
Abstract
Context
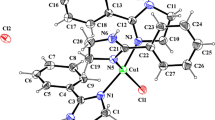
Triazene compounds (-NNN(H)-) exhibit versatility in biological, physical, and chemical applications. In their anionic form (-NNN-)(-), they can act as coordinating sites for metals, forming metallic complexes. In this study, two new isomeric triazene compounds with meta- and para-substituents in their neutral and anionic forms were investigated. A combination of detailed experimental spectroscopic characterization and computational chemistry analyses were employed. The new compounds, 1-(2-benzamide)-3-(3-nitrophenyl) triazene (m-TZN) and 1-(2-benzamide)-3-(4-nitrophenyl) triazene (p-TZN), were compared to 1,3-diphenyltriazene (dph-TZN) to understand the effects of functionalization and targeted triazene deprotonation. The anionic forms are stable, and our investigation suggests that these new compounds are suitable tridentate ligands that can act as chelating agents for metallic cations in stable complexes, similar to those found in vitamin B12.
Methods
The absorption, vibrational, and electronic properties of the newly synthesized triazene compounds were extensively characterized using FT-IR/FT-Raman and UV-Vis spectroscopy. Their distinct molecular properties, intramolecular hydrogen bond effects, stability, and electronic transitions were investigated using the ORCA software. These analyses involved DFT and TD-DFT calculations at the ωB97X-D3/Def2-TZVP level of theory with THF CPCM implicit solvation to determine the molecular topology and electronic structure. The advanced STEOM-DLPNO-CCSD method for excited states was employed, enabling an in-depth analysis of ground and excited-state chemistry, accounting for precise electronic correlation and solvation effects. Explicit THF solvation was tested on the full TD-DFT ωB97X-D3/Def2-TZVP level and using ONIOM on the STEOM calculation. Reactivity was studied using Fukui functions, and action as chelating agents was investigated using GFN-xTB2 and DFT.
Graphical abstract


















Similar content being viewed by others

Data availability
All relevant data generated or analyzed during the work are included in this paper and any additional data can be made available upon reasonable request.
References
Griess P (1866) Ueber eine neue Klasse organischer Verbindungen, in denen Wasserstoff durch Stickstoff vertreten ist. Ann Chem Pharm 137:39–91. https://doi.org/10.1002/jlac.18661370105
Lee W-T, Zeller M, Lugosan A (2018) Bis(triazenide), tris(triazenide), and lantern-type of triazenide iron complexes: synthesis and structural characterization. Inorg Chim Acta 477:109–113. https://doi.org/10.1016/j.ica.2018.02.014
Santos AJRWA, dos Santos Hackbart HC, Giacomini GX et al (2016) Inorganic and organic structures as interleavers among [bis(1-methyl-3-(p-carboxylatephenyl)triazenide 1-oxide)Ni(II)] complexes to form supramolecular arrangements. J Mol Struct 1125:426–432. https://doi.org/10.1016/j.molstruc.2016.07.021
Nunes MS, Garzon LR, Rampelotto RF et al (2017) Synthesis, characterization and biological activity of a gold(I) triazenide complex against chronic myeloid leukemia cells and biofilm producing microorganisms. Braz J Pharm Sci 53. https://doi.org/10.1590/s2175-97902017000400191
Galli P, Moretti P, Cavalleri A et al (2023) Study of the photoreaction of new triazene derivatives in solution and in polymer binder. J Photochem Photobiol A Chem 435:114331. https://doi.org/10.1016/j.jphotochem.2022.114331
Marefat Khah A, Reinholdt P, Nuernberger P et al (2020) Relaxation dynamics of the triazene compound Berenil in DNA-minor-groove confinement after photoexcitation. J Chem Theory Comput 16:5203–5211. https://doi.org/10.1021/acs.jctc.0c00489
Świderski G, Łaźny R, Sienkiewicz M et al (2021) Synthesis, spectroscopic, and theoretical study of copper and cobalt complexes with dacarbazine. Materials 14:3274. https://doi.org/10.3390/ma14123274
Lepre LF, Inoue F, Corio P et al (2013) Spectroscopic characterization of cadion: UV-vis, resonance Raman and DFT calculations of a versatile metal complexing agent. J Raman Spectrosc 44:567–572. https://doi.org/10.1002/jrs.4228
Suleymanov AA, Doll M, Ruggi A et al (2020) Synthesis of tetraarylethene luminogens by C−H vinylation of aromatic compounds with triazenes. Angew Chem 132:10043–10047. https://doi.org/10.1002/ange.201908755
Medrano-Castillo LJ, Collazo-Flores MÁ, Camarena-Díaz JP et al (2020) Base-free transfer hydrogenation of aryl-ketones, alkyl-ketones and alkenones catalyzed by an IrIIICp* complex bearing a triazenide ligand functionalized with pyrazole. Inorg Chim Acta 507:119551. https://doi.org/10.1016/j.ica.2020.119551
Samii R, Zanders D, Fransson A et al (2021) Synthesis, characterization, and thermal study of divalent germanium, tin, and lead triazenides as potential vapor deposition precursors. Inorg Chem 60:12759–12765. https://doi.org/10.1021/acs.inorgchem.1c00695
Randaccio L, Geremia S, Demitri N, Wuerges J (2010) Vitamin B12: unique metalorganic compounds and the most complex vitamins. Molecules 15:3228–3259. https://doi.org/10.3390/molecules15053228
Kruse H, Goerigk L, Grimme S (2012) Why the standard B3LYP/6-31G* model chemistry should not be used in DFT calculations of molecular thermochemistry: understanding and correcting the problem. J Org Chem 77:10824–10834. https://doi.org/10.1021/jo302156p
Lin Y-S, Li G-D, Mao S-P, Chai J-D (2013) Long-range corrected hybrid density functionals with improved dispersion corrections. J Chem Theory Comput 9:263–272. https://doi.org/10.1021/ct300715s
Sous J, Goel P, Nooijen M (2014) Similarity transformed equation of motion coupled cluster theory revisited: a benchmark study of valence excited states. Mol Phys 112:616–638. https://doi.org/10.1080/00268976.2013.847216
Lego C, Neumüller B (2011) Reaktionen von 1,3-Diphenyltriazenid mit Cu+ und Tl+. Z Anorg Allg Chem 637:1784–1789. https://doi.org/10.1002/zaac.201100227
Pracht P, Bohle F, Grimme S (2020) Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys Chem Chem Phys 22:7169–7192. https://doi.org/10.1039/C9CP06869D
Ehlert S, Stahn M, Spicher S, Grimme S (2021) Robust and efficient implicit solvation model for fast semiempirical methods. J Chem Theory Comput 17:4250–4261. https://doi.org/10.1021/acs.jctc.1c00471
Neese F, Wennmohs F, Becker U, Riplinger C (2020). The ORCA quantum chemistry program package 152:224108. https://doi.org/10.1063/5.0004608
Sarkar R, Boggio-Pasqua M, Loos P-F, Jacquemin D (2021) Benchmarking TD-DFT and wave function methods for oscillator strengths and excited-state dipole moments. J Chem Theory Comput 17:1117–1132. https://doi.org/10.1021/acs.jctc.0c01228
Laurent AD, Jacquemin D (2013) TD-DFT benchmarks: a review. Int J Quantum Chem 113:2019–2039. https://doi.org/10.1002/qua.24438
Izsák R (2020) Single-reference coupled cluster methods for computing excitation energies in large molecules: the efficiency and accuracy of approximations. WIREs Comput Mol Sci 10:e1445. https://doi.org/10.1002/wcms.1445
Glendening ED, Landis CR, Weinhold F (2013) NBO 6.0: Natural bond orbital analysis program. J Comput Chem 34:1429–1437. https://doi.org/10.1002/JCC.23266
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.22885
dos Santos AJRWA, Bersch P, de Oliveira HPM et al (2014) Triazene 1-oxide compounds: synthesis, characterization and evaluation as fluorescence sensor for biological applications. J Mol Struct 1060:264–271. https://doi.org/10.1016/j.molstruc.2013.12.055
Masoud MS, Ali AE, Shaker MA, Ghani MA (2004) Solvatochromic behavior of the electronic absorption spectra of some azo derivatives of amino pyridines. Spectrochim Acta A Mol Biomol Spectrosc 60:3155–3159. https://doi.org/10.1016/j.saa.2004.02.030
Paraginski GL, Hörner M, Back DF et al (2016) 1-(2-biphenyl)-3-methyltriazenide-N-oxide as a template for intramolecular copper(II)⋯arene-π interactions. J Mol Struct 1104:79–84. https://doi.org/10.1016/j.molstruc.2015.09.034
McMurry J (2010) Organic chemistry7th edn. Cengage Brooks/Cole, Belmont, CA
Lepre LF, Inoue F, Corio P et al (2013) Spectroscopic characterization of cadion: UV-vis, resonance Raman and DFT calculations of a versatile metal complexing agent: Spectroscopic characterization of cadion. J Raman Spectrosc 44:567–572. https://doi.org/10.1002/jrs.4228
Escudero D, Laurent AD, Jacquemin D (2017) Time-dependent density functional theory: a tool to explore excited states. In: Leszczynski J, Kaczmarek-Kedziera A, Puzyn T et al (eds) Handbook of computational chemistry. Springer International Publishing, Cham, pp 927–961
Casanova-Páez M, Dardis MB, Goerigk L (2019) ωb2PLYP and ωb2GPPLYP: the first two double-hybrid density functionals with long-range correction optimized for excitation energies. J Chem Theory Comput 15:4735–4744. https://doi.org/10.1021/acs.jctc.9b00013
Perdew JP, Schmidt K (2001) Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf Proc 577:1–20. https://doi.org/10.1063/1.1390175
Lechner MH, Neese F, Izsák R (2021) An excited state coupled-cluster study on indigo dyes. Mol Phys 119:e1965235. https://doi.org/10.1080/00268976.2021.1965235
Nooijen M, Bartlett RJ (1997) Similarity transformed equation-of-motion coupled-cluster theory: details, examples, and comparisons. J Chem Phys 107:6812–6830. https://doi.org/10.1063/1.474922
Schmalzbauer M, Marcon M, König B (2021) Excited state anions in organic transformations. Angew Chem Int Ed 60:6270–6292. https://doi.org/10.1002/anie.202009288
Chung LW, Sameera WMC, Ramozzi R et al (2015) The ONIOM method and its applications. Chem Rev 115:5678–5796. https://doi.org/10.1021/cr5004419
Manohara SR, Kumar VU, Shivakumaraiah GL (2013) Estimation of ground and excited-state dipole moments of 1, 2-diazines by solvatochromic method and quantum-chemical calculation. J Mol Liq 181:97–104. https://doi.org/10.1016/j.molliq.2013.02.018
CCCBDB listing of precalculated vibrational scaling factors. https://cccbdb.nist.gov/vibscalejust.asp. Accessed 18 Jan 2023
Smith BC (2023) Infrared spectroscopy of polymers X: polyacrylates. Spectroscopy:10–14. https://doi.org/10.56530/spectroscopy.mi9381w4
Pracht P, Grimme S (2021) Calculation of absolute molecular entropies and heat capacities made simple. Chem Sci 12:6551–6568. https://doi.org/10.1039/D1SC00621E
H⊘yvik IM, Jansik B, J⊘rgensen P (2013) Pipek–Mezey localization of occupied and virtual orbitals. J Comput Chem 34:1456–1462. https://doi.org/10.1002/jcc.23281
Bader RFW, Nguyen-Dang TT (1981) Quantum theory of atoms in molecules - Dalton revisited. Advances in quantum chemistry. Academic Press, pp 63–124
Emamian S, Lu T, Kruse H, Emamian H (2019) Exploring nature and predicting strength of hydrogen bonds: a correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J Comput Chem jcc.26068. https://doi.org/10.1002/jcc.26068
Jezuita A, Szatylowicz H, Krygowski TM (2020) How amino and nitro substituents affect the aromaticity of benzene ring. Chem Phys Lett 753:137567. https://doi.org/10.1016/j.cplett.2020.137567
Suleymanov AA, Le Du E, Dong Z et al (2020) Triazene-activated donor–acceptor cyclopropanes: ring-opening and (3 + 2) annulation reactions. Org Lett 22:4517–4522. https://doi.org/10.1021/acs.orglett.0c01527
Aihara J (1999) Reduced HOMO−LUMO Gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A 103:7487–7495. https://doi.org/10.1021/jp990092i
Pearson RG (1963) Hard and soft acids and bases. J Am Chem Soc 85:3533–3539. https://doi.org/10.1021/ja00905a001
Zhou Z, Parr RG (1990) Activation hardness: new index for describing the orientation of electrophilic aromatic substitution. J Am Chem Soc 112:5720–5724. https://doi.org/10.1021/ja00171a007
Kurtz HA (1984) LUMO energies and negative electron affinities. J Chem Educ 61:580. https://doi.org/10.1021/ed061p580
Parr RG, Yang W (1984) Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc 106:4049–4050. https://doi.org/10.1021/ja00326a036
Beck ME (2005) Do Fukui function maxima relate to sites of metabolism? A critical case study. J Chem Inf Model 45:273–282. https://doi.org/10.1021/ci049687n
Acknowledgements
We thank Dr. Mariana Boneberger Behm (UFFS) and Dr. Manfredo Hörner (UFSM) for providing the m-TZN and p-TZN reactants. Theoretical calculations were performed using the Lobo Carneiro supercomputer from Núcleo Avançado de Computação de Alto Desemprenho (NACAD), under the Project ID a20006 and the Sagarana Cluster from CEPAD - Centro de Processamento de Alto Desempenho ICB/UFMG. The authors would also like to thank the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil) for providing HPC resources of the SDumont supercomputer, which have contributed to the research results reported within this paper. URL: http://sdumont.lncc.br.
Funding
The authors would like to thank the funding from PROAP/CAPES, 001.
Author information
Authors and Affiliations
Contributions
H.C.: theoretical calculations, wrote the main manuscript text, figure preparation. U.A.: experimental FT-Raman/IR measurements, figure preparation. A.J: syntheses, funding, wrote the main manuscript text. E.C.: project management, experimental FT-Raman/IR measurements, FT-Raman/IR discussion, wrote the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 2908 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
de Castro Silva Junior, H., Antunes, U., dos Santos, A.J.R.W.A. et al. Tweaking the conjugation effects on a pair of new triazene compounds by targeted deprotonation: a spectroscopic and theoretical overview. J Mol Model 29, 298 (2023). https://doi.org/10.1007/s00894-023-05685-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05685-3