
Abstract
To investigate the influence of fluid purity on the adsorption properties, adsorption kinetics and adsorption equilibria of two methanol samples with different water content on an activated carbon were studied. The purity of the methanol samples was 98.5% and 99.9%. Measurements were conducted at 298 K and 318 K using a magnetic suspension balance and cover a wide p/p0 range. To determine effective diffusion time constants and mass transfer coefficients, adsorption kinetics were evaluated using an isothermal and a nonisothermal Fickian diffusion model, and the linear driving force model. The pressure dependence of the kinetic parameters was studied and discussed. A small influence of sample purity on the adsorption equilibria was observed, as the purer methanol sample showed slightly higher equilibrium loadings than the less pure sample. However, significantly faster adsorption kinetics were observed for the purer sample at all temperature and pressure conditions. Compared to the less pure sample, the determined effective diffusion time constants and the mass transfer coefficients were up to 98% and 35% higher, respectively.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Adsorption properties of organic solvents are important characteristics for various applications. Since solvents are widely used in chemical industry, e.g., as basic chemicals in synthesis processes, the separation of valuable or environmentally critical gaseous solvents from exhaust gas is an indispensable process step. Through adsorption, the solvents can not only be removed from the exhaust gas stream but also be recycled at the same time [1]. In addition, solvents are used as a working fluid in adsorption refrigeration systems [2]. In both cases, adsorption equilibria and kinetics of the solvent-adsorbent system are essential information for process design. When these properties are investigated, the solvent should be used with the highest possible purity, since impurities in the solvent can influence both the adsorptive and thermophysical properties. First, the impurities can compete with the molecules of the solvent during adsorption, which can lead to distortion of equilibria and kinetics. Second, the impurities in the gaseous phase result in an inaccurate description of the thermophysical properties calculated with an equation of state (EOS) for the pure solvent, which can lead to an incorrect evaluation of the measurement results. Depending on the measurement principle, different thermophysical properties play a role. For example, the density of the gaseous phase ρgas is required for the buoyancy correction during gravimetric adsorption measurements [3] and the compressibility [4] or the non-ideality factor [5] for consideration of real gas behavior during volumetric adsorption measurements. Although it would be possible to determine the gas density using a density sinker according to the Archimedes principle directly in a gravimetric sorption system, this cannot be recommended for solvent adsorption due to the low gas densities of the solvent vapor. For gas densities much lower than 1 kg/m3 (see exemplary values in Table 1), the measurement uncertainty of the density measurement would be significantly larger than the uncertainty of an appropriate EOS. Third, an inaccurately calculated value for the saturated vapor pressure p0 results in a shifted p/p0 plot. To show these discrepancies, the variations between pure and two methanol samples containing water are exemplarily shown for ρgas and p0 in Table 1. For the pure methanol, the EOS by de Reuck and Craven [6] was chosen since it is the recommended EOS by the National Institute of Standard and Technology (NIST), for which the uncertainties in density and saturated vapor pressure are given as 0.1%. For the impure methanol samples, the unpublished mixture model by Blackham and Lemmon as implemented in the software REFRPOP 10.0 [7] was used, for which no uncertainties are given. Deviations of up to 5% can be seen for the relevant values, leading to an inaccurate analysis of the measurement data.
Studies on the influence of impurities on the adsorption properties of CO2 or natural gas are reported in literature [8, 9], but to the best of our knowledge, no investigations regarding the purity of solvents as methanol or toluene were published. However, different purity grades of solvents are frequently used in literature to measure the adsorption properties. In Table 2, several literature studies on the adsorption of methanol on different adsorbents are listed. In most of the studies, methanol samples with purities between 99.5% and 99.95% were used, but in several studies, no information on the purity or the water content of the used methanol sample was given. To degas the liquid methanol sample and to remove residual air components from the methanol reservoir, an additional pretreatment of the methanol sample by several freeze-evacuate-thaw cycles was reported by Passos et al. [10] and Wu et al. [11]. From this brief overview, it becomes apparent that different purities are frequently used and traceability is not ensured in each case.
In order to show that methanol purity has an impact on the adsorption properties, the adsorption of two methanol samples with different purity on a commercial activated carbon was investigated within this study. Adsorption equilibria and kinetics were determined at temperatures of 298 K and 318 K using a magnetic suspension balance. The intention of this study is to show that it is necessary to provide accurate information about the samples used to make the experimental studies more comprehensible. Since experimental adsorption data of pure substances, e.g., are often used for multicomponent modeling [1], impurities such as water can affect the modeling results.
2 Materials and methods
For the gravimetric adsorption measurements, a commercial activated carbon sample of the type “Norit ROZ3” supplied by Cabot Corporation and two different methanol samples were used. By volumetric adsorption measurements with N2 at T = 77.36 K and an analysis according to the theory of Brunauer et al. [12] (BET), a surface area of the activated carbon sample of SBET = 937.1 m2/g was determined. Assuming the Gurvich rule at a relative pressure ratio of p/p0 = 0.99, a pore volume of vBET = 0.491 cm3/g was obtained. Volumetric CO2 measurements at T = 273.15 K were analyzed according to the combination of the theory of Dubinin and Astakhov [13] (DA) with the extended equation by Medek [14], for which the detailed procedure is described by Wedler and Span [15], leading to a microporous surface area of SDA = 661.2 m2/g and a pore volume of vDA = 0.457 cm3/g. In addition to these classical analysis methods, nonlocal density functional theory (NLDFT) calculations were also performed to determine the surface area and the pore volume. For this purpose, the CO2 and the N2 data were analyzed simultaneously using a dual gas analysis, as recommended by Jagiello et al. [16]. The 2D-NLDFT models for porous carbonaceous materials with a heterogeneous pore surface [17, 18] were used, as they are implemented in the analysis tool SAIEUS by Micromeritics. This analysis has the advantage that the entire pore range can be reliably covered by the simultaneous analysis of the measurements with both gases. A surface area of SNLDFT = 603.5 m2/g and a pore volume of vNLDFT = 0.428 cm3/g were obtained, which means that the classical analysis methods might overestimate the pore structure slightly.
The two methanol samples used within this study were supplied by VWR Chemicals and their specifications are listed in Table 3. Sample 1 (M1) is a so-called anhydrous sample with a purity above 99.9%, whereas sample 2 (M2) is of technical grade with a purity above 98.5%. In the analysis certificate provided by the manufacturer for both samples, no information on whether the percentage values are provided in mol%, wt%, or vol% is given. It is only noted that the values are the result of a GC analysis. Therefore, a new GC analysis was performed, for which the samples were decanted in the way described in section 2.1. In the chromatograms, only the peaks for methanol and water were observed, from which it can be concluded that there are no other notable impurities in both samples. However, since no 100% pure methanol sample exists as a reference sample, different dilutions of methanol and water were used as references. Thus, the resulting values should be considered as an estimate rather than a true value. The estimation shows that M2 might be purer than specified by the manufacturer.
2.1 Methanol preparation
Before conducting the adsorption measurements, the methanol samples had to be decanted from the original glass bottle to a small stainless-steel flask, with an inner volume of 25 cm3. For both samples, different decanting and degassing procedures were applied.
Since the anhydrous sample M1 should retain a high purity to minimize the impact of impurities on the adsorption, contact of the sample with humidity in the ambient air during the process of decanting had to be prevented as far as possible. In order to avoid possible contamination, the sample was decanted within the argon atmosphere of a glove-box, which was manufactured in-house. The glove box was continuously flushed with argon and an atmosphere with a relative humidity below 5% was ensured. Beforehand, the flask was evacuated using a rotary vane pump and afterward moved into the glove-box. 20 cm3 of the sample was then filled into the flask and the flask was closed by a valve. To remove the approximately 5 cm3 of argon and other possible contaminations from the flask, the flask was successive frozen by using liquid nitrogen at T = 77.36 K and then evacuated for 45 min [19]. After the thawing of the methanol, the freeze-evacuate-thaw cycle was repeated two times.
To compare the impact of different purity of the methanol sample on the adsorption process, M2 with its lower purity was intentionally decanted at atmospheric conditions. 20 cm3 of the methanol sample M2 was filled into the flask under ambient conditions without evacuating the flask before. Consequently, an input of impurities as water from the ambient air was likely. To enable a required evacuated state in the flask for the adsorption measurement, the 5 cm3 of ambient air had to be removed from the flask. In contrast to M1, the flask was directly frozen by using liquid nitrogen at T = 77.36 K and evacuated only once for around 5 min.
2.2 Gravimetric sorption system and measurement procedure
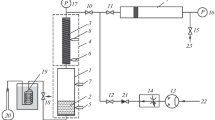
The gravimetric sorption system shown in Fig. 1 is based on a magnetic suspension balance (ISOSORP) of Rubotherm (since 2016 TA Instruments). The mass of the permanent magnet, the lifting cage, the lifting rod, the sample container, the sample, and optionally the sinker in the measuring cell is transmitted contactless via an electromagnetic field to a microbalance, located at ambient conditions. By changing the electromagnetic field, different parts inside the measuring cell are weight: at zero point (ZP), only the permanent magnet (including the sensor core) is lifted. At measuring point 1 (MP1), the permanent magnet lifts the lifting cage, the lifting rod, and the sample container including the solid sample. At measuring point 2 (MP2), a density sinker is lifted as well. For the adsorption measurements, the difference between ZP and MP1 is relevant, whereby the masses and volumes of the individual parts should be determined beforehand in detail. The masses and volumes of these metal parts were determined by precise weighing with an analytical balance at ambient conditions and with a hydrostatic comparator, according to the principle described by McLinden and Splett [20], and are shown in Table 4. For the adsorption measurement of saturated vapors, a vapor–liquid-equilibrium (VLE) cell is temperature-controlled by a bath thermostat and the adsorption measurements are performed with the gaseous phase of the VLE. The methanol-filled stainless-steel flask described in section 2.1 was used as VLE cell. The pressure p of the vapor depends on the temperature in the VLE cell. To ensure a constant temperature, the VLE cell is surrounded by a copper block in the bath. The temperature TVLE of the copper block is measured by a resistance thermometer and is assumed to be the temperature of the VLE. The temperature of the measuring cell is controlled by an additional circulating thermostat and the gas temperature Tads is measured by a resistance thermometer closely to the sample container; the gas temperature is considered the adsorption temperature. To prevent possible condensation of the vapor in the piping between measuring and VLE cell, the pipes are electrically heated. For the vapor adsorption measurements, a pressure sensor with a relatively low-pressure range of up to 0.25 MPa is used. Another pressure sensor with a range of up to 6 MPa can be used for buoyancy measurements with helium to determine the volume of the sample.

Schematic of the gravimetric sorption system, shown at measuring point 1 (MP1)
The data for adsorption equilibria and kinetics were both obtained from the same experiment. The measurements were conducted along adsorption isotherms at 298 K and 318 K. Reproducibility measurements were conducted for a number of measuring points at 318 K. The general measurement procedure was as follows: (a) the sample material was degassed at 473 K for 6 h using a rotary vane pump, (b) TVLE was set to 263.15 K and Tads to 298 K or 318 K, while the valve (V1) between measurement cell and VLE cell was closed, (c) the sample mass ms was determined in the evacuated measuring cell, (d) V1 was opened, the pressure in the measuring cell increased, and the adsorption kinetics were recorded with a data logging interval of 1.5 s, (e) adsorption equilibrium has been reached and recorded, (f) V1 was closed, TVLE was increased (in most cases by 5 K), and (g) the steps (d), (e), and (f) were repeated up to the last pressure point of the isotherm. From this cumulative measurement procedure, it becomes apparent, that the adsorption kinetics are not measured starting in an evacuated state, but are based on the previous adsorption equilibrium.
In order to determine the adsorbed mass mads, the mass balance of the magnetic suspension system in Eq. (1) has to be solved. For this purpose, the weighing signals W in MP1 (1) and ZP (0) are measured. m01 and V01 are mass and volume, respectively, of the additionally lifted parts when switching from ZP to MP1: the sample (s), the sample container (c), the lifting rod (lr), and the lifting cage (lc). The volume of the sample Vs was determined by helium buoyancy measurements at pressures from (1 to 5) MPa and a temperature of 348 K, for which the detailed procedure is described elsewhere [21]. The temperature of 348 K was chosen to minimize the possible adsorption of helium on the sample [22]. The mass of the sample ms was measured in the gravimetric sorption system at evacuated state. The values for Vs and ms are shown in Table 4. For all adsorption measurements within this study, the same sample mass was used since the system was not opened during the investigations.
To consider the buoyancy of the lifted volume V01, the density of gaseous methanol ρgas,EOS is calculated with the equation of state (EOS) by de Reuck and Craven [6] as implemented in the software package TREND 5.0 [23]. In addition, the mass balance must be corrected by the balance calibration factor α and the coupling factor ϕ considering the force transmission error (FTE) of the magnetic suspension coupling. As suggested by Kleinrahm et al. [24], a value of 1.00015 for α was used, which corrects the calibration of the balance in air.
2.3 Determination of the force transmission error
The electromagnetic field used for force transmission is influenced by the magnetic properties of the coupling house and the measuring gas, which are magnetically not entirely neutral. Therefore, the mass balance has to be corrected by the coupling factor ϕ, considering a constant apparatus contribution εvac,01 and a fluid contribution εfluid,01 depending on the density of the gas in the measuring cell (see Eq. 5) [24]. At evacuated state, the apparatus contribution can be determined according to Eq. (6) by weighing the well-known mass of the parts lifted between ZP and MP1, for which the balance calibration factor has to be considered as well (see Eq. 7). However, since the FTE also occurs during the determination of ms and Vs, εvac,01 and εfluid,01 cannot be determined while the sample is inside of the sample container. Instead of the sample, a piece of a non-magnetic metal with a mass mp comparable to the used sample (~ 1 g) and a volume Vp was used, which was calibrated in the same way as the other metal parts.
As shown in Eq. (10), the fluid contribution εfluid,01 depends on an apparatus specific constant ερ,01, the density, and the magnetic susceptibility χS,gas of the measuring gas. As reducing constants, χS,0 = 10–8 m3/kg and ρ0 = 1000 kg/m3 have to be considered. For the diamagnetic methanol, χS,gas = − 8.39327 × 10–9 m3/kg is temperature independent [25]. The apparatus-specific constant ερ,01 describes how the magnetic suspension coupling reacts to the magnetic susceptibility of the measuring gas. Therefore, density measurements with a strongly paramagnetic gas, as oxygen or oxygen mixtures as synthetic air, have to be performed [24]. Valves and sealings of the apparatus are not designed for pure oxygen atmospheres, thus, the measurements for this study were conducted with synthetic air. The measurements were conducted considering the substitutive metal part with known mass and volume. To determine an accurate value for ερ,01, several density measurements were conducted at pressures from (1 to 5) MPa and a temperature of 293.15 K and compared to the density calculated with the GERG-2008 EOS [26] as implemented in TREND 5.0 [23]. As described in Kleinrahm et al. [24], the densities as a function of p and T were determined according to Eq. (11). Therefore, as an initial value for ερ,01 a value given in Kleinrahm et al. [24] was assumed and the magnetic susceptibility of the paramagnetic air was calculated according to Kleinrahm et al. [24] as χS,air = 3.010 × 10–7 m3/kg. The deviation of the experimental density ρair,exp to the density calculated with GERG-2008 EOS is described by εfse,01 (see Eq. 12). With increasing density, εfse,01 increases linearly. The value for ερ,01 is then iteratively adjusted until \(\varepsilon_{{{\text{fse}},01}} (p = 0 \space \text{Pa}) = 0\) since there should be no deviation to the GERG-2008 EOS at zero-density. More details about this procedure can be found in Kleinrahm et al. [24].
2.4 Adsorption data evaluation
To conduct the kinetic adsorption measurements, time-dependent values have to be considered. However, it is counterproductive to switch between MP1 and ZP during the ongoing kinetic measurement since the adsorption kinetic cannot be tracked seamlessly when positions are switched. As shown in Eq. (13), the position remains constant in MP1 during the kinetic measurements and W0,eq is only recorded at the equilibrium state [21]. By rearranging the time-dependent mass balance in Eqs. (14), (15), the time-dependent adsorbed mass mads can be determined.
Considering the molecular mass of the adsorbed gas Mgas and the sample mass, the time depended adsorbed loading q is calculated according to Eq. (16). To describe the kinetics of the adsorption measurements, the fractional uptake F is calculated according to Eq. (17) considering the time-dependent loading at time t, the initial loading at time t0, and the equilibrium loading qeq. For the determination of qeq, Eqs. (15), (16) were used considering the measured values when adsorption equilibrium is reached [cf. procedure step e)]
2.5 Estimation of measurement uncertainty
The uncertainty of the gravimetric adsorption measurements was estimated according to the “Guide of the Expression of Uncertainty in Measurements” [27]. The combined standard uncertainty uc of the adsorption loading q was calculated according to Eq. (18). As described by Yang et al. [3], the uncertainty contribution of the FTE determination is negligible. Since the measurement of p and Tads has no direct influence on the data evaluation, their uncertainty is not considered in the estimation. Therefore, uc depends on the standard uncertainties of the temperature measurement in the VLE cell, the calculated density, the weighing of the balance, and the lifted volumes and the masses. However, p and Tads obviously have an influence on the adsorption loading q(p, Tads) and their standard uncertainty was estimated to be u(p) = 0.125 kPa and u(Tads) = 0.178 K.
For the data evaluation, the individual uncertainty contributions are given in Table 5. Conducting a sensitivity analysis of the individual contributions on the adsorption loading has led to the sensitivity coefficients ∂q/∂x. Under consideration of a coverage factor of k = 2, the combined expanded uncertainty of the adsorption loading Uc(q) was determined and is exemplarily shown for the adsorption of the methanol sample 2 at Tads = 318 K and p = 10.10 kPa with q = 5.377 mmol/g in Table 5. The values for the other equilibrium loadings are listed in Tables 6, 7.
2.6 Equilibrium modeling
The adsorption equilibria are described by using the Dubinin–Astakhov (DA) isotherm model according to Eq. (19), which considers the characteristic energy of adsorption E, the saturated vapor pressure p0 at measuring temperature Tads, the universal gas constant R, the Dubinin coefficient n, the pore volume ν0, and the adsorbed volume νads. The adsorbed volume can be calculated according to Eq. (20) considering the adsorbed loading and the molar density of the adsorbed phase. According to the DA theory, the density of the adsorbed phase was assumed to be the saturated-liquid density ρm,liq,EOS of the measuring fluid at measuring temperature. By adjusting the parameters v0, E, and n, the best-fit values for these adjustable parameters were determined by minimizing the root mean square deviation (RMSD) between the experimental values for νads and the modeled values from Eq. (19).
The DA theory was formulated for the adsorption of pure fluids. Although a fluid as shown in Sect. 2.1 usually contains impurities, this model is used in the literature under the assumption of a pure fluid for the adsorption of solvent vapors. Therefore, it is also assumed for the modeling that pure methanol is used; thus, the EOS by de Reuck and Craven [6] is used to calculate p0 and ρm,liq.
2.7 Kinetic modeling
In order to compare the different kinetic adsorption measurements, the isothermal Fickian diffusion model (IFD) according to Eq. (21) based on Fick’s second law of diffusion, the linear driving force model (LDF) according to Eq. (22), and a nonisothermal solution of Fick’s second law of diffusion (NFD) according to Eq. 25 were used. The IFD model is often referred to as the formulation of [28] or Boyd et al. [29] but was also described earlier by Barrer [30]. It assumes a spherical particle with a constant radius r, a constant diffusion coefficient De, IFD, and that the heat of adsorption has no influence on the adsorption kinetics. To determine the effective diffusion time constant De,IFD/r2, Eq. (21) was fitted to the experimentally determined fractional uptake F. Due to the series expansion, no exact analytical solution of Eq. (21) is feasible. Thus, terms up to n = 10 were considered in the calculations since no influence of the higher terms on the fitting procedure was observed.
The LDF model was formulated by Glueckauf and Coates [31] and describes the adsorption kinetics as a differential equation according to Eq. (22). The solution for an isobaric adsorption process was later developed by Glueckauf [32] and is given in Eq. (23) as a simple exponential function. The fractional uptake is expressed in terms of the mass transfer coefficient kLDF. As discussed by Glueckauf [32] and Ruthven [33], a relation exists between kLDF and De,IFD according to Eq. (24), for which a value of Ω = 15 was suggested as a first approximation. However, it is already known that Ω depends on T, p, and t and therefore differs from the value of 15 [21, 34].
The advantage of the two previous models is that they can be adjusted without difficulty. However, heat effects due to the heat of adsorption are neglected. Especially with larger sample quantities, heat effects can have a significant influence on the adsorption kinetics. The experimental data was therefore also used to fit the more comprehensive NFD model according to Eq. 25, which considers heat effects and diffusion limiting bed effects [35]. The constraints are given in Eqs. (26–28), in which h is the heat transfer coefficient between the external surface area S of the adsorbent and the gas phase, ΔH is the heat of adsorption, cs and ρs are the heat capacity and the density of the adsorbent, and q* is the adsorbed phase concentration at equilibrium state. The number of terms considered for the calculation was also set as n = 10.
3 Results and discussion
3.1 Equilibrium
The results of the adsorption equilibria measurements are shown for both methanol samples at temperatures of 298 K and 318 K in Fig. 2. The numerical values for the adsorption equilibria with the respective combined expanded uncertainty of the data points are also given in Table 6 for sample M1 and in Table 7 for sample M2. In Fig. 2a, equilibrium loadings are plotted in regard to the absolute pressure, showing a pronounced adsorption for both samples at the lower temperature of 298 K. However, if they are plotted in regard to the respective vapor pressure ratio p/(p0 (Tads)) (see Fig. 2b), the differences between the two temperatures become considerably smaller. Regarding the influence of the two methanol samples, no significant difference can be seen in both plots of Fig. 2.

Adsorption equilibria loadings for both methanol samples (M1 and M2), determined isothermally at 298 K and 318 K. Data are shown in regard to absolute pressure p (a) and vapor pressure ratio p/p0 (b)
Nevertheless, by comparing the values listed in Tables 6, 7, differences between the methanol samples become noticeable. For the measurements conducted with M1 at 318 K and pressures up to 10 kPa, adsorption loadings were found to be ~ 0.1 mmol/g higher than for the sample M2, while this deviation is decreasing with a further increase in pressure. Although the deviation seems to be small, they lie outside of the determined measurement uncertainty range and, e.g., result in relative deviations of up to 5% for the measurements at 2 kPa and 3 kPa. Besides, it should be noted that the pressure for the measurements with M1 was in general slightly lower than with M2, which should actually result in lower adsorption loadings for sample M1. The repeated measurements at 318 K show that the data is reproducible. Deviations observed for repeated measurements are within the calculated measurement uncertainty or can be explained by slight differences in pressure. For the adsorption measurements at 298 K, adsorption loadings were found to be (0.04–0.08) mmol/g higher for the M2 sample. Due to higher adsorption at 298 K, the resulting relative deviations are below 1.4%. But in this case, the deviation can be explained by slightly higher pressures at the measurements with M2 (see Fig. 2).
The differences might be related to two different effects. On the one hand, the true saturated vapor pressure p0 of both samples may differ from the value calculated with the EOS. Since both samples contain different amounts of water, the true values of both samples are different (cf. Table 1), leading to a shifted x-axis in Fig. 2b. On the other hand, the adsorption of water might have a stronger temperature dependence than the adsorption of pure methanol, resulting in more pronounced deviations at the higher measurement temperature.
The adsorption data sets were used to adapt the Dubinin-Astakhov isotherm model. Since the model requires a constant adsorption temperature, the arithmetic temperature mean of the data points was used and the resulting best-fit parameters for the pore volume v0, the characteristic energy of adsorption E, and the Dubinin coefficient n are given in Table 8. The deviations of values for v0 and E for both methanol samples differ only slightly, but at both temperatures, the values for M2 are smaller. For the Dubinin coefficient, the deviations are more pronounced, especially comparing the parameters for 298 K. With values for the pore volume v0 between 0.469 cm3/g and 0.481 cm3/g, the results are quite close to the values of the N2-BET analysis (0.491 cm3/g) and the CO2-DA analysis (0.457 cm3/g), and slightly higher than the NLDFT result (0.428 cm3/g). The overall good agreement with these data confirms the reliability of the methanol measurement data.
3.2 Kinetics
The kinetics of adsorption were recorded for the different adsorption measurements. For most of the adsorption experiments, the kinetics could be analyzed and compared. An overview of the evaluated kinetics can be found in Table 9, in which the effective diffusion time constants De/r2 and the mass transfer coefficients kLDF for the different measurements are listed. Due to different reasons, the kinetics of a few measurements could not be properly analyzed. The measurements for the first value of each isotherm were started at an evacuated state in the measuring cell and by cooling down the VLE cell, thus, the data is not comparable with the other measurements. For the last two measurements at 298 K and the last four measurements at 318 K, the difference between Tads and TVLE was rather small, which has led to discontinuities during the increase of pressure and mass. Therefore, no continuous kinetic curve could be recorded and the data were also not comparable to the other measurements. For the kinetic curve at p/p0 = 0.504 at 318 K, a continuous data evaluation was also not possible, thus, the data from the reproduced measurement series were taken for comparison.
The time-dependent fractional uptake (see Eq. 17) is shown for different pressures at 298 K and 318 K in Fig. 2. In all cases, the fractional uptake of sample M1 increases faster than of sample M2. With increasing pressure, the kinetics become in general faster and at the same time the differences between the two samples decrease. The faster kinetics can be explained due to the decreasing values for the adsorption loading differences Δq between the individual pressure steps since less additional adsorption takes place. However, the kinetics at a temperature of 318 K and a pressure of 7.6 kPa and 7.7 kPa, respectively (Fig. 3d), show a slower increase than for the pressure of 3.0 kPa and 3.1 kPa, respectively (see Fig. 3b).

Fractional uptake curves with a logarithmic time scale for the two methanol samples at different temperature and pressure conditions: a Tads = 298 K and p ≈ 3 kPa, b Tads = 318 K and p ≈ 3 kPa, c Tads = 298 K and p ≈ 8 kPa, d Tads = 318 K and p ≈ 8 kPa, e Tads = 298 K and p ≈ 13 kPa, f Tads = 318 K and p ≈ 13 kPa, and g Tads = 318 K and p ≈ 22 kPa
The kinetic data were used to adjust the isothermal Fickian diffusion model (IFD), the linear driving force model (LDF), and the nonisothermal Fickian diffusion model (NFD). For the adsorption of the sample M1 at 318 K, the modeled curves are exemplarily shown for four pressure points in Fig. 4. It can be seen clearly that the LDF model describes the adsorption kinetics by far the worst. The model significantly underestimates the kinetics in the first 500 s; subsequently, it overestimates the kinetics and reaches the adsorption equilibrium too early.

Modeled kinetic curves using the isothermal model (IFD), the linear driving force model (LDF), and the nonisothermal model (NFD) compared with the experimental data for the sample M1 at Tads = 318 K and p ≈ 4.2 kPa (a), p ≈ 7.6 kPa (b), p ≈ 13.2 kPa (c), and p ≈ 22.1 kPa (d)
The IFD and the NFD models describe the kinetics more accurately, but both show different characteristics. The NFD model describes the course of the first 1000 s accurately, while the IFD model shows there some deviations. Since a relatively large sample mass was used for the experiments (see Table 4) and the adsorbed loading is quite large, it can be assumed that the heat of adsorption and the sample loading have a significant influence on the adsorption kinetics. The comparatively strong increase at the beginning of the adsorption process strengthens this assumption. Therefore, the NFD model can describe the initial phase much more accurately than the IFD model. As time progresses (t > 1000 s), the goodness of fit of the IFD model improves. Since the adsorption is less pronounced with increasing time, the heat of adsorption also decreases; thus, the later course of the adsorption kinetics can be well described by the isothermal IFD model, whereas the NFD model partially reaches the adsorption equilibrium too early.
As a result of the kinetic modeling, effective diffusion time constants and mass transfer coefficients were obtained, which are shown in Fig. 5 in regard to the pressure ratio p/p0. These values confirm the observations regarding the differences between the two methanol samples from the kinetic plots in Fig. 3 since the values for De,IFD/r2, kLDF, and De,NFD/r2 are higher for the purer methanol sample M1. The values for De,IFD/r2 are up to 55% higher for sample M1 than for M2, and the values for kLDF and De,NFD/r2 are up to 35% and 98% higher, respectively. For both temperatures, a consistently faster kinetic is found for M1 than for M2 and the differences are in general more pronounced for the kinetics at 298 K. Since the LDF model poorly reproduced the experimental data, the values for kLDF should be taken with caution. Nevertheless, the mass transfer coefficients show with respect to temperature dependence, pressure dependence, and sample purity a similar course as the effective diffusion time constants.

Effective diffusion time constants De,IFD/r2 for the isothermal model (a), mass transfer coefficients kLDF (b), and effective diffusion time constants De,NFD/r2 for the nonisothermal model (c) in regard to the vapor pressure ratio p/p0 for both methanol samples at 298 K and 318 K
In general, higher effective diffusion time constants and mass transfer coefficients were obtained at the higher temperature of 318 K, which agrees with the temperature dependence of the well-known kinetic theory of gases. Regarding the pressure dependence, a more complex behavior with increasing pressures was observed since up to p/p0 = 0.2 the values for the kinetic parameters at 318 K decrease with increasing pressure; at higher relative pressure they increase with pressure. For the measurements at 298 K only one measurement with a pressure ratio below 0.2 was evaluable, thus, this pressure behavior cannot be confirmed based on these data. However, a decrease of the kinetic parameter is not to be expected since in this pressure range the adsorption isotherms have an almost linear course (see Fig. 2). The decrease might indicate that the high sample mass affects the adsorption kinetics even more than can be compensated by the NFD model. This should be investigated in more detail in future experiments with different sample masses, as suggested in the comprehensive review by Wang et al. [36]. Nevertheless, different slopes for the pressure dependence of effective diffusion time constants or mass transfer coefficient were reported in the literature for a variety of gases, but to the best of our knowledge, no study on the dependence of methanol exists for comparison. As some examples, Bae and Lee [37], Saha and Deng [38], Wedler et al. [39], and Maghsoudi et al. [40] measured values increasing with increasing pressure for N2, O2, Ar, CO, CO2, CH4, SO2, NH3, C3H8O2, and C3H8, but decreasing values were obtained by Zhang et al. [41], Saha and Deng [38], and Yang and Liu [42] for CO2, CH4, and N2O. No consistent slope for the pressure dependence of N2, CO, CO2, and CH4 was observed by Xiao et al. [43] and Park et al. [44]. However, the studies mostly investigated very limited absolute pressure ranges and did not analyze them with respect to the p/p0 ratio. Furthermore, in some of the studies, it is not traceably described whether cumulative measurements or measurements starting from vacuum were performed. Both could be a reason for the differing behavior of gases (e.g., for CO2 and CH4) between the studies. The results in Fig. 5 show that the different observations in literature might be explained by different investigated pressure ranges and it becomes apparent that the pressure dependence of kinetic adsorption parameters should be investigated over a wide p/p0 range.
4 Conclusions
The measurements with two different methanol samples within this study show that adsorption equilibria and in particular adsorption kinetics are influenced by the water content of the chosen methanol sample. By comparing adsorption equilibrium isotherms for methanol samples with a purity of 99.9% and 98.5%, deviations for the adsorbed equilibrium loading of up to 5% were found between both samples. For the adsorption kinetics, the differences between the two samples become even more obvious. The kinetics of the less pure methanol sample M2 are noticeably slower, as shown by the curves of the fractional uptake. The effective diffusion time constants De,IFD/r2 and De,NFD/r2 determined by the IFD and NFD model and the mass transfer coefficient kLDF determined by the LDF model are significantly higher for the purer sample M1; for De,IFD/r2 up to 55%, for De,NFD/r2 up to 98%, and for kLDF up to 35%. However, the LDF model could only poorly describe the experimental data of the adsorption kinetics; the results of the IFD and NFD models were more accurate. Especially in the first 1000 s, the NFD model was able to describe the kinetics most accurately since the released heat of adsorption is considered within the model.
The equilibrium and the kinetic results prove that samples should be used as pure as possible, otherwise significant deviations can occur due to the presence of water, which has an impact on the design of the industrial adsorption process. The following two reasons for the deviations should be considered: (a) the water in the sample has different adsorption behavior than pure methanol in regard to adsorption kinetics and temperature and pressure dependence and (b) the true saturated vapor pressure p0 of the samples differ from each other (cf. Table 1) and lead to an incorrect interpretation of the adsorption isotherms. In the future, further studies on the influence of sample purity and water content should be performed using other fluids with low dew points such as toluene, ethanol, or acetone.
In addition to the investigations on the influence of sample purity, the pressure dependence of the kinetic parameters was studied. In literature, the pressure dependence is described in a supposedly contradictory manner, increasing, and decreasing kinetic values with rising pressure are reported. The results from this study showed for p/p0 below 0.2 decreasing values for the kinetic parameter and increasing parameters with increasing pressure. However, the decrease at the low pressures was actually not to be expected since the adsorption isotherms in this pressure range show a linear course. In future studies, the pressure dependence of the kinetic parameters should be further investigated using varying sample masses, to avoid the possible influence of heat and bed effects.
Data availability
Adsorption data is given in the manuscript.
References
Ushiki, I., Ota, M., Sato, Y., Inomata, H.: Prediction of VOCs adsorption equilibria on activated carbon in supercritical carbon dioxide over a wide range of temperature and pressure by using pure component adsorption data: Combined approach of the Dubinin-Astakhov equation and the non-ideal adsorbed solution theory (NIAST). Fluid Phase Equilib. 375, 293–305 (2014). https://doi.org/10.1016/j.fluid.2014.05.004
Choudhury, B., Saha, B.B., Chatterjee, P.K., Sarkar, J.P.: An overview of developments in adsorption refrigeration systems towards a sustainable way of cooling. Appl. Energy 104, 554–567 (2013). https://doi.org/10.1016/j.apenergy.2012.11.042
Yang, X., Kleinrahm, R., McLinden, M.O., Richter, M.: Uncertainty analysis of adsorption measurements using commercial gravimetric sorption analyzers with simultaneous density measurement based on a magnetic-suspension balance. Adsorption (2020). https://doi.org/10.1007/s10450-020-00236-1
Rother, J., Fieback, T., Seif, R., Dreisbach, F.: Characterization of solid and liquid sorbent materials for biogas purification by using a new volumetric screening instrument. Rev. Sci. Instrum. 83(5), 55112 (2012). https://doi.org/10.1063/1.4717681
Wedler, C., Lotz, K., Arami-Niya, A., Xiao, G., Span, R., Muhler, M., May, E.F., Richter, M.: Influence of mineral composition of chars derived by hydrothermal carbonization on sorption behavior of CO2, CH4, and O2. ACS Omega 5(19), 10704–10714 (2020). https://doi.org/10.1021/acsomega.9b04370
de Reuck, K.M., Craven, R.J.B.: International thermodynamic tables of the fluid state-12: Methanol. IUPAC chemical data series, Nr.12. Blackwell Scientific Publications, Oxford (1993)
Lemmon, E.W., Bell, I.H., Huber, M.L., McLinden, M.O.: NIST Standard Reference Database 23: Reference fluid thermodynamic and transport properties-REFPROP, version 10.0, National Institute of Standards and Technology (2018)
Anderson, C.J., Tao, W., Scholes, C.A., Stevens, G.W., Kentish, S.E.: The performance of carbon membranes in the presence of condensable and non-condensable impurities. J. Membr. Sci. 378(1–2), 117–127 (2011). https://doi.org/10.1016/j.memsci.2011.04.058
Bahamon, D., Díaz-Márquez, A., Gamallo, P., Vega, L.F.: Energetic evaluation of swing adsorption processes for CO2 capture in selected MOFs and zeolites: Effect of impurities. Chem. Eng. J. 342, 458–473 (2018). https://doi.org/10.1016/j.cej.2018.02.094
Passos, E., Meunier, F., Gianola, J.C.: Thermodynamic performance improvement of an intermittent solar-powered refrigeration cycle using adsorption of methanol on activated carbon. Heat Recovery Syst. 6(3), 259–264 (1986)
Wu, J.W., Madani, S.H., Biggs, M.J., Phillip, P., Lei, C., Hu, E.J.: Characterizations of activated carbon-methanol adsorption pair including the heat of adsorptions. J. Chem. Eng. Data 60(6), 1727–1731 (2015). https://doi.org/10.1021/je501113y
Brunauer, S., Emmett, P.H., Teller, E.: Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 60(2), 309–319 (1938)
Dubinin, M.M., Astakhov, V.A.: Development of the concepts of volume filling of micropores in the adsorption of gases and vapors by microporous adsorbents: COMMUNICATION 1. Carbon adsorbents. Russ. Chem. Bull. 20(1), 3–7 (1971). https://doi.org/10.1007/BF00849307
Medek, J.: Possibility of micropore analysis of coal and coke from the carbon dioxide isotherm. Fuel 56(2), 131–133 (1977). https://doi.org/10.1016/0016-2361(77)90131-4
Wedler, C., Span, R.: Micropore analysis of biomass chars by CO 2 adsorption: Comparison of different analysis methods. Energy Fuels (2021). https://doi.org/10.1021/acs.energyfuels.1c00280
Jagiello, J., Kenvin, J., Celzard, A., Fierro, V.: Enhanced resolution of ultra micropore size determination of biochars and activated carbons by dual gas analysis using N2 and CO2 with 2D-NLDFT adsorption models. Carbon 144, 206–215 (2019). https://doi.org/10.1016/j.carbon.2018.12.028
Jagiello, J., Olivier, J.P.: 2D-NLDFT adsorption models for carbon slit-shaped pores with surface energetical heterogeneity and geometrical corrugation. Carbon 55, 70–80 (2013). https://doi.org/10.1016/j.carbon.2012.12.011
Jagiello, J., Olivier, J.P.: Carbon slit pore model incorporating surface energetical heterogeneity and geometrical corrugation. Adsorption 19(2–4), 777–783 (2013). https://doi.org/10.1007/s10450-013-9517-4
Scholz, C.W., Frotscher, O., Pohl, S., Span, R., Richter, M.: Measurement and correlation of the (p, ρ, T) behavior of liquid methanol at temperatures from 28315 to 42315 K and pressures up to 90 MPa. Ind. Eng. Chem. Res. 60(9), 3745–3757 (2021). https://doi.org/10.1021/acs.iecr.0c06248
McLinden, M.O., Splett, J.D.: A liquid density standard over wide ranges of temperature and pressure based on toluene. J. Res. Nat. Inst. Stand. Technol. 113(1), 29–67 (2008). https://doi.org/10.6028/jres.113.005
Wedler, C., Span, R.: A pore-structure dependent kinetic adsorption model for consideration in char conversion—Adsorption kinetics of CO2 on biomass chars. Chem. Eng. Sci. 231, 116281 (2021). https://doi.org/10.1016/j.ces.2020.116281
Hwang, J., Joss, L., Pini, R.: Measuring and modelling supercritical adsorption of CO2 and CH4 on montmorillonite source clay. Microporous Mesoporous Mater. 273, 107–121 (2019). https://doi.org/10.1016/j.micromeso.2018.06.050
Span, R., Beckmüller, R., Hielscher, S., Jäger, A., Mickoleit, E., Neumann, T., Pohl, S., Semrau, B., Thol, M.: TREND thermodynamic reference and engineering data 5.0. Ruhr-Universität Bochum, Lehrstuhl für Thermodynamik (2020)
Kleinrahm, R., Yang, X., McLinden, M.O., Richter, M.: Analysis of the systematic force-transmission error of the magnetic-suspension coupling in single-sinker densimeters and commercial gravimetric sorption analyzers. Adsorption 25(4), 717–735 (2019). https://doi.org/10.1007/s10450-019-00071-z
Lide, D.R. (ed.): CRC handbook of chemistry and physics: A ready-reference book of chemical and physical data, 85th edn. CRC Press, Boca Raton (2004)
Kunz, O., Wagner, W.: The GERG-2008 wide-range equation of state for natural gases and other mixtures: An expansion of GERG-2004. J. Chem. Eng. Data 57(11), 3032–3091 (2012). https://doi.org/10.1021/je300655b
ISO/IEC Guide 98–3 Uncertainty of measurement—part 3: Guide to the expression of uncertainty in measurement. International Organization for Standardization (ISO), Geneva (2008)
Crank, J.: The mathematics of diffusion, 2nd edn. Clarendon Press, Oxford (1976)
Boyd, G.E., Adamson, A.W., Myers, L.S.: The exchange adsorption of ions from aqueous solutions by organic zeolites. II. Kinetics 1. J. Am. Chem. Soc. 69(11), 2836–2848 (1947). https://doi.org/10.1021/ja01203a066
Barrer, R.M.: Diffusion in and through solids. Cambridge University Press, Cambridge (1941)
Glueckauf, E., Coates, J.I.: Theory of chromatography; the influence of incomplete equilibrium on the front boundary of chromatograms and on the effectiveness of separation. J. Chem. Soc. (1947). https://doi.org/10.1039/JR9470001315
Glueckauf, E.: Theory of chromatography. Part 10—Formulæ for diffusion into spheres and their application to chromatography. Trans. Faraday Soc. 51, 1540–1551 (1955). https://doi.org/10.1039/TF9555101540
Ruthven, D.M.: Principles of adsorption and adsorption processes. A Wiley-interscience publication. Wiley, New York (1984)
Sircar, S., Hufton, J.R.: Why does the linear driving force model for adsorption kinetics work? Adsorption 6(2), 137–147 (2000). https://doi.org/10.1023/A:1008965317983
Ruthven, D.M., Lee, L.-K.: Kinetics of nonisothermal sorption: Systems with bed diffusion control. AIChE J. 27(4), 654–663 (1981). https://doi.org/10.1002/aic.690270418
Wang, J.-Y., Mangano, E., Brandani, S., Ruthven, D.M.: A review of common practices in gravimetric and volumetric adsorption kinetic experiments. Adsorption 27(3), 295–318 (2021). https://doi.org/10.1007/s10450-020-00276-7
Bae, Y.-S., Lee, C.-H.: Sorption kinetics of eight gases on a carbon molecular sieve at elevated pressure. Carbon 43(1), 95–107 (2005). https://doi.org/10.1016/j.carbon.2004.08.026
Saha, D., Deng, S.: Adsorption equilibrium and kinetics of CO2, CH4, N2O, and NH3 on ordered mesoporous carbon. J. Colloid. Interface Sci. 345(2), 402–409 (2010). https://doi.org/10.1016/j.jcis.2010.01.076
Wedler, C., Arami-Niya, A., Xiao, G., Span, R., May, E.F., Richter, M.: Gas diffusion and sorption in carbon conversion. Energy Procedia 158, 1792–1797 (2019). https://doi.org/10.1016/j.egypro.2019.01.422
Maghsoudi, H., Abdi, H., Aidani, A.: Temperature- and pressure-dependent adsorption equilibria and diffusivities of propylene and propane in pure-silica Si-CHA zeolite. Ind. Eng. Chem. Res. 59(4), 1682–1692 (2020). https://doi.org/10.1021/acs.iecr.9b05451
Zhang, Z., Zhang, W., Chen, X., Xia, Q., Li, Z.: Adsorption of CO2 on zeolite 13× and activated carbon with higher surface area. Sep. Sci. Technol. 45(5), 710–719 (2010). https://doi.org/10.1080/014963909035711
Yang, Y., Liu, S.: Estimation and modeling of pressure-dependent gas diffusion coefficient for coal: A fractal theory-based approach. Fuel 253, 588–606 (2019). https://doi.org/10.1016/j.fuel.2019.05.009
Xiao, G., Li, Z., Saleman, T.L., May, E.F.: Adsorption equilibria and kinetics of CH4 and N2 on commercial zeolites and carbons. Adsorption 23(1), 131–147 (2017). https://doi.org/10.1007/s10450-016-9840-7
Park, Y., Ju, Y., Park, D., Lee, C.-H.: Adsorption equilibria and kinetics of six pure gases on pelletized zeolite 13× up to 10 MPa: CO2, CO, N2, CH4, Ar and H2. Chem. Eng. J. 292, 348–365 (2016). https://doi.org/10.1016/j.cej.2016.02.046
Bandosz, T.J., Jagiełło, J., Schwarz, J.A., Krzyzanowski, A.: Effect of surface chemistry on sorption of water and methanol on activated carbons. Langmuir 12(26), 6480–6486 (1996). https://doi.org/10.1021/la960340r
Linders, M.J.G., van den Broeke, L.J.P., Kapteijn, F., Moulijn, J.A., van Bokhoven, J.J.G.M.: Binary adsorption equilibrium of organics and water on activated carbon. AIChE J. 47(8), 1885–1892 (2001)
Fletcher, A.J., Cussen, E.J., Prior, T.J., Rosseinsky, M.J., Kepert, C.J., Thomas, K.M.: Adsorption dynamics of gases and vapors on the nanoporous metal organic framework material Ni2(4,4′-bipyridine)3(NO3)4: guest modification of host sorption behavior. J. Am. Chem. Soc. 123(41), 10001–10011 (2001). https://doi.org/10.1021/ja0109895
Fletcher, A.J., Cussen, E.J., Bradshaw, D., Rosseinsky, M.J., Thomas, K.M.: Adsorption of gases and vapors on nanoporous Ni2(4,4′-Bipyridine)3(NO3)4 metal-organic framework materials templated with methanol and ethanol: structural effects in adsorption kinetics. J. Am. Chem. Soc. 126(31), 9750–9759 (2004). https://doi.org/10.1021/ja0490267
Fletcher, A.J., Yüzak, Y., Thomas, K.M.: Adsorption and desorption kinetics for hydrophilic and hydrophobic vapors on activated carbon. Carbon 44(5), 989–1004 (2006). https://doi.org/10.1016/j.carbon.2005.10.020
Wang, L.W., Wang, R.Z., Lu, Z.S., Chen, C.J., Wang, K., Wu, J.Y.: The performance of two adsorption ice making test units using activated carbon and a carbon composite as adsorbents. Carbon 44(13), 2671–2680 (2006). https://doi.org/10.1016/j.carbon.2006.04.013
El-Sharkawy, I.I., Hassan, M., Saha, B.B., Koyama, S., Nasr, M.M.: Study on adsorption of methanol onto carbon based adsorbents. Int. J. Refrig. 32(7), 1579–1586 (2009). https://doi.org/10.1016/j.ijrefrig.2009.06.011
Henninger, S.K., Schicktanz, M., Hügenell, P., Sievers, H., Henning, H.-M.: Evaluation of methanol adsorption on activated carbons for thermally driven chillers part I: Thermophysical characterisation. Int. J. Refrig 35(3), 543–553 (2012). https://doi.org/10.1016/j.ijrefrig.2011.10.004
Xiao, Y., He, G., Yuan, M.: Adsorption equilibrium and kinetics of methanol vapor on zeolites NaX, KA, and CaA and activated alumina. Ind. Eng. Chem. Res. 57(42), 14254–14260 (2018). https://doi.org/10.1021/acs.iecr.8b04076
Gao, M., Li, H., Yang, M., Gao, S., Wu, P., Tian, P., Xu, S., Ye, M., Liu, Z.: Direct quantification of surface barriers for mass transfer in nanoporous crystalline materials. Commun. Chem. (2019). https://doi.org/10.1038/s42004-019-0144-1
Acknowledgements
We thank Roland Span for providing the resources for the investigations. We also thank Christin Pflieger and Martin Muhler for the GC analysis of the two methanol samples.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MR: investigation, formal analysis, data curation, writing—original draft. CW: investigation, methodology, conceptualization, validation, visualization, supervision, project administration, writing—original draft, writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The author declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rösler, M., Wedler, C. Adsorption kinetics and equilibria of two methanol samples with different water content on activated carbon. Adsorption 27, 1175–1190 (2021). https://doi.org/10.1007/s10450-021-00341-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10450-021-00341-9