraction index \((-)\)
- \(N^\textrm{pd}\) :
-
Number of primary decomposition reactions \((-)\)
- \(N^\textrm{sd}\) :
-
Number of secondary decomposition reactions \((-)\)
- \(N_\textrm{g}\) :
-
Number of gas phase species \((-)\)
- \(N_\textrm{p}\) :
-
Number of particles \((-)\)
- \(N_\textrm{s}\) :
-
Number of particulate phase species \((-)\)
- P :
-
Thermodynamic pressure (Pa)
- p :
-
Pressure (Pa)
- \(P^*\) :
-
Saturation pressure (Pa)
- R :
-
Universal gas constant = 8.314 J K\(^{-1}\) mol\(^{-1}\)
- r :
-
Radial coordinate (m)
- \(r^\textrm{c}\) :
-
Contact radius between colliding particles (m)
- \(r^\textrm{in}, \, r^\textrm{out}\) :
-
Radial bounds of indirect conduction (m)
- \(S^\mathrm {heat-reac}\) :
-
Heat source/sink term due to reactions (W)
- \(S^\textrm{heat}\) :
-
Interphase heat transfer rate (W)
- \(S^\mathrm {mass-reac}\) :
-
Mass source/sink term due to reactions (kg s\(^{-1}\))
- \(S^\textrm{mass}\) :
-
Interphase mass transfer rate (kg s\(^{-1}\))
- \(S^G\) :
-
Radiation source term (W m\(^{-3}\))
- T :
-
Temperature (K)
- t :
-
Time (s)
- \(T^\textrm{evap}\) :
-
Boiling temperature of water at operating pressure (K)
- V :
-
Volume of a particle (m\(^{3}\))
- \(V^\textrm{pore}\) :
-
Particle pore volume (m\(^{3}\))
- \(V^\textrm{solid}\) :
-
Particle solid volume (m\(^{3}\))
- W :
-
Molar mass (kg mol\(^{-1}\))
- X :
-
Mole fraction \((-)\)
- Y :
-
Mass fraction \((-)\)
- 0:
-
Initial values
- \(\textrm{B}\) :
-
Biomass
- \(\textrm{C}\) :
-
Char
- \(\textrm{g}\) :
-
Gas phase
- \(\textrm{p}\) :
-
Particulate phase
- i :
-
Gas species index
- j :
-
Particle index
- n :
-
Reaction index
- s :
-
Particle species index
References
Abnisa, F., Wan Daud, W.M.A.: A review on co-pyrolysis of biomass: an optional technique to obtain a high-grade pyrolysis oil. Energy Convers. Manag. 87, 71–85 (2014). https://doi.org/10.1016/j.enconman.2014.07.007
Abramzon, B., Sirignano, W.: Droplet vaporization model for spray combustion calculations. Int. J. Heat Mass Transfer 32(9), 1605–1618 (1989). https://doi.org/10.1016/0017-9310(89)90043-4
Akkus, Y.: The effect of Stefan flow on the models of droplet evaporation. Isı Bilimi ve Tekniği Dergisi 40(2), 309–318 (2020)
Al-Salem, S., Lettieri, P.: Kinetic study of high density polyethylene (HDPE) pyrolysis. Chem. Eng. Res. Des. 88(12), 1599–1606 (2010). https://doi.org/10.1016/j.cherd.2010.03.012
Anca-Couce, A., Scharler, R.: Modelling heat of reaction in biomass pyrolysis with detailed reaction schemes. Fuel 206, 572–579 (2017). https://doi.org/10.1016/j.fuel.2017.06.011
Anderson, T.B., Jackson, R.: Fluid mechanical description of fluidized beds. Equations of motion. Indus. Eng. Chem. Fundam. 6(4), 527–539 (1967). https://doi.org/10.1021/i160024a007
Anuar Sharuddin, S.D., Abnisa, F., Wan Daud, W.M.A., Aroua, M.K.: A review on pyrolysis of plastic wastes. Energy Convers. Manag. 115, 308–326 (2016). https://doi.org/10.1016/j.enconman.2016.02.037
Aramideh, S., Xiong, Q., Kong, S.C., Brown, R.C.: Numerical simulation of biomass fast pyrolysis in an auger reactor. Fuel 156, 234–242 (2015)
Armenise, S., Wong, S., Ramírez-Velásquez, J.M., Launay, F., Wuebben, D., Nyakuma, B.B., Rams, J., Muñoz, M.: Application of computational approach in plastic pyrolysis kinetic modelling: a review. React. Kinet. Mech. Catal. 134(2), 591–614 (2021). https://doi.org/10.1007/s11144-021-02093-7
Basile, L., Tugnoli, A., Stramigioli, C., Cozzani, V.: Influence of pressure on the heat of biomass pyrolysis. Fuel 137, 277–284 (2014). https://doi.org/10.1016/j.fuel.2014.07.071
Basu, P., Kaushal, P.: Modeling of Pyrolysis and Gasification of Biomass in Fluidized Beds: A Review. Chem. Prod. Process. Model. (2009). https://doi.org/10.2202/1934-2659.1338
Batchelor, G., O’Brien, R.: Thermal or electrical conduction through a granular material. Proc. R. Soc. A Math. Phys. Eng. Sci. 355, 313–333 (1977). https://doi.org/10.1098/rspa.1977.0100
Beetham, S., Capecelatro, J.: Biomass pyrolysis in fully-developed turbulent riser flow. Renew. Energy 140, 751–760 (2019). https://doi.org/10.1016/j.renene.2019.03.095
Bellais, M.: Modelling of the pyrolysis of large wood particles. PhD thesis, KTH Royal Institute of Technology, Stockholm. http://urn.kb.se/resolve?urn=urn:nbn:se:kth:diva-4422 (2007)
Blum, J.: Dust agglomeration. Adv. Phys. 55(7), 881–947 (2006). https://doi.org/10.1080/00018730601095039
Bradbury, A.G.W., Sakai, Y., Shafizadeh, F.: A kinetic model for pyrolysis of cellulose. J. Appl. Polym. Sci. 23(11), 3271–3280 (1979). https://doi.org/10.1002/app.1979.070231112
Broido, A.: Kinetics of solid-phase cellulose pyrolysis. In: Shafizadeh F, Sarkanen KV, Tillman DA (eds) Thermal Uses and Properties of Carbohydrates and Lignins. Academic Press, pp 19–36 (1976) https://doi.org/10.1016/B978-0-12-637750-7.50006-6
Bruchmüller, J., van Wachem, B., Gu, S., Luo, K.H.: Modelling discrete fragmentation of brittle particles. Powder Technol. 208(3), 731–739 (2011). https://doi.org/10.1016/j.powtec.2011.01.017
Bruchmüller, J., van Wachem, B., Gu, S., Luo, K., Brown, R.: Modeling the thermochemical degradation of biomass inside a fast pyrolysis fluidized bed reactor. AIChE J. 58(10), 3030–3042 (2012). https://doi.org/10.1002/aic.13705
Bruchmüller, J., Luo, K.H., van Wachem, B.G.M.: Tar formation variations during fluidised bed pyrolytic biomass conversion. Proc. Combust. Inst. 34(2), 2373–2381 (2013). https://doi.org/10.1016/j.proci.2012.06.054
Bryden, K.M., Hagge, M.J.: Modeling the combined impact of moisture and char shrinkage on the pyrolysis of a biomass particle. Fuel 82(13), 1633–1644 (2003). https://doi.org/10.1016/S0016-2361(03)00108-X
Cai, J., Wu, W., Liu, R.: An overview of distributed activation energy model and its application in the pyrolysis of lignocellulosic biomass. Renew. Sustain. Energy Rev. 36, 236–246 (2014). https://doi.org/10.1016/j.rser.2014.04.052
Cao, Z., Tafti, D.K.: Fluid forces and torques in suspensions of oblate cylinders with aspect ratio 1:4. Int. J. Multiph. Flow 131(103), 394 (2020). https://doi.org/10.1016/j.ijmultiphaseflow.2020.103394
Capecelatro, J., Desjardins, O.: An Euler–Lagrange strategy for simulating particle-laden flows. J. Comput. Phys. 238, 1–31 (2013). https://doi.org/10.1016/j.jcp.2012.12.015
Caposciutti, G., Almuina-Villar, H., Dieguez-Alonso, A., Gruber, T., Kelz, J., Desideri, U., Hochenauer, C., Scharler, R., Anca-Couce, A.: Experimental investigation on biomass shrinking and swelling behaviour: particles pyrolysis and wood logs combustion. Biomass Bioenerg. 123, 1–13 (2019)
Çepelioğullar, Ö., Pütün, A.E.: Utilization of two different types of plastic wastes from daily and industrial life. J. Selcuk Univ. Nat. Appl. Sci. 2(2), 694–706 (2013)
Chaloupková, V., Ivanova, T., Krepl, V.: Particle size and shape characterization of feedstock material for biofuel production. Agron. Res. 17(5), 1861–1873 (2019)
Chandrasekhar, S.: Radiative Transfer. Oxford University Press, Dover (1960)
Chaudhary, A., Lakhani, J., Dalsaniya, P., Chaudhary, P., Trada, A., Shah, N.K., Upadhyay, D.S.: Slow pyrolysis of low-density Poly-Ethylene (LDPE): a batch experiment and thermodynamic analysis. Energy 263(125), 810 (2023)
Chen, T., Ku, X., Lin, J., Ström, H.: CFD-DEM simulation of biomass pyrolysis in fluidized-bed reactor with a multistep kinetic scheme. Energies 13(20), 5358 (2020). https://doi.org/10.3390/en13205358
Chen, X., Che, Q., Li, S., Liu, Z., Yang, H., Chen, Y., Wang, X., Shao, J., Chen, H.: Recent developments in lignocellulosic biomass catalytic fast pyrolysis: Strategies for the optimization of bio-oil quality and yield. Fuel Process. Technol. 196(106), 180 (2019)
Chen, Y., Charpenay, S., Jensen, A., Wójtowicz, M.A., Serio, M.A.: Modeling of biomass pyrolysis kinetics. Symp. (Int.) Combust. 27(1), 1327–1334 (1998). https://doi.org/10.1016/S0082-0784(98)80537-7
Cordiner, S., Manni, A., Mulone, V., Rocco, V.: Biomass pyrolysis modeling of systems at laboratory scale with experimental validation. Int. J. Numer. Methods Heat Fluid Flow 28(2), 413–438 (2018). https://doi.org/10.1108/HFF-11-2016-0459
Costa, P., Pinto, F., Ramos, A., Gulyurtlu, I., Cabrita, I., Bernardo, M.: Study of the pyrolysis kinetics of a mixture of polyethylene, polypropylene, and polystyrene. Energy Fuels 24(12), 6239–6247 (2010)
Crowe, C.T., Sharma, M.P., Stock, D.E.: The particle-source-in cell (PSI-CELL) model for gas-droplet flows. J. Fluids Eng. 99(2), 325–333 (1977)
Cundall, P., Strack, O.D.L.: A discrete numerical model for granular assemblies. Géotechnique 29(1), 47–65 (1979). https://doi.org/10.1680/geot.1979.29.1.47
Davidsson, K.O., Pettersson, J.B.: Birch wood particle shrinkage during rapid pyrolysis. Fuel 81(3), 263–270 (2002)
Debiagi, P., Gentile, G., Cuoci, A., Frassoldati, A., Ranzi, E., Faravelli, T.: A predictive model of biochar formation and characterization. J. Anal. Appl. Pyrolysis 134, 326–335 (2018). https://doi.org/10.1016/j.jaap.2018.06.022
Debiagi, P.E.A., Gentile, G., Pelucchi, M., Frassoldati, A., Cuoci, A., Faravelli, T., Ranzi, E.: Detailed kinetic mechanism of gas-phase reactions of volatiles released from biomass pyrolysis. Biomass Bioenerg. 93, 60–71 (2016). https://doi.org/10.1016/j.biombioe.2016.06.015
Di Blasi, C.: Numerical simulation of cellulose pyrolysis. Biomass Bioenerg. 7(1), 87–98 (1994). https://doi.org/10.1016/0961-9534(94)00040-Z
Di Blasi, C.: Heat, momentum and mass transport through a shrinking biomass particle exposed to thermal radiation. Chem. Eng. Sci. 51(7), 1121–1132 (1996). https://doi.org/10.1016/S0009-2509(96)80011-X
Di Blasi, C., Signorelli, G., Di Russo, C., Rea, G.: Product distribution from pyrolysis of wood and agricultural residues. Indus. Eng. Chem. Res. 38(6), 2216–2224 (1999). https://doi.org/10.1021/ie980711u
Di Blasi, C., Branca, C., Sarnataro, F.E., Gallo, A.: Thermal runaway in the pyrolysis of some lignocellulosic biomasses. Energy Fuels 28(4), 2684–2696 (2014). https://doi.org/10.1021/ef500296g
Di Blasi, C., Branca, C., Galgano, A.: On the experimental evidence of exothermicity in wood and biomass pyrolysis. Energ. Technol. 5(1), 19–29 (2017). https://doi.org/10.1002/ente.201600091
Di Blasi, C., Branca, C., Galgano, A.: Role of the potassium chemical state in the global exothermicity of wood pyrolysis. Indust. Eng. Chem. Res. 57(34), 11561–11571 (2018). https://doi.org/10.1021/acs.iecr.8b02047
Di Felice, R.: The voidage function for fluid-particle interaction systems. Int. J. Multiph. Flow 20(1), 153–159 (1994)
Diebel, J.: Representing attitude: Euler angles, unit quaternions, and rotation vectors. Stanford University, California, USA, Tech. rep. (2006)
Eaton, J.K.: Two-way coupled turbulence simulations of gas-particle flows using point-particle tracking. Int. J. Multiph. Flow 35(9), 792–800 (2009)
Evrard, F., Denner, F., van Wachem, B.: A multi-scale approach to simulate atomisation processes. Int. J. Multiph. Flow 119, 194–216 (2019). https://doi.org/10.1016/j.ijmultiphaseflow.2019.07.005
Evrard, F., Denner, F., van Wachem, B.: Quantifying the errors of the particle-source-in-cell Euler-Lagrange method. Int. J. Multiph. Flow 135(103), 535 (2021). https://doi.org/10.1016/j.ijmultiphaseflow.2020.103535
Eze, W.U., Umunakwe, R., Obasi, H.C., Ugbaja, M.I., Uche, C.C., Madufor, I.C., Eze, W.U., Umunakwe, R., Obasi, H.C., Ugbaja, M.I., Uche, C.C., Madufor, I.C.: Plastics waste management: A review of pyrolysis technology. Clean Technol. Recycl. 1(1), 50–69 (2021). https://doi.org/10.3934/ctr.2021003
Fahmi, R., Bridgwater, A.V., Donnison, I., Yates, N., Jones, J.: The effect of lignin and inorganic species in biomass on pyrolysis oil yields, quality and stability. Fuel 87(7), 1230–1240 (2008)
Fan, H., Gu, J., Wang, Y., Yuan, H., Chen, Y., Luo, B.: Effect of potassium on the pyrolysis of biomass components: pyrolysis behaviors, product distribution and kinetic characteristics. Waste Manag. 121, 255–264 (2021)
Faravelli, T., Pinciroli, M., Pisano, F., Bozzano, G., Dente, M., Ranzi, E.: Thermal degradation of polystyrene. J. Anal. Appl. Pyrol. 60(1), 103–121 (2001)
Finnie, I.: Erosion of surfaces by solid particles. Wear 3(2), 87–103 (1960)
Fitzpatrick, R.: Newtonian Dynamics: An Introduction. CRC Press, Boca Raton (2021)
Fletcher, T.H., Pond, H.R., Webster, J., Wooters, J., Baxter, L.L.: Prediction of tar and light gas during pyrolysis of black liquor and biomass. Energy Fuels 26(6), 3381–3387 (2012). https://doi.org/10.1021/ef300574n
Flor-Barriga, L.A.D.l., Rodríguez-Zúñiga, U.F.: Numerical analysis on a catalytic pyrolysis reactor design for plastic waste upcycling using CFD modelling. RSC Adv. 12(20), 12436–12445 (2022). https://doi.org/10.1039/D2RA01407F
Ganser, G.H.: A rational approach to drag prediction of spherical and nonspherical particles. Powder Technol. 77(2), 143–152 (1993)
Gao, X., Li, T., Sarkar, A., Lu, L., Rogers, W.A.: Development and validation of an enhanced filtered drag model for simulating gas-solid fluidization of geldart a particles in all flow regimes. Chem. Eng. Sci. 184, 33–51 (2018)
Gao, X., Lu, L., Shahnam, M., Rogers, W.A., Smith, K., Gaston, K., Robichaud, D., Brennan Pecha, M., Crowley, M., Ciesielski, P.N., Debiagi, P., Faravelli, T., Wiggins, G., Finney, C.E.A., Parks, J.E.: Assessment of a detailed biomass pyrolysis kinetic scheme in multiscale simulations of a single-particle pyrolyzer and a pilot-scale entrained flow pyrolyzer. Chem. Eng. J. 418(129), 347 (2021). https://doi.org/10.1016/j.cej.2021.129347
Gao, X., Yu, J., Lu, L., Rogers, W.A.: Coupling particle scale model and SuperDEM-CFD for multiscale simulation of biomass pyrolysis in a packed bed pyrolyzer. AIChE J. 67(4), e17139 (2021). https://doi.org/10.1002/aic.17139
Geldart, D.: Types of gas fluidization. Powder Technol. 7(5), 285–292 (1973)
Gellerstedt, G., Ek, M., Henriksson, G.: Wood Chemistry and Wood Biotechnology. Walter de Gruyter (2009)
Gentile, G., Debiagi, P.E.A., Cuoci, A., Frassoldati, A., Ranzi, E., Faravelli, T.: A computational framework for the pyrolysis of anisotropic biomass particles. Chem. Eng. J. 321, 458–473 (2017). https://doi.org/10.1016/j.cej.2017.03.113
Geyer, R., Jambeck, J.R., Law, K.L.: Production, use, and fate of all plastics ever made. Sci. Adv. 3(7), e1700782 (2017)
Gidaspow, D., Bezburuah, R., Ding, J.: Hydrodynamics of circulating fluidized beds: kinetic theory approach. Tech. rep., Illinois Institute of Technology, Chicago, IL (United States). Department of Chemical Engineering (1991)
Grant, D.M., Pugmire, R.J., Fletcher, T.H., Kerstein, A.R.: Chemical model of coal devolatilization using percolation lattice statistics. Energy Fuels 3(2), 175–186 (1989). https://doi.org/10.1021/ef00014a011
Grønli, M.G., Melaaen, M.C.: Mathematical model for wood pyrolysis comparison of experimental measurements with model predictions. Energy Fuels 14(4), 791–800 (2000)
Guizani, C., Valin, S., Billaud, J., Peyrot, M., Salvador, S.: Biomass fast pyrolysis in a drop tube reactor for bio oil production: experiments and modeling. Fuel 207, 71–84 (2017)
Gunn, D.J.: Transfer of heat or mass to particles in fixed and fluidised beds. Int. J. Heat Mass Transfer 21(4), 467–476 (1978). https://doi.org/10.1016/0017-9310(78)90080-7
Hirschfelder, J.O., Curtiss, C.F., Bird, R.B.: The Molecular Theory of Gases and Liquids, revised, edition Wiley-Interscience, Hoboken, NJ (1964)
Hölzer, A., Sommerfeld, M.: New simple correlation formula for the drag coefficient of non-spherical particles. Powder Technol. 184(3), 361–365 (2008)
Hooshdaran, B., Haghshenasfard, M., Hosseini, S.H., Esfahany, M.N., Lopez, G., Olazar, M.: CFD modeling and experimental validation of biomass fast pyrolysis in a conical spouted bed reactor. J. Anal. Appl. Pyrol. 154(105), 011 (2021)
Houston, R., Oyedeji, O., Abdoulmoumine, N.: Detailed biomass fast pyrolysis kinetics integrated to computational fluid dynamic (CFD) and discrete element modeling framework: predicting product yields at the bench-scale. Chem. Eng. J. 444(136), 419 (2022). https://doi.org/10.1016/j.cej.2022.136419
Hu, C., Luo, K., Wang, S., Sun, L., Fan, J.: Computational fluid dynamics/discrete element method investigation on the biomass fast pyrolysis: the influences of shrinkage patterns and operating parameters. Ind. Eng. Chem. Res. 58(3), 1404–1416 (2019). https://doi.org/10.1021/acs.iecr.8b05279
Hulburt, H.M., Katz, S.: Some problems in particle technology: A statistical mechanical formulation. Chem. Eng. Sci. 19(8), 555–574 (1964)
Jeans, J.H.: Stars, Gaseous, radiative transfer of energy. Mon. Not. R. Astron. Soc. 78, 28–36 (1917)
Jing, X., Zhao, Y., Wen, H., Xu, Z.: Interactions between low-density polyethylene (LDPE) and polypropylene (PP) during the mild cracking of polyolefin mixtures in a closed-batch reactor. Energy Fuels 27(10), 5841–5851 (2013)
Kayacan, I., Doğan, Ö.: Pyrolysis of low and high density polyethylene. Part I: non-isothermal pyrolysis kinetics. Energy Sources A Recov. Utilization Environ. Effects 30(5), 385–391 (2008)
Krause, B., Liedmann, B., Wiese, J., Wirtz, S., Scherer, V.: Coupled three dimensional DEM-CFD simulation of a lime shaft kiln-Calcination, particle movement and gas phase flow field. Chem. Eng. Sci. 134, 834–849 (2015). https://doi.org/10.1016/j.ces.2015.06.002
Krause, B., Liedmann, B., Wiese, J., Bucher, P., Wirtz, S., Piringer, H., Scherer, V.: 3D-DEM-CFD simulation of heat and mass transfer, gas combustion and calcination in an intermittent operating lime shaft kiln. Int. J. Therm. Sci. 117, 121–135 (2017). https://doi.org/10.1016/j.ijthermalsci.2017.03.017
Ku, X., Li, T., Løvås, T.: CFD-DEM simulation of biomass gasification with steam in a fluidized bed reactor. Chem. Eng. Sci. 122, 270–283 (2015)
Ku, X., Li, T., Løvås, T.: Effects of particle shrinkage and devolatilization models on high-temperature biomass pyrolysis and gasification. Energy Fuels 29(8), 5127–5135 (2015). https://doi.org/10.1021/acs.energyfuels.5b00953
Ku, X., Shen, F., Jin, H., Lin, J., Li, H.: Simulation of biomass pyrolysis in a fluidized bed reactor using thermally thick treatment. Indus. Eng. Chem. Res. 58(4), 1720–1731 (2019). https://doi.org/10.1021/acs.iecr.8b04778
Kuerten, J.G.M.: Point-particle DNS and LES of particle-laden turbulent flow—a state-of-the-art review. Flow Turbul. Combust. 97(3), 689–713 (2016). https://doi.org/10.1007/s10494-016-9765-y
Kuipers, J.A.M., Prins, W., Van Swaaij, W.P.M.: Numerical calculation of wall-to-bed heat-transfer coefficients in gas-fluidized beds. AIChE J. 38(7), 1079–1091 (1992). https://doi.org/10.1002/aic.690380711
Kumar, R.R., Kolar, A.K., Leckner, B.: Shrinkage characteristics of casuarina wood during devolatilization in a fluidized bed combustor. Biomass Bioenerg. 30(2), 153–165 (2006)
Kunii, D., Levenspiel, O.: Fluidization Engineering. Krieger Publishing Company, R. E (1977)
Lathouwers, D., Bellan, J.: Modeling of dense gas-solid reactive mixtures applied to biomass pyrolysis in a fluidized bed. Int. J. Multiph. Flow 27, 2155–22187 (2001)
Li, T., Ku, X., Løvås, T.: CFD simulation of devolatilization of biomass with shrinkage effect. Energy Procedia 105, 505–510 (2017). https://doi.org/10.1016/j.egypro.2017.03.348
Lin, Y.H., Yen, H.Y.: Fluidised bed pyrolysis of polypropylene over cracking catalysts for producing hydrocarbons. Polym. Degrad. Stab. 89(1), 101–108 (2005)
Liu, B., Papadikis, K., Gu, S., Fidalgo, B., Longhurst, P., Li, Z., Kolios, A.: CFD modelling of particle shrinkage in a fluidized bed for biomass fast pyrolysis with quadrature method of moment. Fuel Process. Technol. 164, 51–68 (2017)
Liu, S., Xing, Y., Chen, H., Tang, P., Jiang, J., Tang, S., Liang, B.: Sustainable reactors for biomass conversion using pyrolysis and fermentation. Reference Module in Earth Systems and Environmental Sciences (2017b)
Lu, G., Third, J., Müller, C.: Discrete element models for non-spherical particle systems: From theoretical developments to applications. Chem. Eng. Sci. 127, 425–465 (2015). https://doi.org/10.1016/j.ces.2014.11.050
Lu, L., Gao, X., Shahnam, M., Rogers, W.A.: Bridging particle and reactor scales in the simulation of biomass fast pyrolysis by coupling particle resolved simulation and coarse grained CFD-DEM. Chem. Eng. Sci. 216(115), 471 (2020). https://doi.org/10.1016/j.ces.2020.115471
Lu, L., Gao, X., Dietiker, J.F., Shahnam, M., Rogers, W.: MFiX based multi-scale CFD simulations of biomass fast pyrolysis: a review. Chem. Eng. Sci. 248(117), 131 (2021). https://doi.org/10.1016/j.ces.2021.117131
Lu, L., Gao, X., Gel, A., Wiggins, G.M., Crowley, M., Pecha, B., Shahnam, M., Rogers, W.A., Parks, J., Ciesielski, P.N.: Investigating biomass composition and size effects on fast pyrolysis using global sensitivity analysis and CFD simulations. Chem. Eng. J. 421(127), 789 (2021). https://doi.org/10.1016/j.cej.2020.127789
Lu, L., Gao, X., Shahnam, M., Rogers, W.A.: Open source implementation of glued sphere discrete element method and nonspherical biomass fast pyrolysis simulation. AIChE J. 67(6), e17211 (2021). https://doi.org/10.1002/aic.17211
Lu, L., Gao, X., Shahnam, M., Rogers, W.A.: Simulations of biomass pyrolysis using glued-sphere CFD-DEM with 3-D intra-particle models. Chem. Eng. J. (2021). https://doi.org/10.1016/j.cej.2021.129564
Lun, C., Savage, S.B., Jeffrey, D., Chepurniy, N.: Kinetic theories for granular flow: inelastic particles in couette flow and slightly inelastic particles in a general flowfield. J. Fluid Mech. 140, 223–256 (1984)
Luo, H., Lin, W., Dam-Johansen, K., Wu, H.: Heat-transfer-corrected isothermal model for devolatilization of thermally thick biomass particles. Energy Fuels 34(8), 9620–9631 (2020). https://doi.org/10.1021/acs.energyfuels.0c01155
Luo, H., Wang, X., Liu, X., Wu, X., Shi, X., Xiong, Q.: A review on CFD simulation of biomass pyrolysis in fluidized bed reactors with emphasis on particle-scale models. J. Anal. Appl. Pyrol. 162(105), 433 (2022). https://doi.org/10.1016/j.jaap.2022.105433
Machmudah, S., Wicaksono, D., Happy, M., Winardi, S., Diono, W., Kanda, H., Goto, M.: Water removal from wood biomass by liquefied dimethyl ether for enhancing heating value. Energy Rep. 6, 824–831 (2020). https://doi.org/10.1016/j.egyr.2020.04.006
Mahmoudi, A.H., Hoffmann, F., Peters, B.: Detailed numerical modeling of pyrolysis in a heterogeneous packed bed using XDEM. J. Anal. Appl. Pyrol. 106, 9–20 (2014). https://doi.org/10.1016/j.jaap.2013.12.001
Mahmoudi, A.H., Hoffmann, F., Peters, B.: Semi-resolved modeling of heat-up, drying and pyrolysis of biomass solid particles as a new feature in XDEM. Appl. Therm. Eng. 93, 1091–1104 (2016). https://doi.org/10.1016/j.applthermaleng.2015.10.033
Mahmoudi, A.H., Hoffmann, F., Peters, B., Besseron, X.: Numerical study of the influence of particle size and packing on pyrolysis products using XDEM. Int. Commun. Heat Mass Transfer 71, 20–34 (2016). https://doi.org/10.1016/j.icheatmasstransfer.2015.12.011
Marchisio, D.L., Fox, R.O.: Computational Models for Polydisperse Particulate and Multiphase Systems. Cambridge Series in Chemical Engineering, Cambridge University Press, Cambridge (2013)
Martín-Gullón, I., Esperanza, M., Font, R.: Kinetic model for the pyrolysis and combustion of poly-(ethylene terephthalate)(PET). J. Anal. Appl. Pyrol. 58, 635–650 (2001)
Mastral, F., Esperanza, E., Garcıa, P., Juste, M.: Pyrolysis of high-density polyethylene in a fluidised bed reactor influence of the temperature and residence time. J. Anal. Appl. Pyrol. 63(1), 1–15 (2002)
Matayeva, A., Basile, F., Cavani, F., Bianchi, D., Chiaberge, S.:x Development of upgraded bio-oil via liquefaction and pyrolysis. In: Studies in Surface Science and Catalysis, vol. 178, pp. 231–256. Elsevier (2021)
Mehrabian, R., Zahirovic, S., Scharler, R., Obernberger, I., Kleditzsch, S., Wirtz, S., Scherer, V., Lu, H., Baxter, L.L.: A CFD model for thermal conversion of thermally thick biomass particles. Fuel Process. Technol. 95, 96–108 (2012). https://doi.org/10.1016/j.fuproc.2011.11.021
Miller, R., Bellan, J.: A generalized biomass pyrolysis model based on superimposed cellulose, hemicellulose and lignin kinetics. Combust. Sci. Technol. 126(1–6), 97–137 (1997). https://doi.org/10.1080/00102209708935670
Modest, M.F., Mazumder, S.: Radiative Heat Transfer. Academic Press, Cambridge (2021)
Mok, W.S.L., Antal, M.J.: Effects of pressure on biomass pyrolysis II Heats of reaction of cellulose pyrolysis. Thermochim. Acta 68(2), 165–186 (1983). https://doi.org/10.1016/0040-6031(83)80222-6
Niksa, S.: Predicting the rapid devolatilization of diverse forms of biomass with bio-flashchain. Proc. Combust. Inst. 28(2), 2727–2733 (2000). https://doi.org/10.1016/S0082-0784(00)80693-1
Papadikis, K., Gu, S.: CFD modelling of the fast pyrolysis of biomass in fluidised bed reactors: modelling the impact of biomass shrinkage. Chem. Eng. J. 149(1–3), 417–427 (2009). https://doi.org/10.1016/j.cej.2009.01.036
Papadikis, K., Bridgwater, A.V., Gu, S.: CFD modelling of the fast pyrolysis of biomass in fluidised bed reactors, Part A: Eulerian computation of momentum transport in bubbling fluidised beds. Chem. Eng. Sci. 63(16), 4218–4227 (2008). https://doi.org/10.1016/j.ces.2008.05.045
Papadikis, K., Gerhauser, H., Bridgwater, A.V., Gu, S.: CFD modelling of the fast pyrolysis of an in-flight cellulosic particle subjected to convective heat transfer. Biomass Bioenerg. 33(1), 97–107 (2009). https://doi.org/10.1016/j.biombioe.2008.04.021
Papadikis, K., Gu, S., Bridgwater, A.V.: CFD modelling of the fast pyrolysis of biomass in fluidised bed reactors Part B: Heat, momentum and mass transport in bubbling fluidised beds. Chem. Eng. Sci. 64(5), 1036–1045 (2009). https://doi.org/10.1016/j.ces.2008.11.007
Papadikis, K., Gu, S., Bridgwater, A.V., Gerhauser, H.: Application of CFD to model fast pyrolysis of biomass. Fuel Process. Technol. 90(4), 504–512 (2009). https://doi.org/10.1016/j.fuproc.2009.01.010
Papari, S., Hawboldt, K.: A review on the pyrolysis of woody biomass to bio-oil: focus on kinetic models. Renew. Sustain. Energy Rev. 52, 1580–1595 (2015). https://doi.org/10.1016/j.rser.2015.07.191
Park, W.C., Atreya, A., Baum, H.R.: Experimental and theoretical investigation of heat and mass transfer processes during wood pyrolysis. Combust. Flame 157(3), 481–494 (2010). https://doi.org/10.1016/j.combustflame.2009.10.006
Paulauskas, R., Džiugys, A., Striūgas, N.: Experimental investigation of wood pellet swelling and shrinking during pyrolysis. Fuel 142, 145–151 (2015)
Pelucchi, M., Cavallotti, C., Cuoci, A., Faravelli, T., Frassoldati, A., Ranzi, E.: Detailed kinetics of substituted phenolic species in pyrolysis bio-oils. React. Chem. Eng. 4(3), 490–506 (2019). https://doi.org/10.1039/C8RE00198G
Poinsot, T., Veynante, D.: Theoretical and Numerical Combustion. R.T. Edwards, Inc. (2005)
Qi, F., Wright, M.M.: A DEM modeling of biomass fast pyrolysis in a double auger reactor. Int. J. Heat Mass Transf. 150(119), 308 (2020). https://doi.org/10.1016/j.ijheatmasstransfer.2020.119308
Rabinovich, O.S., Korban, V.V., Pal’chenok, G.I., Khorol’skaya, O.P.: Modeling of fast pyrolysis of a single biomass particle in an inert boiling bed. J. Eng. Phys. Thermophys. 82(4), 611 (2009). https://doi.org/10.1007/s10891-009-0244-3
Rabinovich, O.S., Borodulya, V.A., Vinogradov, L.M., Korban, V.V.: Fast pyrolysis of an ensemble of biomass particles in a fluidized bed. J. Eng. Phys. Thermophys. 83(4), 742–752 (2010). https://doi.org/10.1007/s10891-010-0393-4
Ranganathan, P., Gu, S.: Computational fluid dynamics modelling of biomass fast pyrolysis in fluidised bed reactors, focusing different kinetic schemes. Biores. Technol. 213, 333–341 (2016). https://doi.org/10.1016/j.biortech.2016.02.042
Ranz, W., Marshall, W.: Evaporation from drops. Chem. Eng. Prog. 48(3), 141–146 (1952)
Ranzi, E., Frassoldati, A., Stagni, A., Pelucchi, M., Cuoci, A., Faravelli, T.: Reduced kinetic schemes of complex reaction systems: fossil and biomass-derived transportation fuels. Int. J. Chem. Kinet. 46(9), 512–542 (2014). https://doi.org/10.1002/kin.20867
Rigopoulos, S.: Population balance modelling of polydispersed particles in reactive flows. Prog. Energy Combust. Sci. 36(4), 412–443 (2010)
Rong, D., Horio, M.: DEM simulation of char combustion in a fluidized bed. In: Second International Conference on CFD in the Minerals and Process Industries CSIRO, pp. 65–70. Melbourne, Australia (1999)
Sadaka, S., Boateng, A.A.: Pyrolysis and Bio-Oil. University of Arkansas, US Department of Agriculture and county governments cooperating (2009)
Safar, M., Lin, B.J., Chen, W.H., Langauer, D., Chang, J.S., Raclavska, H., Pétrissans, A., Rousset, P., Pétrissans, M.: Catalytic effects of potassium on biomass pyrolysis, combustion and torrefaction. Appl. Energy 235, 346–355 (2019)
Septien, S., Valin, S., Dupont, C., Peyrot, M., Salvador, S.: Effect of particle size and temperature on woody biomass fast pyrolysis at high temperature (1000–1400 c). Fuel 97, 202–210 (2012)
Shen, J., Wang, X.S., Garcia-Perez, M., Mourant, D., Rhodes, M.J., Li, C.Z.: Effects of particle size on the fast pyrolysis of oil mallee woody biomass. Fuel 88(10), 1810–1817 (2009)
Solomon, P.R., Fletcher, T.H.: Impact of coal pyrolysis on combustion. Symp. (Int.) Combust. 25(1), 463–474 (1994)
Solomon, P.R., Hamblen, D.G., Carangelo, R.M., Serio, M.A., Deshpande, G.V.: General model of coal devolatilization. Energy Fuels 2(4), 405–422 (1988). https://doi.org/10.1021/ef00010a006
Spalding, D.: The combustion of liquid fuels. In: Symposium (International) on Combustion, vol. 4, pp. 847–864. Elsevier (1953)
Stelmachowski, M.: Thermal conversion of waste polyolefins to the mixture of hydrocarbons in the reactor with molten metal bed. Energy Convers. Manag. 51(10), 2016–2024 (2010)
Ström, H., Thunman, H.: CFD simulations of biofuel bed conversion: a submodel for the drying and devolatilization of thermally thick wood particles. Combust. Flame 160(2), 417–431 (2013). https://doi.org/10.1016/j.combustflame.2012.10.005
Syamlal, M., O’Brien, T.J., et al.: Computer simulation of bubbles in a fluidized bed. AIChE Symp. Ser. 85(270), 22–31 (1989)
Tavassoli, H., Peters, E.A.J.F., Kuipers, J.A.M.: Direct numerical simulation of fluid-particle heat transfer in fixed random arrays of non-spherical particles. Chem. Eng. Sci. 129, 42–48 (2015). https://doi.org/10.1016/j.ces.2015.02.024
Tenneti, S., Subramaniam, S.: Particle-resolved direct numerical simulation for gas-solid flow model development. Annu. Rev. Fluid Mech. 46(1), 199–230 (2014). https://doi.org/10.1146/annurev-fluid-010313-141344
Thurner, F., Mann, U.: Kinetic investigation of wood pyrolysis. Indus. Eng. Chem. Process Design Dev. 20(3), 482–488 (1981). https://doi.org/10.1021/i200014a015
Turns, S.R.: An Introduction to Combustion: Concepts and Applications, 3rd edn. McGraw-Hill (2012)
Uddin, M.N., Techato, K., Taweekun, J., Rahman, M.M., Rasul, M.G., Mahlia, T.M.I., Ashrafur, S.M.: An overview of recent developments in biomass pyrolysis technologies. Energies 11(11), 3115 (2018). https://doi.org/10.3390/en11113115
Vijayakumar, A., Sebastian, J.: Pyrolysis process to produce fuel from different types of plastic—a review. IOP Conf. Ser. Mater. Sci. Eng. 396(012), 062 (2018). https://doi.org/10.1088/1757-899X/396/1/012062
van Wachem, B., Thalberg, K., Nguyen, D., Martin de Juan, L., Remmelgas, J., Niklasson-Bjorn, I.: Analysis, modelling and simulation of the fragmentation of agglomerates. Chem. Eng. Sci. 227(115), 944 (2020). https://doi.org/10.1016/j.ces.2020.115944
Wachs, A.: Particle-scale computational approaches to model dry and saturated granular flows of non-Brownian, non-cohesive, and non-spherical rigid bodies. Acta Mech. 230(6), 1919–1980 (2019). https://doi.org/10.1007/s00707-019-02389-9
Wang, J., Ku, X., Lin, J., Yang, S.: Impact of the reactor structure on biomass pyrolysis in fluidized-bed reactors: a coarse-grained CFD-DEM study. Energy Fuels 35(12), 10035–10050 (2021). https://doi.org/10.1021/acs.energyfuels.1c01107
Wang, J., Ku, X., Yang, S.: Simulation of biomass pyrolysis in a rotary drum by coupling cfd-dem with a one-dimensional thermally thick model. Energy Fuels 36(7), 3665–3679 (2022). https://doi.org/10.1021/acs.energyfuels.2c00050
Wang, W., Lu, Y., Xu, K., Wu, K., Zhang, Z., Duan, J.: Experimental and simulated study on fluidization characteristics of particle shrinkage in a multi-chamber fluidized bed for biomass fast pyrolysis. Fuel Process. Technol. 216(106), 799 (2021). https://doi.org/10.1016/j.fuproc.2021.106799
Wang, Z., Burra, K.G., Lei, T., Gupta, A.K.: Co-pyrolysis of waste plastic and solid biomass for synergistic production of biofuels and chemicals-a review. Prog. Energy Combust. Sci. 84(100), 899 (2021)
Wei, G., Zhang, H., An, X., Xiong, B., Jiang, S.: CFD-DEM study on heat transfer characteristics and microstructure of the blast furnace raceway with ellipsoidal particles. Powder Technol. 346, 350–362 (2019)
Wen, C.Y.: Mechanics of fluidization. In: Fluid Particle Technology, Chem. Eng. Progress. Symposium Series, vol. 62, pp. 100–111 (1966)
Wilke, C.R.: Diffusional properties of multicomponent gases. Chem. Eng. Prog. 46(2), 95–104 (1950)
Williams, C.L., Westover, T.L., Emerson, R.M., Tumuluru, J.S., Li, C.: Sources of biomass feedstock variability and the potential impact on biofuels production. BioEnergy Res. 9(1), 1–14 (2016)
World Bioenergy Association: Global Bioenergy Statistics 2020. https://www.worldbioenergy.org/uploads/201210%20WBA%20GBS%202020.pdf (2020)
Wurzenberger, J.C., Wallner, S., Raupenstrauch, H., Khinast, J.G.: Thermal conversion of biomass: comprehensive reactor and particle modeling. AIChE J. 48(10), 2398–2411 (2002). https://doi.org/10.1002/aic.690481029
Xiong, Q., Zhang, J., Xu, F., Wiggins, G., Stuart Daw, C.: Coupling DAEM and CFD for simulating biomass fast pyrolysis in fluidized beds. J. Anal. Appl. Pyrol. 117, 176–181 (2016). https://doi.org/10.1016/j.jaap.2015.11.015
Xiong, Q., Yang, Y., Xu, F., Pan, Y., Zhang, J., Hong, K., Lorenzini, G., Wang, S.: Overview of computational fluid dynamics simulation of reactor-scale biomass pyrolysis. ACS Sustain. Chem. Eng. 5(4), 2783–2798 (2017). https://doi.org/10.1021/acssuschemeng.6b02634
Xiu, S., Wang, N., Yi, W., Li, B., Shahbazi, G.: Validation of kinetic parameter values for prediction of pyrolysis behaviour of corn stalks in a horizontal entrained-flow reactor. Biosys. Eng. 100(1), 79–85 (2008)
Xue, Q., Fox, R.O.: Computational modeling of biomass thermochemical conversion in fluidized beds: particle density variation and size distribution. Indust. Eng. Chem. Res. 54(16), 4084–4094 (2015)
Yang, N., Wang, W., Ge, W., Li, J.: CFD simulation of concurrent-up gas-solid flow in circulating fluidized beds with structure-dependent drag coefficient. Chem. Eng. J. 96(1–3), 71–80 (2003)
Yang, S., Wan, Z., Wang, S., Wang, H.: Reactive MP-PIC investigation of heat and mass transfer behaviors during the biomass pyrolysis in a fluidized bed reactor. J. Environ. Chem. Eng. 9(2), 105047 (2021). https://doi.org/10.1016/j.jece.2021.105047
Yildiz, G., Ronsse, F., Venderbosch, R., Rv, Duren, Kersten, S.R.A., Prins, W.: Effect of biomass ash in catalytic fast pyrolysis of pine wood. Appl. Catal. B Environ. 168–169, 203–211 (2015). https://doi.org/10.1016/j.apcatb.2014.12.044
Yin, L.J., Chen, D.Z., Wang, H., Ma, X.B., Zhou, G.M.: Simulation of an innovative reactor for waste plastics pyrolysis. Chem. Eng. J. 237, 229–235 (2014). https://doi.org/10.1016/j.cej.2013.09.114
Zastawny, M., Mallouppas, G., Zhao, F., van Wachem, B.: Derivation of drag and lift force and torque coefficients for non-spherical particles in flows. Int. J. Multiph. Flow 39, 227–239 (2012). https://doi.org/10.1016/j.ijmultiphaseflow.2011.09.004
Zhang, M., Zhang, Y., Ma, D., Li, A., Fu, W., Ji, G., Dong, J.: Numerical investigation on the heat transfer of plastic waste pyrolysis in a rotary furnace. Chem. Eng. J. 445(136), 686 (2022). https://doi.org/10.1016/j.cej.2022.136686
Zhang, X., Wang, W., Rt, Guo, Wg, Pan, Xj, Li.: Study on the effect of pyrolysis conditions on the combustion behavior and char structure evolution. ACS Omega 7(27), 23,634-23,642 (2022)
Zhao, F., van Wachem, B.: A novel Quaternion integration approach for describing the behaviour of non-spherical particles. Acta Mech. 224(12), 3091–3109 (2013). https://doi.org/10.1007/s00707-013-0914-2
Zhou, C., Zhang, Q., Arnold, L., Yang, W., Blasiak, W.: A study of the pyrolysis behaviors of pelletized recovered municipal solid waste fuels. Appl. Energy 107, 173–182 (2013)
Zhou, Z.Y., Yu, A.B., Zulli, P.: A new computational method for studying heat transfer in fluid bed reactors. Powder Technol. 197(1), 102–110 (2010). https://doi.org/10.1016/j.powtec.2009.09.002
Acknowledgements
This work is part of the Research Initiative “SmartProSys: Intelligent Process Systems for the Sustainable Production of Chemicals” funded by the Ministry for Science, Energy, Climate Protection and the Environment of the State of Saxony-Anhalt. We gratefully acknowledge this funding.
Funding
Open Access funding enabled and organized by Projekt DEAL. This work has been funded by the Ministry for Science, Energy, Climate Protection and the Environment of the State of Saxony-Anhalt.
Author information
Authors and Affiliations
Contributions
DDA, FS, and BvW wrote the main manuscript. BvW and AD-A submitted the project proposal and received the funding. AD and SK provided input for the kinetic modelling. All authors have provided input to the manuscript and reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Attanayake, D.D., Sewerin, F., Kulkarni, S. et al. Review of Modelling of Pyrolysis Processes with CFD-DEM. Flow Turbulence Combust 111, 355–408 (2023). https://doi.org/10.1007/s10494-023-00436-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10494-023-00436-z