
Abstract
Available knowledge on the European white oaks (Quercus, sect. Quercus) plastome still exhibits large gaps, mostly in respect of detailed phylogeny and information from southern Europe and adjacent Near East. We investigated DNA sequence polymorphism at two plastid loci on 270 individuals representing 13 white oak species/taxa and retrieved several hundred sequences of the same two DNA regions from GenBank to fill the above gaps, and contribute to a better understanding of white oak diversity and evolution. The modern European sect. Quercus species exhibits a poorly differentiated plastid DNA, despite its current highly diversified taxonomy. Twenty-nine haplotypes were identified across the Euro-Mediterranean and Near East regions, including two ancestral variants that are westerly and south-easterly partitioned. Other high- and low-frequency haplotypes also showed a well-structured geographic distribution consistent with the current phytogeographic framework of the European continent. The climatic and geological events that characterized the end of the Tertiary and Quaternary periods played a crucial role in triggering haplotype mixing, isolation, and in offering niche opportunities for more recent diversification. Euro-Mediterranean southern territories host a high and as yet poorly studied genetic variation; the role of the Italian Peninsula as a crossroad and threshold for haplotype diversity and distribution clearly emerges from this study.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The genus Quercus is one of the main woody components of the forests in the boreal hemisphere. Its ecological dominance and the remarkable heterogeneity and biodiversity asset of its habitats endorse the importance of multidisciplinary studies to integrate ecology and evolution for a better comprehension of community assembly and adaptation processes in a changing world (Cavender-Bares et al. 2016; Kremer and Hipp 2020).
One of the Quercus major clades, section Quercus (the white oaks), includes nearly 150 species distributed throughout North America, western Eurasia, East Asia, and North Africa (Denk et al. 2017). In the American continent, where over 100 species occur, the white oaks exhibit an extremely wide morpho-physiological variation including sclerophyllous shrubby species inhabiting desert zones and dry savannahs, and lobe-leaved large dominant tree species in alluvial flatwoods, bottomlands and cold intermontane woodlands (Manos and Hipp 2021).
In Eurasia as well, shrubby and tree oak species do occur on extremely diversified niches and act as guide species in a very high number of syntaxa within all the ranks of phytosociological classification (Mucina et al. 2016). For instance, species that are more widespread, such as Quercus robur or Q. mongolica, are dominant throughout the majority of the temperate broad-leaved deciduous forest Biome and extend to the boundary of Taiga boreal forest or cold steppe Biomes (Kubitzki 1993; Menitsky 2005). Instead, xerothermic forms belonging to the Q. pubescens s.l. complex give rise to thermophilous forests adjacently to primary Oleo-Ceratonion thermo-Mediterranean shrublands (Brullo and Marcenò 1985; Blasi and Di Pietro 1998).
In addition to this great ecological amplitude, West Eurasian white oaks are characterized by a significant taxonomic complexity. In fact, the debate about the makeup of the white oaks’ species list and the definition of a shared taxonomic framework based on morpho-ecological descriptors is still very lively, especially as regards the Q. pubescens and Q. petraea collective species groups (Trinajstić 2007; Fortini et al. 2009, 2015a, 2022; Di Pietro et al. 2016; Denk et al. 2017). In addition, longstanding partially unsolved nomenclatural issues (see Amaral-Franco 1990; Di Pietro et al. 2012) and the limits that affect the concept of biological species in such a notoriously interfertile genus (Burger 1975; Antonecchia et al. 2015; Hipp 2015), greatly complicate the assessment of taxonomic and syntaxonomic frameworks at national and international scales (Wellstein and Spada 2015; Pasta et al. 2016; Grossoni et al. 2021; Kaplan et al. 2022).What is beyond doubt is that the divisive and sometimes inhomogeneous taxonomic classification and the consequent nomenclatural complexity characterizing the West Eurasian white oaks have negative repercussions for studies of living matter at various level (gene, species, population, ecosystem) involving diverse fields of research (genetics, ecology phytosociology, landscape planning and design, conservation etc.). Taxonomic contradictions and consequent nomenclatural disputes are therefore to be viewed as outcomes of the difficulties in classifying the peculiar molecular, morphological and ecological variability that white oak species and communities express at present, especially in the Euro-Mediterranean zone (Guarino et al. 2015; Fortini et al. 2015b; Piredda et al. 2021).
Biogeographic history and evolutionary legacies had a strong influence on oak species differentiation and local adaptation worldwide (Cavender-Bares 2019). It is generally assumed that the complex palaeogeographic and palaeoclimatic vicissitudes that affected western Eurasia during the Miocene and throughout the ‘turbulent’ Quaternary played a key role in setting the scene for the high white oaks’ diversity we can presently observe. It remains to be established the extent to which the aforementioned vicissitudes influenced the original amount of genetic diversity and how they combined in the different lineages and territories to drive diversification to produce such a varied and puzzling species group.
In this view, robust plastid phylogenies are essential to reveal phylogeographic patterns in closely related species, highlighting complex evolutionary phenomena, gene pools deserving attention and offering the opportunity to correlate the obtained data with the historical reconstruction of biomes, niche evolution and future landscape planning (Cavender-Bares et al. 2016; Blair 2023). However, any direct involvement of plastid phylogenies could be challenged by inaccurate methodologies (Blair 2023). For instance, when only one or few representatives of each species are investigated, thus providing little insight into the multifold issues pertinent to a species’ diversity (Backs and Ashley 2021).
Studies on the plastid DNA of the European white oaks began in the closing decade of the last millennium (Dumolin et al. 1995) and culminated with the fundamental research of Petit et al. (2002b) where six main PCR–RFLP lineages were identified across a wide extent of the European continent. From the general results presented in that large-scale research, further insights were subsequently derived, focusing on single European countries or well-defined geographic areas (e.g. Cottrell et al. 2002; Fineschi et al. 2002; Olalde et al. 2002, for Great Britain, Italy and Spain, respectively; Csaikl et al. 2002 for the Alpine Region). Since then, the majority of phylogenetic research has concentrated on the genus nucleome, with the European white oaks being only partially included (e.g. Hubert et al. 2014; see also McVay et al. 2017; Hipp et al. 2020; Denk et al. 2023), while plastid DNA investigation has been relegated to DNA barcoding projects or small-scale phylogeographic studies (e.g. Simeone et al. 2013; Ekhvaia et al. 2018; Douaihy et al. 2020). Curiously, complete plastid genome sequences of the West Eurasian white oaks has also received little attention in phylogenetic investigations. Instead, they have lagged well behind the large amount of data gathered on North American and East Asian oaks (e.g. Pham et al. 2017; Pang et al. 2019; Liu et al. 2021; Li et al. 2022). Furthermore, in contrast to sects. Ilex and Cerris, the white oaks do not even have a tentative framework phylogeny to direct well-aimed samplings for deeper genomic studies. This information gap represents a serious limitation for any attempts to outline the evolutionary history of European white oaks and propose phylogeographic patterns that might explain the highly variable taxonomical and phytosociological patterns of white oak communities. Such a limitation takes on greater significance if we consider that the majority of the quaternary glacial refugia for the temperate forest vegetation and the highest coenological diversity for the white oaks are concentrated in southern Europe (Blasi et al. 2004; Mucina et al. 2016). All this reinforces the opinion that full genome sequencing from one or just a few individuals per species should not eclipse studies conducted using few but well-defined marker regions, especially if the latter are phylogenetically informative and the sampling design is well aimed and dense. For example, all the major intra- and interclade relationships that have recently been highlighted by means of complete plastome sequence of sect. Ilex oaks (Yang et al. 2021; Zhou et al. 2022) were already disclosed in previous studies based on just two markers (trnH-psbA, trnK-matK) and exhaustive samplings (Simeone et al. 2016; Vitelli et al. 2017).
In this work, we have tried to tackle this issue by maximizing taxonomic and geographic sampling in order to: (i) reconstruct phylogenetic relationships of the West Eurasian white oaks plastid DNA; (ii) improve our understanding of the phylogeography of species, populations and areas; (iii) capture rare genetic variants that could subtend divergent evolutionary lineages. With this aim, we have selected two marker regions that proved their efficacy in previous oak studies and partially compensating for the absence of wider samplings taking advantage of the data available in the GenBank repository, thereby expanding the taxonomic and biogeographic breadth of the investigated dataset.
Materials and methods
Sampling design
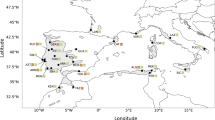
The study was carried out over a vast area of the Euro-Mediterranean region with some extensions in central Europe and North Africa (Fig. 1). Largely focused on Italy, the sampling (‘primary dataset’) also included ten additional countries: Austria, Bulgaria, Croatia, Czech Republic, France, Greece, Romania, Serbia, Spain and Morocco. Where possible, the collection sites were selected prioritizing stands of taxonomic or biological relevance (e.g. loci classici, protected areas and sites where phytosociological descriptions of forest vegetation were available). The following accepted (www.ipni.org; www.powo.science.kew.org) oak species were investigated: Quercus robur L., Q. petraea (Matt.) Liebl. (including Q. petraea subsp. austrotyrrhenica Brullo, Guarino & Siracusa), Q. frainetto Ten., Q. pubescens Willd., Q. dalechampii Ten., Q. faginea Lam., Q. pyrenaica Willd., Q. congesta C.Presl and Q. ichnusae Mossa, Bacch. & Brullo. In addition three other species were included in our dataset although these are considered as synonyms of other taxa in POWO. These are Quercus banatus P.Kucera (name recently proposed as replacing the name Q. dalechampii Ten. in SE Europe in the collective group of Q. petraea), Q. virgiliana (Ten.) Ten. (name largely used in the, national floras checklists, phytosociological descriptions and syntaxonomic frameworks of several S European countries) and Q. leptobalana Guss (accepted as valid name in Pignatti et al. 2017 last edition of Flora of Italy and guide species of the association Quercetum leptobalanae Brullo & Marcenò 1985). In fact, diverse papers published in the last decade (Bock and Tison 2012; Di Pietro et al. 2012, 2016, 2020b; Von Raab-Straube and Raus 2013; Kučera 2018; Fortini et al. 2022) aimed at a critical analysis of the taxonomy of Q. pubescens and Q. petraea collective groups, exhibited a tendency towards a reduction in the number of oak taxa. On the other hand, the taxonomic arrangement within these two collective groups is far from being fully defined. For this reason, in order not to lose information on local oak forest diversity, we have preferred to make reference to the names currently reported in the national floras or in phytosociological synthesis already published in the sites of collection or in surrounding areas (see Table 2). However a detailed description of the investigated dataset together with a direct reference to the IPNI/POWO nomenclature is reported in supplementary file 1. Three oak individuals per population collected at least 30 m apart from each other and 90 total populations were sampled. These populations are divided into the following taxa: Quercus banatus (1), Q. congesta (9), Q. dalechampii (8), Q. faginea (3), Q. frainetto (6), Q. ichnusae (3), Q. leptobalana (1), Q. petraea (12), Q. petraea subsp. austrotyrrhena (2), Q. pubescens (26), Q. pyrenaica (1), Q. robur (11), Q. virgiliana (7). The collected specimens were identified through the use of analytical keys present in the national floras. In the case of dubious or particularly critical specimens, reference was made to already published floristic-phytosociological papers concerning the collection sites (where present) and the expert knowledge of the authors. Voucher specimens (ID number reported in Supplementary file S1) are deposited at the herbarium of the University of Molise (IS; Thiers 2016).

Geographical distribution of the collection sites of white oaks populations (black dots). Countries from which accessible sequences on GenBank were retrieved and used in our analyses are filled in light grey colour (China, Japan and S-Korea are not reported in this map)
DNA extraction, sequencing and editing
DNA (270 total samples) was extracted with the NucleoSpin™ Plant II Kit (Macherey–Nagel) from silica gel dried leaves, following the manufacturer’s instructions. TrnH-psbA intergenic spacer and a portion of the trnK-matK region (3’ intron and partial gene sequence) were chosen because of their high number of accessible sequences on GenBank, and the variability displayed in previous studies (e.g. Okaura et al. 2007; Manos et al. 2008; Simeone et al. 2013, 2016). Primers and PCR conditions were as in Piredda et al. (2011). PCR products were purified with Illustra GFX PCR DNA Purification Kit (GE Healthcare) and standardized aliquots were sent to Macrogen Europe (https://www.macrogen-europe.com/) for bi directional sequencing. Electropherograms were edited with Chromas 2.6.2 (https://www.technelysium.com.au) and checked visually. Multiple alignments of the single and combined plastid regions were generated with MEGA X (Kumar et al. 2018) and adjusted manually.
In order to set our data in a broader diversity and phylogenetic context, GenBank was explored for all members of subgenus Quercus and West Eurasian subgen. Cerris sequenced with trnH-psbA and trnK-matK. Only single individuals sequenced with both markers were retrieved and used in the downstream analyses, with the exception of the oaks of sect. Cerris, for which the scarcity of trnK-matK sequences did not allow the creation of a consistent dataset. Haplotype lists and the main diversity parameters of the investigated markers were computed with DnaSP v.6 (Rozas et al. 2017).
Finally, the generated haplotypes were blasted against 244 Quercus complete chloroplast genomes available on GenBank (accessed on Jan. 31, 2023).
Data analyses
Phylogenetic tree inference and bootstrap analyses were performed under maximum likelihood with RAxML v.8.2.11 (Stamatakis 2014). We used the GTR + CAT approximation model and the ‘extended majority-rule consensus’ criterion as bootstopping option (Pattengale et al. 2009), with up to 1000 bootstrap (BS) pseudoreplicates to assess branch support (BS). The CAT model is a computational work–around for the widely used General Time Reversible model of nucleotide substitution under the Gamma model of rate heterogeneity (GTR-Γ). Compared to GTR-Γ, it has the advantage of significantly lower memory consumption, faster inference times and superior likelihood values in the obtained trees (Stamatakis 2006, 2014). The output (78-tip tree, after the removal of the identical sequences) included six accessions of subgen. Cerris (sects. Cerris, Cyclobalanopsis and Ilex; this latter including all the currently identified main lineages: East Asian, West Asia–Himalaya–East Asia ('WAHEA’) and Euro-Med; Simeone et al. 2016), was rooted between the two subgenera following Zhou et al. (2022) and imported in iTOL (www.itol.embl.de) for visualization and labelling. A planar (equal angle, parameters set to default) split graph was generated with the Neighbor-net (NNet) algorithm (Bryant and Moulton 2004) implemented in SplitsTree4 (Huson and Bryant 2006), based on the pairwise uncorrected-p (‘Hamming’) distance matrix estimated with the same program. Median Joining (MJ) haplotype networks were run with Network 4.6.1.1 (http://www.fluxus-engineering.com/), treating gaps as 5th state and using the MJ algorithm with default parameters (equal weight of transversion/transition).
Results
trnH-psbA and trnK-matK sequence data
The primary data (Supplementary file S1) comprised 270 individuals belonging to the Euro-Mediterranean sect. Quercus newly sequenced with trnH-psbA and trnK-matK. Sequence quality was high for both marker regions and unambiguous electropherograms were obtained for 100% of the investigated samples. GenBank searches extended the West Eurasian sect. Quercus dataset to 425 individuals (22 species, see supplementary files S1, S2) sequenced with the same markers, including 89 Caucasian (Ekhvaia et al. 2018), 23 Lebanese (Douaihy et al. 2020) and further 43 European, Mediterranean and Near East accessions from works mostly focused on DNA barcoding or phylogenetic studies on other Quercus sections (Simeone et al. 2013, 2016).
A wider evolutionary contextualization of the white oaks was achieved by expanding the phylogenetic analyses with 81 GenBank accessions of Asian sect. Quercus, eight Caucasian samples belonging to sect. Ponticae and 31 accessions of American members of subgen. Quercus (sects. Quercus, Lobatae, Protobalanus, Virentes and Ponticae; Simeone et al. 2016; Ekhvaia et al. 2018; Yang et al. 2020). Over 500 trnH-psbA and trnK-matK GenBank sequences of West Eurasian sects. Cerris and Ilex oaks (14–16 species) were retrieved to compare the molecular differentiation estimates found in the Eurasian white oaks’ dataset.
All generated multiple alignments were straightforward. The two-marker combination in the primary dataset was 1147 bp long. Trimming uneven GenBank sequence ends and coding indels longer than 1 bp as single binary characters produced a 1068 bp long matrix in the Eurasian oaks of sect. Quercus. A trnH-psbA inversion of 34 bp occurring in 14 North American oak individuals was replaced with its reverse complementary sequence and a binary character was inserted to keep record of it. The final alignment including all members of subgen. Quercus and six accessions of Eurasian subgen. Cerris was 1252 bp long.
The newly sequenced Euro-Mediterranean white oak dataset produced 14 haplotypes, scaling up to 29 after the inclusion of the GenBank West Eurasian samples. Eleven haplotypes out of the total were inter-specifically shared (up to 13 species), 16 were singlets (12 derived from GenBank) and only two were restricted to one or more populations of a single species (Q. pubescens from Croatia) or species complex (Q. ichnusae, Q. virgiliana, Q. congesta from Sardinia). The relative haplotype frequency was very variable: besides the 16 singlets, five haplotypes appeared in between 46 and 94 individuals each. The East Eurasian oaks of sect. Quercus produced 14 haplotypes, two of which were in common with the West Eurasian white oaks, four were inter-specifically shared (up to six species) and eight were intra-specifically shared. In total, 41 trnH-psbA + trnK-matK haplotypes were detected in the Eurasian sect. Quercus. Members of North American subgen Quercus, Eurasian sect. Ponticae and subgen. Cerris generated 37 additional haplotypes. All GenBank accessions, taxonomic identities, geographic origins, and haplotypes are reported in Supplementary file S2.
Phylogenetic setting of the Eurasian white oaks
The 78-tip RAxML tree (Fig. 2), rooted between subgen. Cerris (here represented by members of sections Cyclobalanopsis, Cerris and Ilex) and subgen. Quercus, reports the well-acknowledged sectional and lineage differentiation within each subgenus. In subgen. Cerris, the two Euro-Med members of sect. Ilex (Q. ilex and Q. coccifera from Spain and North Africa) are included in the Euro-Med lineage, slightly diverging from the other members of the same section (Q. alnifolia and Q. baroni, belonging to the WAHEA and the East Asian lineages, respectively), and from the two members of sect. Cerris (Q. cerris) and sect. Cyclobalanopsis (Q. acuta) forming a minor subclade (BS = 57–62). In subgen. Quercus, five clades are produced, with medium high support (BS = 60–100). Four clades include all North American oaks of subgen. Quercus (sects. Quercus, Lobatae, Protobalanus, Virentes, Ponticae) and the last one includes their Eurasian counterparts (sects. Quercus and Ponticae).

Maximum likelihood phylogram of the trnH-psbA + trnK-matK concatenated regions of the investigated West Eurasian sect. Quercus dataset, integrated with GenBank haplotypes of subgen. Quercus and representatives of West Eurasian subgen. Cerris. Bootstrap support (> 50) values are reported above branches. Colouration refers to the major taxonomic and geographic affiliations of specimens
Besides the major geographic split within sects. Quercus and Ponticae, some sectional misplacements emerged. These involve a few members of sect. Virentes and Lobatae (placed in the sect. Quercus subclade), and the two only American and Eurasian surviving members of sect. Ponticae (respectively inserted in sects. Protobalanus and Eurasian Quercus). The Eurasian white oak clade (BS = 99) is highly unresolved: with the only exceptions of five minor subclades including Q. pontica (BS = 96, 58), two East Asian oaks (BS = 80), and two local species groups from Lebanon and south–central Italy (BS = 64–67), no consistent differentiation could be observed at the taxonomic or geographic level, even between Western and Eastern Eurasian samples. Differentiation of a clade-basal group of sequences collecting both single and shared West and East Eurasian haplotypes is possible but low-supported (BS < 50).
The obtained RAxML topology is clearly mirrored by the Neighbor net graph shown in Fig. 3. All members of subgen. Cerris are connected to the North American cluster, with the two diverging Euro-Med members of sect. Ilex showing more affinity with the Eurasian sect. Quercus. Except for a few divergent sequences, all the Eurasian white oak haplotypes are highly mixed and organized in reticulated and little diverging clusters, with a huge trunk confirming the main split between North American and Eurasian sections Quercus and Ponticae. A composite cluster, corresponding to the low-supported clade-basal group of sequences observed in the RAxML tree, acts as the most direct connection between the two split groups.

Neighbor-Net splits graph of the trnH-psbA + trnK-matK concatenated regions of the investigated West Eurasian sect. Quercus dataset, integrated with GenBank haplotypes of subgen. Quercus and representatives of West Eurasian subgen. Cerris. Average Bootstrap support (> 50) percentages of main clusters are reported. Colouration as in Fig. 2
trnH-psbA and trnK-matK variation in Eurasian white oaks
A more detailed look into the plastid differentiation patterns of West Eurasian white oaks could be obtained with a closer inspection of the molecular diversity of the sequenced markers (Table 1) and the resulting haplotype network (Fig. 4). The two marker sequences produced in this study and retrieved from GenBank (Table 1) allow a good comparison among sections and subgenera, totalling nearly 1000 individuals belonging to 47 oak taxa. In particular, all the currently accepted species belonging to Eurasian section Quercus and West Eurasian sections Cerris and Ilex were included in our evaluation. The markers’ variation was moderate-to-low, and trnH-psbA showed higher diversity than trnK-matK across all lineages, except sect. Ilex. Both markers displayed remarkably lower values of molecular diversity in sect. Quercus than in sect. Ilex, and comparable estimates with sect. Cerris (characterized by lower numbers of individuals included).

Median Joining haplotype network of the trnH-psbA + trnK-matK concatenated regions, combining the investigated Euro-Mediterranean samples (primary dataset) and all available GenBank haplotypes of Eurasian subgen. Quercus. Colouration and symbols identify major lineages (sects. Quercus and Ponticeae) and the geographic distribution of the haplotypes (detailed in the Supplementary files S1, S2). Line thickness proportional to the number of mutations separating each haplotype (1, 3–4); dashed line corresponds to > 10 mutations. Asterisks indicate the haplotypes detected in the primary dataset (those with the highest frequency are in red)
Within sect. Quercus, the here investigated Euro-Mediterranean dataset (‘primary dataset’) was the least variable; its variation was only partially increased after the combination with the West Eurasian sequences from GenBank, that included samples from all over Europe, the Middle East, and the Caucasus region. In contrast, the East Eurasian dataset showed higher diversity, especially with trnH-psbA, and prefigures to host higher plastome diversity than the West Eurasian counterparts, due to the comparable values scored by a lower number of individuals included in the analysis. The relationships of the 41 Eurasian white oak haplotypes are reported in the haplotype network (Fig. 4). Only single mutations separate every West Eurasian sect. Quercus haplotype from the nearest, preventing the identification of any lineage, contrarily to the patterns detected in sects. Ilex and Cerris (Simeone et al. 2016, 2018). The network is rather intricate and unfolds around two major variants (labelled #1 and 2). Haplotype #1 is found in 46 individuals from 13 species and extends from Japan to Portugal, across China, Bulgaria, Greece, Cyprus, C and S Italy, France, England, Spain, Algeria and Morocco. Haplotype #2 has a lower frequency (13 individuals) and was detected in five species across Korea, Georgia (E Caucasus), Lebanon, S Italy and Sicily. Both haplotypes correspond to the basal group of sequences identified in the Eurasian white oak clade of Figs. 1 and 2. It is interesting to note that a BLAST search performed against all Quercus complete chloroplast genomes available on GenBank revealed 100% sequence identity of haplotype #1 with three accessions of Q. robur (OW028778, LT996900, England; MN562095, not specified) and one accession of Q. fabri (MK105456). Likewise, haplotype #2 matched one accession of Q. fabri (MK922346) and six accessions of Q. mongolica and Q. dentata (MK089571, MK105460, NC_043858, MK105453-105455). The closest connection with the North American sect. Quercus Ilex and Cerris clades (Figs. 1 and 2), the location at the core of the network and the wide taxonomic and geographic distribution of these two haplotypes suggest their possible ancestry (Posada and Crandall 2001). Several variants/haplotype groups directly depart from either one of these two haplotypes. Ten haplotypes from China and Korea (#4–13) form a discrete, divergent lineage, possibly comprising further sublineages (e.g. haplotypes #4, 5–12, 13); two other East Asian haplotypes are either very close (#14) or divergent (#3) from the putative ancestral variants.
The geographic distribution of shared or related West Eurasian haplotypes cover either very large or narrow regions and appear quite intermingled. For instance, haplotypes #23–28 extend from Lebanon to Turkey and Iran, and are only distantly related to the sympatric haplotypes #38–40. Haplotypes #15–19 cover a region spanning from S Italy to Georgia across Bulgaria, Turkey and Ukraine; interestingly, two haplotypes found in Q. pontica are directly connected to this cluster, whereas two, more divergent, depart from haplotype #1, and two further are included in haplotype # 18. However, other diverging Caucasian white oaks samples are placed in haplotypes #32, 33. The latter two haplotypes and #36 cover various territories in central, south, east Europe and the Near East, whereas haplotype #31 extends westwards from peninsular Italy, Sicily, and Sardinia to Spain. Together with #1, this highly related group of four haplotypes (#31, 32, 33, 36) collects the highest number of individuals across the entire dataset (up to 94 individuals each). Haplotype #33 also matched a complete chloroplast genome of Q. petraea (LT996899, England). Other haplotypes with remarkable frequencies, but distributed on narrower regions are #17, 18 and 24, respectively collecting 21, 26 and 17 individuals from the Caucasus, the Black Sea coasts and Lebanon, and #19 and 41, each one collecting 12 individuals from Sardinia. Some derivate haplotypes located at the tips of the network were scored from the Mediterranean major islands (e.g., #20, 30, 41), south central Spain (#21, 22) and central Anatolia (#38, 39); except for haplotype #41, they are all singlets. No further 100% identity scores with the GenBank complete Quercus genomes were identified.
Geographic structuring of the West Eurasian white oak plastid DNA variation
Dissecting the network into haplotype clusters based on molecular identity, affinity, scored frequency, and geographic positioning revealed some interesting geographic patterns (Fig. 5a–d). The two potentially ancestral variants (Fig. 5a; haplotypes #1, 2) are rather uniformly distributed across the Euro-Mediterranean region, all located within 35° and 44° Lat. North, except for the British (52° Lat. North) and one Caucasian sample. The four most frequent (and closely related) haplotypes (Fig. 5b; haplotypes #31, 32, 33, 36) identify a west central Mediterranean distribution (#31: Iberia and Italy, major islands included), contrasted to a central east European (#33: England, France, Italian Peninsula, Austria, Czech Republic, north east Croatia, Romania, Bulgaria, Georgia, Armenia), and two south eastern Mediterranean distributions (#32: Italian Peninsula and Sicily, west Croatia, Bulgaria, Georgia and Lebanon; #36: Italian Peninsula, south Croatia, Serbia, Bulgaria, Greece). Figure 5c shows the distribution of the less frequent, disjunct haplotypes: one (#15) derives from the ancestral variants (Fig. 4), is related with the Caucasian haplotypes (#16, 17) and connects southeast Italy and Bulgaria; likewise, one haplotype group (#23–28) departs from the ancestral variants and connects Lebanon to Iran across Turkey. Haplotypes #19 (connecting Sardinia and south Italy), and #18 (connecting Ukraine, Turkey and Georgia) identify well-known biogeographical links within each region; however, the two haplotypes result, unexpectedly, closely related. Finally, Fig. 5d shows the distribution of the single, narrowly distributed or highly divergent haplotypes: two haplotypes (#41 and 35) identified four populations in Sardinia (12 individuals) and one in Croatia (three individuals), all other were just singlets. As shown in Fig. 4, haplotypes #41, 35, 20, 30, 34, 37 and 40 are directly derived or closely related to co-occurring main (#1, 32, 33 and 36) or disjunct (#19) haplotypes. In contrast, haplotypes #21, 22 (South Spain) and #38, 39 (Turkey) are related to haplotypes from geographically very distant regions (#18 and 31, respectively).

(a–d) Geographic patterns of the plastid haplotype variation in West Eurasian sect. Quercus, (investigated and GenBank retrieved datasets), including 100% identical trnH-psbA + trnK-matK haplotypes detected in the complete chloroplast genomes of Eurasian subgen. Quercus. Connections between haplotypes are reported (see Fig. 4). a: Potential ancestral haplotypes; b: most frequent West Eurasian haplotypes; c: less frequent, disjunct haplotypes; d: local, unique haplotypes
Discussion
This work complements the current knowledge on plastid DNA phylogeography of West Eurasian oaks (sects. Ilex and Cerris, Simeone et al. 2016; Vitelli et al. 2017; Simeone et al. 2018), filling the large gap represented in particular by sect. Quercus and extending and improving upon the only available diversity studies that were conducted more than 20 years ago with dated molecular tools (Petit et al. 2002a and references therein). The two markers used herein acted synergistically, enabling identification of congruent widespread patterns and additional derived haplotypes with a narrower distribution (Supplementary file S3). Moreover, our analyses took advantage of the sequences available on GenBank to reveal molecular patterns consistent with major (genus level) and circumstantial (regional level) oak phylogenies, and outline a compelling framework of the white oaks evolution in Europe which will deserve new attention in future studies.
Phylogenetic patterns
The RAxML tree topology shown in Fig. 2 perfectly matches the most recent inter and intrasectional differentiation obtained with both more numerous and/or more powerful plastid markers (five DNA regions: Yang et al. 2020; over 200 coding and non-coding loci from RNA-seq data: Yang et al. 2021; whole genome sequencing: Zhou et al. 2022), and complements the evidence recently derived from nuclear data (RAD-sequencing; Hipp et al. 2020) of the complex genus evolution. As a side result, the peculiar relationships of the sect. Ilex Euro-Med lineage in relation to the Eurasian sect. Quercus is highlighted (see Fig. 3). As originally suggested in Simeone et al. (2016), the Euro-Med lineage may have been among the earliest diverging Quercus plastomes and represent the legacy of an ancient cross-sectional oak lineage (cf. Yang et al. 2021) established in the West Mediterranean area. Our phylogenetic reconstructions (Figs. 1 and 2) also separate New World and Old World oaks of the same evolutionary lineage, i.e. sect. Quercus and Ponticeae, complying with a geographic differentiation in their primordial members predating divergence and subsequent manifestation in modern taxa (Denk and Grimm 2010). In fact, the observed sectional plastome non-monophyly has already been documented in American and Eurasian oaks (Pham et al. 2017; Crowl et al. 2020; Manos and Hipp 2021; Zhou et al. 2022), and explained with chloroplast capture via hybridization in the early diversification of the genus. In sect. Ponticae (see Figs. 1 and 3) it has been postulated that the relict species Q. sadleriana survived by introgressing plastomes from sympatric Protobalanus members in the past (McVay et al. 2017; Hipp et al. 2020). Similarly, its Caucasian sister (Q. pontica) introgressed plastomes of sympatric (Caucasian) members of sect. Quercus, likely from different sources and at different times in the past. On the other hand, the intra-sectional deep incongruence between plastid data and taxonomic identity is well-acknowledged across the entire genus (Yang et al. 2021). However, the lack of resolution we observed within the Eurasian white oaks, even between members from western and eastern Eurasia, was somewhat unexpected and is in sharp contrast with sects. Cerris and Ilex (c.f. Simeone et al. 2018; Yang et al. 2021).
The molecular diversity values reported in Table 1, collected from a vast and comprehensive Eurasian oak dataset, help to explain the low resolution of our phylogenetic reconstructions. In particular, we can see that despite the higher numbers of species and individuals investigated, the West Eurasian white oaks (both those investigated here and the GenBank expanded dataset) showed: (1) fewer, and (2) less variable haplotypes (in terms of sequence divergence) than West Eurasian members of sect. Ilex, and only slightly higher variation than West Eurasian members of sect. Cerris, of which there were far fewer individuals included in the comparison. Interestingly, Yang et al. (2020) found the same differences comparing five plastid markers across East Asian members of the three sections, and suggested different evolutionary dynamics associated with their distinct origins (New/Old World) as a possible explanation. In our work, the variation found suggests the identification of a single West Eurasian white oak haplotype lineage (largely unresolved in the RAxML tree and Neighbour-Net graph), in sharp contrast with the three lineages found in (sympatric) sect. Ilex (WAHEA, Cerris-Ilex, West-Med; Simeone et al. 2016) and the (at least) two in sect. Cerris (L1, L2; Simeone et al. 2018). In these cited studies, the different intra-sectional lineages were identified based on their relative positions in the phylogeographic reconstructions (e.g., high-supported clades and subclades in the RAxML tree, congruent geographic distribution) and the high number of mutations (up to five) separating the closest haplotypes of each lineage. Instead, the poorly resolved West Eurasian white oak haplotypes (Figs. 1 and 2) are each separated by single mutations (with the only exception of two singlets likely corresponding to geographically isolated samples; Fig. 4). The East Eurasian samples showed some divergent haplotype clusters and singlets that might suggest the occurrence of further lineages (c.f. Yan et al. 2019; Yang et al. 2020). However, differences in the representativeness of the sampling designs across West and East Eurasia may have contributed to revealing a sort of genetic continuum in the more densely sampled Western regions, in contrast to the more punctuated diversity in the less covered Asian region.
The Northeast Asian origins of sect. Ilex (first) and sect. Cerris (soon after) have been traced back to the Eocene (40–50 million years ago; Denk et al. 2023). Subsequent range expansions lead their progenitors to penetrate and colonize Western Eurasia along two different routes (Ilex: via the Tibet-Himalayan Corridor; Cerris: across Northern and Central Asia; Jiang et al. 2019; Denk et al. 2023) by > 20 million years ago (Early-Middle Miocene), causing major East–West Eurasian splits, and soon after differentiating their main intra-sectional lineages and species groups. All gathered data exhibited by the plastid markers in the Euro-Mediterranean members of these two sections (Vitelli et al. 2017; Simeone et al. 2018) show clear phylogeographical patterns that are indicative of reticulation, lineage sorting, diversification and quick dispersal not involving severe bottlenecks.
In contrast, colonization of Eurasia by a stock of North American sect. Quercus oaks can be dated considerably later (ca. 10 to 20 million years ago; Hipp et al. 2020), with the major intra-sectional split between the plastomes of the two white oak groups explained on the basis of their long-term geographic isolation due to submersion of the North Atlantic and Bering land bridges since the late Neogene (Denk et al. 2017). According to some authors, species differentiation and the division into eastern and western Eurasian lineages would have taken place in the late Miocene (ca. 10 Ma), as the likely result of decreasing temperatures and an intensification of the Asian monsoon system (Yan et al. 2019; Hipp et al. 2020). By the Late Miocene – Early Pliocene, the West Eurasian white oak progenitors were established in most European regions investigated in the present study (Jiménez-Moreno and Suc 2007; Kvaček et al. 2008, 2020; Velitzelos et al. 2014; Barrón et al. 2017; Niccolini et al. 2022; Vieira et al. 2023). During this period, these oak populations experienced multiple extirpation, biome shifts and complex repopulation phases over time (Kremer and Hipp 2020). Heavy vegetation changes with continuous remodelling in the structure and composition of both local and regional floras were caused by considerable geo-morphological and climate changes along with the Messinian Salinity Crisis (Krijgsman et al. 1999; Krijgsman 2002), the late Pliocene development of the Mediterranean climate (Suc 1984), the Pleistocene Glacial/Interglacial cycles (Pons et al. 1995) and the concurrent uplift of major mountain systems associated with intense volcanic activity, and repeated, temporary land connections across sea straits (Blondel et al. 2010; Nieto Feliner 2014). As a result, a mosaic of local/regional conditions varying with latitude, longitude, altitude and sea proximity (Suc 1984; Suc and Popescu 2005) occurred especially in the Mediterranean regions, where temperate Quercus forests became progressively dominant around 1.4–1.3 Ma (Combourieu-Nebout et al. 2015; Magri et al. 2017) and found effective shelters from the widespread tree extinctions across much of Northern and Central Europe during the ice ages (Brewer et al. 2002).
The preliminary climatic deterioration of the late Pliocene, and especially the upheavals linked to the Pleistocene glacial and interglacial cycles, probably affected the newly arrived mesophylous white oaks more severely than the long-established, xero-thermic Ilex and pre-adapted Cerris oaks (Denk et al. 2023), further depleting their plastid diversity. The current geographical distribution of the West Eurasian species belonging to sect. Quercus and those of sect. Cerris and Ilex, as well as their ecological features, suggest some interpretative keys to explain the haplotype depletion characterizing white oaks. Sects. Ilex and Cerris oaks show a strictly steno-Mediterranean distribution (the former) and Euro-Mediterranean-Pontic (the latter). The European species of Sect. Quercus show a much wider northern distribution, seemingly related to their ability to cope with microthermic climates (e.g., Q. petraea s.l. and Q. robur, exhibiting their centre of distribution in Central Europe and extending westwards to the whole British Archipelago and northwards to southern Scandinavia and Siberia). It is therefore probable that sects. Cerris and Ilex species underwent only limited range contractions during the glaciations or were able to reach glacial refugia with relative ease by virtue of their privileged long-established southern distribution, thus conserving a huge portion of the populations and their genetic diversity. In contrast, a large part of white oaks populations (i.e., those of central and northern Europe) were constrained between the ice caps advancing from the north and the southern Pyrenees-Alps-Carpathians longitudinal mountainous alignment and were drastically reduced. This dramatic entrapment, which almost totally prevented oak forests migration southwards causing a virtual mass extinction (see Bennett et al. 1991), had the consequence that significant portions of the genetic diversity brought by the “northern” white oaks populations was irretrievably lost. The highly heterogeneous and changing landscapes of the post-glacial periods then allowed preservation of the genetic pools that survived in the earlier inhabited southern regions, and promoted diversification triggering adaptation, isolation and drift in the backup sources for subsequent recolonization (Hipp et al. 2018).
Phylogeographic patterns
The two haplotypes located at the centre of the Network (Fig. 4, haplotypes #1, 2), closer to the connection with the North American Cerris and Ilex sister lineages, can be considered the ancestral Eurasian white oak haplotypes. These haplotypes are exhibited by up to ten East samples and a large number of West Eurasian ones, with a fourth Asian haplotype separated by just one mutation. Yan et al. (2019) also found one shared haplotype between two East and three West Eurasian white oaks (far east Russia, North Europe) by using trnH-psbA and three other plastid marker sequences, strengthening the suggestion of extremely ancient genetic imprints still surviving in some Eurasian white oak plastomes. Given the geographical distribution of the samples included in our study, #1 appears to be dominant in the west-central European regions, whereas #2 is restricted to southern (Sicily), southeastern (Lebanon) and eastern (Georgia) latitudes, with a higher frequency in East Asia. Such a spatial separation could be explained by a different (western/eastern), possibly coeval, origin of the two ancestral haplotypes. Subsequent molecular differentiation (suggested by the relatively high number of derived lineages (Table 1) seems to have occurred with different efficacies on the two sides of the continent, probably reflecting the different ecological opportunities offered by the two regions or promoted by the different topography and survival rate of these oaks during the Pleistocene (c.f. Li et al. 2019). In West Eurasia, all the most frequent European haplotypes (# 31–33, 36) are directly linked to the ancestral variants; the white oaks pronounced ability for rapid migration (Kremer and Hipp 2020) likely worked in concert with the complex palaeogeology and palaeoclimatology of Europe and the Mediterranean basin to allow a highly efficient recolonization across Europe, but resulting at the same time in a phylogeographic structure which is difficult to dissect. Nevertheless, some basic patterns can be identified.
The ancestral haplotypes H01 and H02 are concentrated in southern Europe, whereas they are almost totally absent from central-northern Europe. We cannot exclude the possibility that the asymmetric North/South distribution may partially arise from the preponderance of southern European populations in our dataset. However, the two haplotypes occur in a wide number of Euro-Mediterranean territories (Fig. 5a), where regions able to host them are known to have existed for prolonged periods during both the Miocene and Pleistocene (Médail and Diadema 2009). It is therefore much more likely that this asymmetric distribution is to be linked to the survival of original gene pools and the role played by the Quaternary glaciations in repeatedly resetting the genetic memory of the European white oak populations. The most accredited theory is that the vegetation landscape of the Central and northern Europe, currently occupied by the deciduous temperate forests Biome, was almost completely covered by the Artemisia sp. steppe-like grasslands and the cushion-like Tundra during the Quaternary cold periods (Magri 2010; Tzedakis et al. 2013). Although it cannot be excluded that isolated stands of conifers could have survived in refugia in central Europe during the last glaciation (Fickert et al. 2007; Parducci et al. 2012a) a topic on which the debate is still open, (see Birks et al. 2012 and Parducci et al. 2012b),) the hypothesis of the occurrence of northern refugia for the significantly more thermophilous white oaks seems untenable. Accordingly, the absence of ancestral haplotypes of white oaks from the European territories north of the Alps is somewhat to be expected. In contrast, the absence of ancestral haplotypes within a large belt of central and southern Europe including Italian Po valley, northern Croatia and Serbia was not expected. It is probable that the cold and semi-arid conditions which characterized the Late Dryas (12,500 years BP) and that persisted in the first two millennia of the Holocene played a crucial role. Pollen data reports that in northern Italy such prolonged harsh climate events pushed the boreal coniferous forests to the foothills of the southern slopes of the Alps and the cold steppic grasslands to the Adriatic Sea coasts, causing complete extinction of deciduous forests or their confinement within a few small-size enclaves (Ravazzi et al. 2007; Kaltenrieder et al. 2009; Pini et al. 2022).
The presence of haplotype # H01 in Great Britain might seem out of context, considering that the ancestral haplotypes are absent from countries located much further south than Great Britain, such as France, Austria, and Serbia. However, this British occurrence is not surprising (see Cottrell et al. 2002; Nocchi et al. 2022), being explainable by an efficient migration of Iberian white oaks in the post-glacial period (even though the lack of basic information on the provenance of the British sequences downloaded from GenBank means that anthropic plantations of southern European germplasm cannot be excluded). As far as Southern Europe is concerned, in contrast to Sicily (see below), no ancestral haplotypes were found in Sardinia, despite our extensive sampling and the well-known richness in systematically isolated plant paleo-endemisms of this island (Mansion et al. 2009; Schmitt et al. 2021; Fois et al. 2022). In fact, the conservation and evolution of the genetic heritage of the white oaks in Sardinia and Sicily differed according to the differing paleogeographic histories of these two islands. The Sardinian block detached from the Catalan-Provençal plate (southern France and north-eastern Spain at present) in the early Miocene (ca. 30–15 Ma) and started a backward migration that brought it to the centre of the proto-Tyrrhenian Sea during the Messinian age. Since then, no further land connections occurred with the mainland, with possible exceptions during the Messinian salinity crisis, when it is assumed Sardinia was connected to the Apulian platform and to north Africa (Hsü et al. 1977), and during the LGM when a connection with the Tuscan Archipelago has been hypothesized (Médail and Diadema 2009; Schmitt et al. 2021).
Consequently, Sardinia displays four differently derived plastid variants which can be related to the paleogeographic events mentioned above. The lack of ancestral haplotypes may reflect the Early Miocene geological detachment of the Sardinian block from the Western European landmass (i.e., before the arrival of the original North American white oak plastomes); haplotype # 19 (Q. congesta, Q. ichnusae, Q. virgiliana) might be indicative of the ancient (Messinian) links with the Southern Italian Peninsula, coupled with more recent (Pleistocene glacial periods) immigrations from Northern Italy (haplotype # 31; Q. congesta, Q. pubescens), likely facilitated by land connection via the Tuscan Archipelago. Further recent diversification (derived haplotypes # 20, 41; Q. congesta, Q. ichnusae, Q. virgiliana) could have been triggered subsequently owing to isolation.
In contrast, Sicily (with an equally ancient history and a more complex orography) was less isolated. Both at the Miocene-Pliocene boundary and during the Pleistocene, there were no barriers to oak migration from northern Italian and Balkan territories via adjacent Calabria and Apulia (see alsoVitelli et al. 2017; Simeone et al. 2018). According to some authors (Brullo et al. 1999; Pignatti et al. 2017), in Sicily at present there are no Q. petraea subsp. petraea and Q. pubescens s.s., but rather their southern xerothermic forms (Q. petraea subsp. austrotyrrhenica, Q. amplifolia, Q. congesta, Q. dalechampii, Q. leptobalanos, Q. virgiliana). Although the most recent bio-systematic revisions do not seem to confirm this taxonomic and nomenclatural variability (Di Pietro et al. 2020a, 2021), we can only hypothesize that the pronounced paleogeographic and paleoclimatic vicissitudes that Sicily experienced over geological history allowed an early island colonization, local preservation of ancient haplotypes, subsequent diversification and exchange with the mainland. Morphologically only slightly observable, this variability is expressed in a phylogeographic key by four different haplotypes, including the potentially ancestral #2 (at its westernmost occurrence; Q. congesta), the early derived #31, 32 (in all the taxa occurring in Sicily) and an isolated derivate from the ancestral haplotype #1 (#30; Q. virgiliana).
Finally, the geographical distribution of the most frequent haplotypes identified in our study (# 31- 33, 36) also seems to be well structured and consistent with the current biogeographical map of Europe (Rivas-Martínez et al. 2004). The major western/eastern separation is demonstrated by the geo-vicariance of haplotypes # 31 and # 36, while the overall distribution of haplotype # 36 perfectly matches the boundaries of the Apennine-Balkan Province. The wide distributions of haplotypes # 32 and 33 (extending from southern Italy to the Caucasus and Lebanon, and from central Europe to Great Britain) likely provide evidence of biogeographic connections prior to Quaternary glaciations and may reflect ancient East/West vicariance. The evolutionary interpretations of the haplotypes found in the most densely sampled area of our study (the Italian Peninsula and its major islands), are presented in Table 2.
Phylogeography of the European white oak plastome revisited
Despite the unbalanced sampling designs, we may sketch a comparison of our results with the distribution patterns proposed by (Petit et al. 2002a, b), representing the latest synthesis and the current state of the art for the European white oak plastome diversity. In their seminal works conducted on over 2600 white oak populations across Europe (overlooking, however, the southeast Mediterranean and Near east areas), Petit et al. (2002a) detected 32 chloroplast PCR–RFLP haplotypes grouped in six major lineages (A–F). In contrast, our data include the overlooked regions but are somewhat deficient as regards the Northern European regions. However, we detected a comparable number of genetic variants (29) and we can expect the greater part of the molecular plastome diversity has been captured by our investigation. In fact, Petit et al. (2002a) also identified fewer chloroplast variants in Northern Europe and all present in the southern regions.
Our results match several of the previously assessed findings and provide additional insights. We confirm that many haplotypes have an extremely large distribution while others are more delimited, and that non-coeval molecular signatures coexist in the three Mediterranean peninsulas and major islands (Sicily and Sardinia). In addition, the same major areas of genetic similarity are found in our work too, confirming the previously acknowledged refugial role and the subsequent recolonization routes (Petit et al. 2002b). These certainly include the patterns of the most widespread haplotypes #31, 32, 33 and 36, identifying clear phylogeographic relationships between North Spain, Italy and major Tyrrhenian islands (#31), the Italian Peninsula and the southern Balkans (#36), Italy and Central-East Europe (#32, 33). Other congruent relationships are also evidenced by less widespread haplotypes such as #15 (South Italy and the Balkans), #18 (Black Sea coasts) and #19 (South Italy and Sardinia), whereas haplotypes #23–28 identify the previously overlooked Near East region. The occurrence of unique Iberian and Italian haplotypes (#20–22, 29, 30, 37) is also confirmed (cf. Olalde et al. 2002; Fineschi et al. 2002), together with the new evidence of further rare variants in the Southeast Mediterranean area (#16, 23, 25–28, 35, 38–40). Finally, not a single species showed an exclusive haplotype composition: the most densely sampled species (22 to 68 individuals each of Q. robur, Q. petraea, Q. frainetto, Q. iberica) displayed six to eight highly shared haplotypes, the only exception being a peculiar form of Q. robur from Anatolia (Q. haas).
However, we also document some significant novelties as compared to the generally accepted scenario. Our extensive dataset, combining new sampling points with a high number of GenBank sequences from the whole of Eurasia, enabled us to sketch out for the first time a suggestive phylogenetic backbone of the white oak plastome, to identify ancestral signatures and highlight a well-structured phylogeographic distribution. Evidence of ancestral haplotypes possibly dating back to the Miocene, i.e. prior to the white oaks species’ differentiation and separation between the Eastern and Western Eurasian, opens up new interpretative scenarios on the evolution of white oaks and their spread across Europe. Furthermore, 17 unique variants were found, all exhibited by isolated (Sardinian, Sicilian) or disjunct (Anatolian, Lebanese, South Iberian) populations of species otherwise sharing the largest part of their plastome signatures with numerous congenerics, independently of geographic proximity or taxonomic affinity. The only exceptions are the Anatolian endemic Q. vulcanica and a couple of undefinable hybrids from Lebanon. In fact, the majority of the unique haplotypes (either ancient or derived) are displayed by xerophylous species, such as Q. faginea, Q. ichnusae, Q. infectoria, Q. pubescens, Q. virgiliana. This finding can be explained by means of a combination of ancestral legacies, ability to adapt to Mediterranean ecological extremes and isolation (i.e., ‘phylogenetic conservatism and ecological opportunity’ Cavender-Bares 2019; Hipp et al. 2020). As such, they may provide new input for future, more informative studies on the white oak phylogenomics.
Another newly emerging result is the multitasking phylogeographic role played by the Italian Peninsula, which hosted numerous glacial refugia, and did not simply act as a crossroad for widespread haplotypes but rather as a real biogeographical threshold. This is evident observing the structured distribution of the ancestral haplotypes, as well as the distribution of their derived variants that find their easternmost (#31) or westernmost (#32, 36 boundary precisely in the Italian Peninsula. Among these, and with a sharpness that has never emerged in previous works on European white oaks, a well-defined Amphi-Adriatic haplotype area has here been outlined. This creates interesting parallels with what has recently emerged from other fields of research, for example the new insight on the coenological and synchorological features of the Quercus frainetto and Q. cerris Apennine-Balkan forests (Di Pietro et al. 2020c), and the biosystematics studies regarding some other diagnostic (in phytogeographical terms) genera, such as Campanula, Salvia, Sesleria (Kuzmanović et al. 2017; Janković et al. 2019; Radosavljević et al. 2022). Obviously, we are aware of the paucity of data from Greece, Anatolia, and Eastern Europe from which it would be reasonable to expect a greater diversity than that detected by means of the relatively few samples included in our analyses. Greater availability of data may have enabled us to obtain results similar to those obtained for Q. cerris (Bagnoli et al. 2016), also considering the remarkable occurrence of ancestral (Bulgaria, Georgia), inter-sectionally shared (Georgia), disjunct (Ukraine), highly diverse (Bulgaria) and unique (Croatia, Georgia) haplotypes found in our study.
A final comment is also due regarding the genetic variation found in Q. pubescens s.l., the most xero-thermic white oak in western Eurasia. This species revealed the highest number of haplotypes (18) in the entire dataset, including both ancient and derived variants. Five haplotypes, uniquely located in Spain, Italy (2), Croatia and Greece, were found solely in its typical form (Q. pubescens Willd.), whereas seven haplotypes can be assigned to its (largely Italian) taxonomically critical species (e.g. Q. congesta, Q. virgiliana etc.). The remaining were interspecifically and geographically highly shared. This high variability of Q. pubescens s.l. plastid DNA matches other findings highlighted in previous papers, such as this species’ wide morphological variability (probably the widest in the whole genus) which was demonstrated as deriving from a genetic base (Viscosi et al. 2009; Curtu et al. 2011; Fortini et al. 2015b) and its pronounced tendency to introgressive hybridization which, according to some authors, represented one of the most effective mechanisms of its postglacial expansion (Lepais and Gerber 2011).
All this indicates the long, complex evolutionary history and the high tendency to local adaptation and differentiation of the downy oaks, which, although it is still far from being completely understood, bodes well for the adaptation of this xerothermic oak to the current ongoing climatic changes.
Conclusion
The rise of phylogenomic analysis over the last few years has provided superior tools for clarifying the evolutionary diversification of Quercus. However, the recent boost in the availability of genomic data deriving from one to few individuals per species may not provide reliable and complete information, especially if samples from biogeographically important areas are neglected and diagnostic natural populations are not considered. Findings resulting from our methodological trade-off (few markers, many samples, biogeographical diagnosis, GenBank exploitation, comprehensive comparative analyses) are complex or even puzzling, but at the same time confirm basic geographic patterns and highlight important variants bearing notable biogeographic or evolutionary signatures.
Our reconstructions are congruent with the general framework provided by Petit et al. (2002a, b). We gathered evidence that the West Eurasian white oaks, which originated from a limited genetic source and were only recently distributed across the continent after passing through a long series of impacting events in highly heterogeneous landscapes, have preserved a poorly differentiated plastid DNA, where signatures of a distant past abide with more recent, still unfixed genetic diversity. Finally, we provide a phylogenetic inference in which geographical areas (and their oak populations) preserving very ancient molecular signatures, overlapping combination of extensive Pliocene to Pleistocene migration waves, and local, more recent differentiation, are highlighted. Such a prolonged preservation, diversification and cohabitation dynamics of the white oak plastome certainly requires more in-depth multidisciplinary studies aimed at identifying the drivers of species evolution, identity and community assembly, in order to implement adequate conservation strategies. Some Eurasian regions have never been included in a thorough phylogenetic work and these were largely overlooked in recent phylogenomic studies; they include the focal area of our study (Central and South Italy, Sardinia, Sicily), together with central Iberia, Anatolia, the Middle East and the Caucasus. More specifically, the Italian Peninsula emerges as a clear phylogeographic threshold for lineages of different origins and provenance. Despite extensive blurring of specific molecular footprints via admixing over time and incomplete reproductive barriers, ancient and derived plastid variants co-occur in Italy, intriguingly encompassing some “major” species (Q. frainetto, Q. petraea s.s., Q. pubescens s.s., Q. robur s.s.) and peculiar, endemic, and still enigmatic phenotypes that urge a better assessment. Beyond the taxonomic and nomenclatural issues on their specific binomials, our results stress the potential biological and conservation significance of Q. petraea subsp. austrothyrrenica, Q. congesta, Q. virgiliana and Q. ichnusae in Italy, Sardinia and Sicily (this work), together with Q. faginea in Andalucia, Q. vulcanica and Q. macranthera subsp. syspirensis in Anatolia, Q. kotschyana and Q. cedrorum in the Middle East, Q. hartwissiana, Q. iberica and Q. robur subsp. imeretina in the Caucasian region (Ekhvaia et al. 2018; Hipp et al. 2020; Douaihy et al. 2020; Piredda et al. 2021). Future research on genomic variants with potential phylogenetic importance or exploitable (adaptive) traits capable of increasing the European forest cover, withstand the ongoing climate change and reverse the biodiversity crisis, should be focused more on oaks from Southern latitudes and should be inclusive of all peculiar xero-types having still unresolved taxonomy.
Data availability
Used alignments and inference files are included in the Online Data Archive available at: https://10.0.23.196/m9.figshare.22787504. TrnH-psbA and trnK-matK sequences are deposited in the NCBI archive under accession numbers OQ990790–OQ990817. Other relevant data are contained within the manuscript and its supporting materials.
References
Amaral-Franco J (1990) Quercus. In: Castroviejo, S. et al. Flora Ibérica II CSIC Madrid
Antonecchia G, Fortini P, Lepais O, Gerber S, Legér P, Scippa, G S, Viscosi V (2015) Genetic structure of a natural oak community in central Italy: Evidence of gene flow between three sympatric white oak species (Quercus, Fagaceae). Ann for Res (2): 205–216
Backs JR, Ashley MV (2021) Quercus conservation genetics and genomics: past, present, and future. Forests 12:882. https://doi.org/10.3390/f12070882
Barrón E, Averyanova A, Kvaček Z et al (2017) The Fossil History of Quercus. In: Gil-Pelegrín E, Peguero-Pina JJ, Sancho-Knapik D (eds) Oaks Physiological Ecology. Springer International Publishing, Cham, Exploring the Functional Diversity of Genus Quercus L, pp 39–105
Bennett KD, Tzedakis PC, Willis KJ (1991) Quaternary refugia of north European trees. J Biogeogr 18:103–115
Birks HH, Giesecke T, Hewitt GM et al (2012) Comment on “glacial survival of boreal trees in Northern Scandinavia.” Science 338:742–742. https://doi.org/10.1126/science.1225345
Blair C (2023) Organellar DNA continues to provide a rich source of information in the genomics era. Mol Ecol 32:2144–2150. https://doi.org/10.1111/mec.16872
Blasi C, Di Pietro R (1998) Two new phytosociological types of Quercus pubescens s.l. woodland communities in southern Latium. Plant Biosyst 132(3):207–223. https://doi.org/10.1080/11263504.1998.10654205
Blasi C, Di Pietro R, Filesi L (2004) Syntaxonomical revision of Quercetalia pubescenti-petraeae in the Italian Peninsula. Fitosociologia 41:87–164
Blondel J, Aronson J, Bodiou J-Y, Boeuf G (2010) The Mediterranean region: biological diversity in space and time. Oxford University Press Inc., New York
Bock B, Tison J-M (2012) Revisions nomenclaturales et taxonomiques (note n 2). Bull De La Societe Botanique Du Centre-Ouest (nouv Ser) 43:209–220
Brewer S, Cheddadi R, de Beaulieu JL, Reille M (2002) The spread of deciduous Quercus throughout Europe since the last glacial period. For Ecol Manage 156:27–48. https://doi.org/10.1016/S0378-1127(01)00646-6
Brullo S, Marcenò C (1985) Contributo alla conoscenza della classe Quercetea ilicis in Sicilia. Not Fitosoc 19(1):183–229
Brullo S, Guarino R, Siracusa G (1999) Taxonomical revision about the deciduous oaks of Sicily. Webbia 54:1–72. https://doi.org/10.1080/00837792.1999.10670670
Bryant D, Moulton V (2004) Neighbor-Net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21:255–265. https://doi.org/10.1093/molbev/msh018
Burger WC (1975) The species concept in Quercus. Taxon 24:45–50. https://doi.org/10.2307/1218998
Cavender-Bares J (2019) Diversification, adaptation, and community assembly of the American oaks (Quercus), a model clade for integrating ecology and evolution. New Phytol 221:669–692. https://doi.org/10.1111/nph.15450
Cavender-Bares J, Ackerly DD, Hobbie SE, Townsend PA (2016) Evolutionary legacy effects on ecosystems: biogeographic origins, plant traits, and implications for management in the era of global change. Annu Rev Ecol Evol Syst 47:433–462. https://doi.org/10.1146/annurev-ecolsys-121415-032229
Combourieu-Nebout N, Bertini A, Russo-Ermolli E et al (2015) Climate changes in the central Mediterranean and Italian vegetation dynamics since the Pliocene. Rev Palaeobot Palynol 218:127–147. https://doi.org/10.1016/j.revpalbo.2015.03.001
Cottrell JE, Munro RC, Tabbener HE et al (2002) Distribution of chloroplast DNA variation in British oaks (Quercus robur and Q. petraea): the influence of postglacial colonisation and human management. For Ecol Manage 156:181–195
Crowl AA, Manos PS, McVay JD et al (2020) Uncovering the genomic signature of ancient introgression between white oak lineages (Quercus). New Phytol 226:1158–1170. https://doi.org/10.1111/nph.15842
Csaikl UM, Burg K, Fineschi S et al (2002) Chloroplast DNA variation of white oaks in the alpine region. For Ecol Manage 156:131–145
Curtu AL, Moldovan IC, Enescu CM, Craciunesc I, Sofletea N (2011) Genetic differentiation between Quercus frainetto Ten. and Q. pubescens Willd. in Romania. Not Bot Horti Agrobo 39:275–282
Denk T, Grimm GW (2010) The oaks of western Eurasia: Traditional classifications and evidence from two nuclear markers. Taxon 59:351–366
Denk T, Grimm GW, Manos PS et al (2017) An updated infrageneric classification of the oaks: review of previous taxonomic schemes and synthesis of evolutionary patterns. In: Gil-Pelegrín E, Peguero-Pina JJ, Sancho-Knapik D (eds) Oaks Physiological Ecology. Springer International Publishing, Cham, Exploring the Functional Diversity of Genus Quercus L, pp 13–38
Denk T, Grimm GW, Hipp AL et al (2023) Niche evolution in a northern temperate tree lineage: biogeographical legacies in cork oaks (Quercus section Cerris). Annals of Botany 131 (5): 769–787 https://doi.org/10.1093/aob/mcad032
Di Pietro R, Viscosi V, Peruzzi L, Fortini P (2012) A review of the application of the name Quercus dalechampii. Taxon 61:1311–1316. https://doi.org/10.1002/tax.616012
Di Pietro R, Di Marzio P, Medagli P et al (2016) Evidence from multivariate morphometric study of the Quercus pubescens complex in southeast Italy. Botanica Serbica 40:83–100
Di Pietro R, Conte AL, Di Marzio P et al (2020a) A multivariate morphometric analysis of diagnostic traits in southern Italy and Sicily pubescent oaks. Folia Geobot 55:163–183. https://doi.org/10.1007/s12224-020-09378-0
Di Pietro R, Di Marzio P, Antonecchia G et al (2020b) Preliminary characterization of the Quercus pubescens complex in southern Italy using molecular markers. Acta Bot Croat 79(1):15–25
Di Pietro R, Fortini P, Ciaschetti G et al (2020c) A revision of the syntaxonomy of the Apennine-Balkan Quercus cerris and Q. frainetto forests and correct application of the name Melittio-Quercion frainetto. Plant Biosyst 154:887–909. https://doi.org/10.1080/11263504.2019.1701127
Di Pietro R, Conte AL, Di Marzio P et al (2021) Does the genetic diversity among pubescent white oaks in southern Italy, Sicily and Sardinia islands support the current taxonomic classification? Eur J Forest Res 140:355–371. https://doi.org/10.1007/s10342-020-01334-z
Douaihy B, Saliba C, Stephan J et al (2020) Tracking diversity and evolutionary pathways of Lebanese oak taxa through plastome analyses. Botany Letters 167:315–330. https://doi.org/10.1080/23818107.2020.1765197
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theoret Appl Genetics 91:1253–1256. https://doi.org/10.1007/BF00220937
Ekhvaia J, Simeone MC, Silakadze N, Abdaladze O (2018) Morphological diversity and phylogeography of the Georgian durmast oak (Q. petraea subsp. iberica) and related Caucasian oak species in Georgia (South Caucasus). Tree Genetics & Genomes 14:17. https://doi.org/10.1007/s11295-018-1232-6
Fickert T, Friend D, Grüninger F, Molnia B, Richter M (2007) Did debris-covered glaciers serve as pleistocene refugia for plants? a new hypothesis derived from observations of recent plant growth on glacier surfaces. Arct Antarct Alp Res 39(2):245–257. https://doi.org/10.1657/1523-0430(2007)39[245:DDGSAP]2.0.CO;2
Fineschi S, Taurchini D, Grossoni P et al (2002) Chloroplast DNA variation of white oaks in Italy. For Ecol Manage 156:103–114. https://doi.org/10.1016/S0378-1127(01)00637-5
Fois M, Farris E, Calvia G et al (2022) The endemic vascular flora of sardinia: a dynamic checklist with an overview of biogeography and conservation status. Plants 11:601. https://doi.org/10.3390/plants11050601
Fortini P, Viscosi V, Maiuro L et al (2009) Comparative leaf surface morphology and molecular data of five oaks of the subgenus Quercus Oerst (Fagaceae). Plant Biosyst 143:543–554. https://doi.org/10.1080/11263500902722980
Fortini P, Antonecchia G, Di Marzio P et al (2015a) Role of micromorphological leaf traits and molecular data in taxonomy of three sympatric white oak species and their hybrids (Quercus L.). Plant Biosyst 149:546–558. https://doi.org/10.1080/11263504.2013.868374
Fortini P, Di Marzio P, Di Pietro R (2015b) Differentiation and hybridization of Quercus frainetto, Q. petraea, and Q. pubescens (Fagaceae): insights from macro-morphological leaf traits and molecular data. Plant Syst Evol 301:375–385. https://doi.org/10.1007/s00606-014-1080-2
Fortini P, Di Marzio P, Conte AL et al (2022) Morphological and molecular results from a geographical transect focusing on Quercus pubescens/Q. virgiliana ecological-altitudinal vicariance in peninsular Italy. Plant Biosyst 156:1498–1511. https://doi.org/10.1080/11263504.2022.2131923
Grossoni P, Bruschi P, Bussotti F et al (2021) The taxonomic interpretation of Mediterranean oaks of Quercus sect. Quercus (Fagaceae): uncertainties and diverging concepts. Flora Mediterranea 31:271–278. https://doi.org/10.7320/FlMedit31SI.271
Guarino R, Bazan G, Paura B (2015) Downy-oak woods of italy: phytogeographical remarks on a controversial taxonomic and ecologic issue. In: Box EO, Fujiwara K (eds) Warm-Temperate Deciduous Forests around the Northern Hemisphere. Springer International Publishing, Cham, pp 139–151
Hipp AL (2015) Should hybridization make us skeptical of the oak phylogeny. Int Oaks 26:9–17
Hipp AL, Manos PS, González-Rodríguez A et al (2018) Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytol 217:439–452. https://doi.org/10.1111/nph.14773
Hipp AL, Manos PS, Hahn M et al (2020) Genomic landscape of the global oak phylogeny. New Phytol 226:1198–1212. https://doi.org/10.1111/nph.16162
Hsü KJ, Montadert L, Bernoulli D et al (1977) History of the Mediterranean salinity crisis. Nature 267:399–403. https://doi.org/10.1038/267399a0
Hubert F, Grimm GW, Jousselin E et al (2014) Multiple nuclear genes stabilize the phylogenetic backbone of the genus Quercus. Syst Biodivers 12:405–423. https://doi.org/10.1080/14772000.2014.941037
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. https://doi.org/10.1093/molbev/msj030
Janković I, Satovic Z, Liber Z et al (2019) Genetic and morphological data reveal new insights into the taxonomy of Campanula versicolor s.l. (Campanulaceae). Taxon 68:340–369. https://doi.org/10.1002/tax.12050
Jiang X-L, Hipp AL, Deng M et al (2019) East Asian origins of European holly oaks (Quercus section Ilex Loudon) via the Tibet-Himalaya. J Biogeogr 46:2203–2214. https://doi.org/10.1111/jbi.13654
Jiménez-Moreno G, Suc J-P (2007) Middle Miocene latitudinal climatic gradient in Western Europe: evidence from pollen records. Palaeogeogr Palaeoclimatol Palaeoecol 253:208–225. https://doi.org/10.1016/j.palaeo.2007.03.040
Kaltenrieder P, Belis CA, Hofstetter S et al (2009) Environmental and climatic conditions at a potential Glacial refugial site of tree species near the Southern Alpine glaciers. New insights from multiproxy sedimentary studies at Lago della Costa (Euganean Hills, Northeastern Italy). Quatern Sci Rev 28:2647–2662. https://doi.org/10.1016/j.quascirev.2009.05.025
Kaplan Z, Danihelka J, Chrtek J et al (2022) Distributions of vascular plants in the Czech Republic Part 11. Preslia 94:335–427
Kremer A, Hipp AL (2020) Oaks: an evolutionary success story. New Phytol 226:987–1011. https://doi.org/10.1111/nph.16274
Krijgsman W (2002) The mediterranean: mare nostrum of earth sciences. Earth Planet Sci Lett 205:1–12. https://doi.org/10.1016/S0012-821X(02)01008-7
Krijgsman W, Hilgen FJ, Raffi I et al (1999) Chronology, causes and progression of the Messinian salinity crisis. Nature 400:652–655. https://doi.org/10.1038/23231
Kubitzki K (1993) Fagaceae. In: Kubitzki K, Rohwer JG, Bittrich V (eds) Flowering plants dicotyledons: magnoliid, hamamelid and caryophyllid families. Springer, Berlin, Heidelberg, pp 301–309
Kučera P (2018) New name for Central Europaean oak formerly labelled as Quercus dalechampii. Biologia 73:313–317. https://doi.org/10.2478/s11756-018-0048-z
Kumar S, Stecher G, Li M et al (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Kuzmanović N, Lakušić D, Frajman B et al (2017) Phylogenetic relationships in Seslerieae (Poaceae) including resurrection of Psilathera and Sesleriella, two monotypic genera endemic to the Alps. Taxon 66:1349–1370
Kvaček Z, Teodoridis V, Gregor H-J (2008) The Pliocene leaf flora of Auenheim, Northern Alsace (France). Documenta Naturae 155:1–108
Kvaček Z, Teodoridis V, Denk T (2020) The Pliocene flora of Frankfurt am Main, Germany: taxonomy, palaeoenvironments and biogeographic affinities. Palaeobio Palaeoenv 100:647–703. https://doi.org/10.1007/s12549-019-00391-6
Lepais O, Gerber S (2011) Reproductive patterns shape introgression dynamics and species succession within the European white oak species complex. Evolution 65:156–170
Li Y, Zhang X, Fang Y (2019) Landscape features and climatic forces shape the genetic structure and evolutionary history of an oak species (Quercus chenii) in East China. Front Plant Sci 10:1060. https://doi.org/10.3389/fpls.2019.01060
Li Y, Wang T-R, Kozlowski G et al (2022) Complete chloroplast genome of an endangered species quercus litseoides, and its comparative, evolutionary, and phylogenetic study with other quercus section cyclobalanopsis species. Genes 13:1184. https://doi.org/10.3390/genes13071184
Liu X, Chang E, Liu J, Jiang Z (2021) Comparative analysis of the complete chloroplast genomes of six white oaks with high ecological amplitude in China. J for Res 32:2203–2218. https://doi.org/10.1007/s11676-020-01288-3
Magri D (2010) Persistence of tree taxa in Europe and Quaternary climate changes. Quatern Int 219:145–151. https://doi.org/10.1016/j.quaint.2009.10.032
Magri D, Di Rita F, Aranbarri J et al (2017) Quaternary disappearance of tree taxa from Southern Europe: timing and trends. Quatern Sci Rev 163:23–55. https://doi.org/10.1016/j.quascirev.2017.02.014
Manos PS, Hipp AL (2021) An updated infrageneric classification of the North American Oaks (Quercus Subgenus Quercus): review of the contribution of phylogenomic data to biogeography and species diversity. Forests 12:786. https://doi.org/10.3390/f12060786
Manos PS, Cannon CH, Oh S-H (2008) Phylogenetic relationships and taxonomic status of the paleoendemic fagaceae of western North America: recognition of a new genus. Notholithocarpus Madr 55:181–190. https://doi.org/10.3120/0024-9637-55.3.181
Mansion G, Selvi F, Guggisberg A, Conti E (2009) Origin of Mediterranean insular endemics in the Boraginales: integrative evidence from molecular dating and ancestral area reconstruction. J Biogeogr 36:1282–1296. https://doi.org/10.1111/j.1365-2699.2009.02082.x
McVay JD, Hipp AL, Manos PS (2017) A genetic legacy of introgression confounds phylogeny and biogeography in oaks. Proc R Soc Lond B 284:20170300. https://doi.org/10.1098/rspb.2017.0300
Médail F, Diadema K (2009) Glacial refugia influence plant diversity patterns in the Mediterranean Basin. J Biogeogr 36:1333–1345. https://doi.org/10.1111/j.1365-2699.2008.02051.x
Menitsky YL (2005) Oaks of Asia (Translated from Russian). Science Publishes, Inc, Enfield, New Hampshire, USA, p 549
Mucina L, Bültmann H, Dierßen K et al (2016) Vegetation of Europe: hierarchical floristic classification system of vascular plant, bryophyte, lichen, and algal communities. Appl Veg Sci 19:3–264
Niccolini G, Martinetto E, Lanini B et al (2022) Late Messinian flora from the post-evaporitic deposits of the Piedmont Basin (Northwest Italy). Fossil Imprint 78:189–216
Nieto Feliner G (2014) Patterns and processes in plant phylogeography in the mediterranean basin. a review. Perspect Plant Ecol Evolut Syst 16:265–278. https://doi.org/10.1016/j.ppees.2014.07.002
Nocchi G, Brown N, Coker TLR et al (2022) Genomic structure and diversity of oak populations in British parklands. Plants, People, Planet 4:167–181. https://doi.org/10.1002/ppp3.10229
Okaura T, Quang ND, Ubukata M, Harada K (2007) Phylogeographic structure and late Quaternary population history of the Japanese oak Quercus mongolica var. crispula and related species revealed by chloroplast DNA variation. Genes Genet Syst 82:465–477. https://doi.org/10.1266/ggs.82.465
Olalde M, Herrán A, Espinel S, Goicoechea PG (2002) White oaks phylogeography in the Iberian Peninsula. For Ecol Manage 156:89–102. https://doi.org/10.1016/S0378-1127(01)00636-3
Pang X, Liu H, Wu S et al (2019) Species identification of oaks (Quercus L., Fagaceae) from gene to genome. Int J Mol Sci 20:5940. https://doi.org/10.3390/ijms20235940
Parducci L, Jørgensen T, Tollefsrud MM et al (2012a) Glacial survival of boreal trees in northern Scandinavia. Science 335:1083–1086. https://doi.org/10.1126/science.1216043
Parducci L, Edwards M, Bennett K, Alm T, Elverland E et al (2012b) Response to comment on “glacial survival of boreal trees in Northern Scandinavia.” Science 338(6108):742–2012
Pasta S, De Rigo D, Caudullo G (2016) Quercus pubescens in Europe: distribution, habitat, usage and threats. In: San Miguel Ayanz J, de Rigo D, Caudullo G, Houston Durrant T, Mauri A (eds) European atlas of forest tree species. Publ. Off. EU, Luxembourg, e019e5c+European Atlas of forest tree species:156–157
Pattengale ND, Alipour M, Bininda-Emonds ORP et al (2009) How Many Bootstrap Replicates Are Necessary? In: Batzoglou S (ed) Research in Computational Molecular Biology. Springer, Berlin, Heidelberg, pp 184–200
Petit RJ, Csaikl UM, Bordács S et al (2002a) Chloroplast DNA variation in European white oaks: Phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manage 156:5–26. https://doi.org/10.1016/S0378-1127(01)00645-4
Petit RJ, Brewer S, Bordács S et al (2002b) Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. For Ecol Manage 156:49–74. https://doi.org/10.1016/S0378-1127(01)00634-X
Pham KK, Hipp AL, Manos PS, Cronn RC (2017) A time and a place for everything: phylogenetic history and geography as joint predictors of oak plastome phylogeny. Genome 60:720–732. https://doi.org/10.1139/gen-2016-0191
Pignatti S, Guarino R, La Rosa M (2017) Flora d’Italia, 2nd edn. Edagricole, Bologna
Pini R, Furlanetto G, Vallé F et al (2022) Linking North Atlantic and Alpine Last Glacial Maximum climates via a high-resolution pollen-based subarctic forest steppe record. Quatern Sci Rev 294:107759. https://doi.org/10.1016/j.quascirev.2022.107759
Piredda R, Simeone MC, Attimonelli M et al (2011) Prospects of barcoding the Italian wild dendroflora: oaks reveal severe limitations to tracking species identity. Mol Ecol Resour 11:72–83. https://doi.org/10.1111/j.1755-0998.2010.02900.x
Piredda R, Grimm GW, Schulze E-D et al (2021) High-throughput sequencing of 5S-IGS in oaks: Exploring intragenomic variation and algorithms to recognize target species in pure and mixed samples. Mol Ecol Resour 21:495–510. https://doi.org/10.1111/1755-0998.13264
Pons A, Suc J-P, Reille M, Combourieu-Nebout N (1995) The history of dryness in regions with a mediterranean climate. In: Roy J, Aronson J, di Castri F (eds) Time Scales of Biological Responses to Water Constraints. Publishing, Amsterdam, Acad, pp 169–188
Posada D, Crandall KA (2001) Intraspecific gene genealogies: trees grafting into networks. Trends Ecol Evol 16:37–45. https://doi.org/10.1016/S0169-5347(00)02026-7
Von Raab-Straube E, Raus T (2013) Euro+ Med-Checklist Notulae, 1. Willdenowia 43:
Radosavljević I, Satovic Z, Di Pietro R et al (2022) Phylogeographic structure of common sage (Salvia officinalis L.) reveals microrefugia throughout the Balkans and colonizations of the Apennines. Sci Rep 12:15726. https://doi.org/10.1038/s41598-022-20055-4
Ravazzi C, Peresani M, Pini R, Vescovi E (2007) Il Tardoglaciale nelle Alpi italiane e in Pianura Padana. Evoluzione stratigrafica, storia della vegetazione e del popolamento antropico [The Late Glacial in the Italian Alps and in the Po Valley. Stratigraphic evolution, history of vegetation and anthropic population]. Alpine Med Quat 20:163–184
Rivas-Martínez S, Penas A, Díaz TE (2004) Bioclimatic and biogeographic maps of Europe. University of León URL: www.globalbioclimatics.org/form/maps.htm
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Schmitt T, Fritz U, Delfino M et al (2021) Biogeography of Italy revisited: genetic lineages confirm major phylogeographic patterns and a pre-Pleistocene origin of its biota. Front Zool 18:34. https://doi.org/10.1186/s12983-021-00418-9
Simeone MC, Piredda R, Papini A et al (2013) Application of plastid and nuclear markers to DNA barcoding of Euro-Mediterranean oaks (Quercus, Fagaceae): problems, prospects and phylogenetic implications. Bot J Linn Soc 172:478–499. https://doi.org/10.1111/boj.12059
Simeone MC, Grimm GW, Papini A et al (2016) Plastome data reveal multiple geographic origins of Quercus Group Ilex. PeerJ 4:e1897. https://doi.org/10.7717/peerj.1897
Simeone MC, Cardoni S, Piredda R et al (2018) Comparative systematics and phylogeography of Quercus Section Cerris in western Eurasia: inferences from plastid and nuclear DNA variation. PeerJ 6:e5793. https://doi.org/10.7717/peerj.5793
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stamatakis A (2006) Phylogenetic models of rate heterogeneity: a high performance computing perspective. In: Proceedings 20th IEEE International Parallel & Distributed Processing Symposium. 1639535
Suc J-P (1984) Origin and evolution of the Mediterranean vegetation and climate in Europe. Nature 307:429–432. https://doi.org/10.1038/307429a0
Suc J-P, Popescu S-M (2005) Pollen records and climatic cycles in the North Mediterranean region since 2.7 Ma. Geol Soc, London, Special Pub 247:147–158
Thiers B (2016) Index Herbariorum: A global directory of public herbaria and associated staff. New York Botanical Garden’s Virtual Herbarium. https://sweetgum.nybg.org/science/ih/
Trinajstić I (2007) About the problem of differentiation between the oaks quercus pubescens willd. and Quercus virgiliana. (Ten.) Ten. Šumarski List 131:57–60
Tzedakis PC, Emerson BC, Hewitt GM (2013) Cryptic or mystic? Glacial tree refugia in northern Europe. Trends Ecol Evol 28:696–704. https://doi.org/10.1016/j.tree.2013.09.001
Velitzelos D, Bouchal JM, Denk T (2014) Review of the Cenozoic floras and vegetation of Greece. Rev Palaeobot Palynol 204:56–117. https://doi.org/10.1016/j.revpalbo.2014.02.006
Vieira M, Zetter R, Grímsson F, Denk T (2023) Niche evolution versus niche conservatism and habitat loss determine persistence and extirpation in late Neogene European Fagaceae. Quatern Sci Rev 300:107896. https://doi.org/10.1016/j.quascirev.2022.107896
Vitelli M, Vessella F, Cardoni S et al (2017) Phylogeographic structuring of plastome diversity in Mediterranean oaks (Quercus Group Ilex, Fagaceae). Tree Genet Genomes 13:3. https://doi.org/10.1007/s11295-016-1086-8
Wellstein C, Spada F (2015) The Status of Quercus pubescens Willd. in Europe. In: Box EO, Fujiwara K (eds) Warm-Temperate Deciduous Forests around the Northern Hemisphere. Springer International Publishing, Cham, pp 153–163
Yan M, Liu R, Li Y et al (2019) Ancient events and climate adaptive capacity shaped distinct chloroplast genetic structure in the oak lineages. BMC Evol Biol 19:202. https://doi.org/10.1186/s12862-019-1523-z
Yang J, Guo Y-F, Chen X-D et al (2020) Framework phylogeny, evolution and complex diversification of Chinese oaks. Plants 9:1024. https://doi.org/10.3390/plants9081024
Yang Y-Y, Qu X-J, Zhang R et al (2021) Plastid phylogenomic analyses of Fagales reveal signatures of conflict and ancient chloroplast capture. Mol Phylogenet Evol 163:107232. https://doi.org/10.1016/j.ympev.2021.107232
Zhou B-F, Yuan S, Crowl AA et al (2022) Phylogenomic analyses highlight innovation and introgression in the continental radiations of Fagaceae across the Northern Hemisphere. Nat Commun 13:1320. https://doi.org/10.1038/s41467-022-28917-1
Acknowledgements
The authors wish to acknowledge two anonymous referees and the Editor for valuable feedbacks on an earlier version of the manuscript. Special thanks go to the colleagues who have helped in collecting specimens of white oaks in different places in Europe. They are J. Loidi (Spain), K. Chytrý (Czech Republic), B. Vincent (France), W. Willner (Austria), P. Dimopoulos (Greece), Ž. Škvorc and N. Jasprika (Croatia), R. Tzonev (Bulgaria); A.L. Conte, F. Conti, E. Farris, L. Filesi, L. Gianguzzi, D. Iamonico, G. Misano, L. Rosati, G. Spampinato (Italy) I. Jankovic, N. Kuzmanovic, D. Lakusic, (Serbia). For the same reason we thank the Park Rangers D. Di Fazio and colleagues (Ausoni Mountains and Fondi Lake Regional Park) and M. Patriarca, P. Perrella and F. Soscia (Aurunci Mountains Regional Park). We thank P. Di Marzio and C. Ravazzi for the useful suggestions for statistical analysis procedures and palaeobotanical information. Jim McManus kindly improved our English.
Funding
Open access funding provided by Università degli Studi di Roma La Sapienza within the CRUI-CARE Agreement. The research was supported by the National Recovery and Resilience Plan (NRRP), Mission 4 Component 2: Investment 1.4—Call for tender No. 3138 of 16 December 2021, rectified by Decree n.3175 of 18 December 2021 of Italian Ministry of University and Research; Award Number: Project code CN_00000033, Concession Decree No. 1034 of 17 June 2022 adopted by the Italian Ministry of University and Research, Project funded by the European Union—‘NextGenerationEU NBFC’. R. Di Pietro: CUP B83C22002950007, Project title “National Biodiversity Future Center”; P. Fortini: Project: “Strengthening of research structures and creation of ‘National R&S samples on some Key Enabling Technologies”. CUP H73C22000300001: M.C. Simeone: Project ECS 0000024 Rome Technopole CUP B83C22002820006, Investment 1.5.
Author information
Authors and Affiliations
Contributions
R.D.P., P.F. and M.C.S. designed the study and wrote the first draft of the paper; E.P., P.F., R.D.P. and M.C.S. (together with colleagues mentioned in the acknowledgments) collected the plant material; E.P., S.C. and L.Q. generated and analysed the molecular data, R.D.P., P.F. and M.C.S. analysed the phylogeographic comparative data, R.D.P., P.F., E.P., S.C., L.Q. and M.C.S. discussed the results and finalized the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
The Authors declare no potential sources of conflict of interest.
Additional information
Communicated by Oliver Gailing.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fortini, P., Di Pietro, R., Proietti, E. et al. Dissecting the continuum and unravelling the phylogeographic knot of plastid DNA in European white oaks (Quercus sect. Quercus): ancient signatures and multiple diversity reservoirs. Eur J Forest Res 143, 107–127 (2024). https://doi.org/10.1007/s10342-023-01610-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10342-023-01610-8