
Abstract
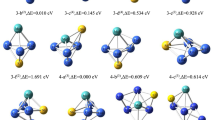
Small atomic clusters with exotic stability, bonding, aromaticity, and reactivity properties can be made use of for various purposes. In this work, we revisit the trapping of noble gas atoms (He–Kr) by the triatomic H3+ and Li3+ species by using some analytical tools from density functional theory, conceptual density functional theory, and the information-theoretic approach. Our results showcase that though similar in geometry, H3+ and Li3+ exhibit markedly different behavior in bonding, aromaticity, and reactivity properties after the addition of noble gas atoms. Moreover, the exchange–correlation interaction and steric effect are key energy components in stabilizing the clusters. This study also finds that the origin of the molecular stability of these species is due to the spatial delocalization of the electron density distribution. Our work provides an additional arsenal towards a better understanding of small atomic clusters capturing noble gases.




Similar content being viewed by others

Data Availability
All data in this work are in the text.
Code Availability
No new code is generated from this work.
References
Fan Y, Liu S, Yi Y, Rong H, Zhang J (2021) Catalytic nanomaterials toward atomic levels for biomedical applications: from metal clusters to single-atom catalysts. ACS Nano 15:2005–2037
Philip R, Chantharasupawong P, Qian H, Jin RC, Thomas J (2012) Evolution of nonlinear optical properties: from gold atomic clusters to plasmonic nanocrystals. Nano Lett 12:4661–4667
Jena P, Sun Q (2018) Super atomic clusters: design rules and potential for building blocks of materials. Chem Rev 118:5755–5870
Bartlett N (1962) Xenon hexafluoroplatinate(V) Xe+[PtF6]-. Proc Chem Soc 115:218
Miller S, Tennyson J, Geballe TR, Stallard T (2020) Thirty years of H3+ astronomy. Rev Mod Phys 92:035003
Borocci S, Giordani M, Grandinetti F (2011) Cationic noble gas hydrides-2: a theoretical investigation on HNgHNgH+ (Ng = Ar, Kr, Xe). Comput Theor Chem 964:318–323
McDonald DC II, Rittgers BM, Theis RA, Fortenberry RC, Marks JH, Leicht D, Duncan MA (2020) Infrared spectroscopy and anharmonic theory of H3+Ar2,3 complexes: the role of symmetry in solvation. J Chem Phys 153:134305
Pauzat F, Ellinger Y, Pilmé J, Mousis O (2009) H3+ as a trap for noble gases-3: multiple trapping of neon, argon, and krypton in XnH3+ (n=1–3). J Chem Phys 130:174313
Chakraborty A, Giri S, Chattaraj PK (2010) Trapping of noble gases (He–Kr) by the aromatic H3+ and Li3+ species: a conceptual DFT approach. New J Chem 34:1936–1945
Parr RG, Yang WT (1989) Density functional theory for atoms and molecules. Oxford University Press, New York
Liu SB (2007) Steric effect: a quantitative description from density functional theory. J Chem Phys 126:244103
Liu SB, Govind N (2008) Toward understanding the nature of internal rotation barriers with a new energy partition scheme: ethane and n-butane. J Phys Chem A 112:6690–6699
Liu SB, Govind N, Pedersen LG (2008) Exploring the origin of the internal rotational barrier for molecules with one rotatable dihedral angle. J Chem Phys 129:094104
Liu SB, Hu H, Pedersen LG (2010) Steric, quantum, and electrostatic effects on SN2 reaction barriers in gas phase. J Phys Chem A 114:5913–5918
Ess DH, Liu SB, De Proft F (2010) Density functional steric analysis of linear and branched alkanes. J Phys Chem A 114:12952–12957
Liu SB (2013) Origin and nature of bond rotation barriers: a unified view. J Phys Chem A 117:962–965
Huang Y, Zhong AG, Yang QS, Liu SB (2011) Origin of anomeric effect: a density functional steric analysis. J Chem Phys 134:084103
Zhou XY, Yu DH, Rong CY, Lu T, Liu SB (2017) Anomeric effect revisited: perspective from information-theoretic approach in density functional reactivity theory. Chem Phys Lett 694:97–102
Cao XF, Liu SQ, Rong CY, Lu T, Liu SB (2017) Is there a generalized anomeric effect? Analyses from energy components and information-theoretic quantities from density functional reactivity theory. Chem Phys Lett 687:131–137
Zhao DB, Rong CY, Jenkins S, Kirk SR, Yin DL, Liu SB (2013) Origin of the cis-effect: a density functional theory study of doubly substituted ethylenes. Acta Phys-Chim Sin 29:43–54
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874
Chattaraj PK, Sarkar U, Roy DR (2006) Electrophilicity index. Chem Rev 106:2065–2091
Liu SB (2009) Conceptual density functional theory and some recent developments. Acta Phys-Chim Sin 25:590–600
Parr RG, Yang WT (1984) Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc 106:4049–4050
Ayers PW, Levy M (2000) Perspective on “Density functional approach to the frontier-electron theory of chemical reactivity.” Theor Chem Acc 103:353–360
Guo CN, He X, Rong CY, Lu T, Liu SB, Chattaraj PK (2021) Local temperature as a chemical reactivity descriptor. J Phys Chem Lett 12:5623–5630
Garcia-Aldea D, Alvarellos JE (2007) Kinetic energy density study of some representative semilocal kinetic energy functionals. J Chem Phys 127:144109
Thakkar (1992) Comparison of kinetic-energy density functionals. Phys Rev A: At Mol Opt Phys 46:6920–6924
Salazar EX, Guarderas PF, Ludena EV, Cornejo MH, Karasiev VV (2016) Study of some simple approximations to the non-interacting kinetic energy functional. Int J Quantum Chem 116:1313–1321
Laricchia S, Fabiano E, Constantin LA, Della Sala F (2011) Generalized gradient approximations of the noninteracting kinetic energy from the semiclassical atom theory: rationalization of the accuracy of the frozen density embedding theory for nonbonded interactions. J Chem Theory Comput 7:2439–2451
Constantin LA, Fabiano E, Della Sala F (2017) Modified fourth-order kinetic energy gradient expansion with hartree potential-dependent coefficients. J Chem Theory Comput 13:4228–4239
Nafziger J, Jiang K, Wasserman A (2017) Accurate reference data for the nonadditive, noninteracting kinetic energy in covalent bonds. J Chem Theory Comput 13:577–586
Ghosh SK, Berkowitz M, Parr RG (1984) Transcription of ground-state density-functional theory into a local thermodynamics. Proc Natl Acad Sci U S A 81:8028–8031
Ghosh SK, Parr RG (1986) Phase-space approach to the exchange-energy functional of density-functional theory. Phys Rev A 34:785–791
Becke AD, Edgecombe K (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686
Liu SB, Rong CY, Lu T, Hu H (2018) Identifying strong covalent interactions with Pauli energy. J Phys Chem A 122:3087–3095
Holas A, March NH (1991) Construction of the Pauli potential, Pauli energy, and effective potential from the electron density. Phys Rev A: At Mol Opt Phys 44:5521–5536
Huang Y, Liu LH, Rong CY, Lu T, Ayers PW, Liu SB (2018) SCI: a robust and reliable density-based descriptor to determine multiple covalent bond orders. J Mol Model 24:213
Gaillard MJ, Gemmell DS, Goldring G, Levine I, Pietsch WJ, Poizat JC, Ratkowski AJ, Remillieux J, Vager Z, Zabransky BJ (1978) Experimental determination of the structure of H3+. Phys Rev A 17:1797–1803
Alexandrova AN, Boldyrev AI (2003) σ-Aromaticity and σ-antiaromaticity in alkali metal and alkaline earth metal small clusters. J Phys Chem A 107:554–560
Frisch MJ, Trucks GW, Schlegel HB et al (2016) Gaussian 16, revision A.03. Gaussian Inc, Wallingford
Becke AD (1993) Density-functional thermochemistry. III the role of exact exchange. J Chem Phys 98:5648–5652
Lee C, Yang WT, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Dunning TH Jr (1989) Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J Chem Phys 90:1007–1023
Lu T, Chen FW (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Schleyer PvR, Maerker C, Dransfeld A, Jiao H, van Eikema Hommes NJR (1996) Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J Am Chem Soc 118:6317–6318
Juselius J, Sundholm D, Gauss J (2004) Calculation of current densities using gauge-including atomic orbitals. J Chem Phys 121:3952–3963
He X, Yu DH, Wu JY, Wang B, Rong CY, Chattaraj PK, Liu SB (2020) Towards understanding metal aromaticity in different spin states: a density functional theory and information-theoretic approach analysis. Chem Phys Lett 761:138065
Yu DH, Rong CY, Lu T, Chattaraj PK, De Proft F, Liu SB (2017) Aromaticity and antiaromaticity of substituted fulvene derivatives: perspectives from the information-theoretic approach in density functional reactivity theory. Phys Chem Chem Phys 19:18635–18645
Yu DH, Rong CY, Lu T, De Proft F, Liu SB (2018) Baird’s rule in substituted fulvene derivatives: an information-theoretic study on triplet-state aromaticity and antiaromaticity. ACS Omega 3:18370–18379
Yu DH, Rong CY, Lu T, De Proft F, Liu SB (2018) Aromaticity study of benzene-fused fulvene derivatives using the information-theoretic approach in density functional reactivity theory. Acta Phys-Chim Sin 34:639–649
Yu DH, Rong CY, Lu T, Geerlings P, De Proft F, Alonso M, Liu SB (2020) Switching between Hückel and Möbius aromaticity: a density functional theory and information-theoretic approach study. Phys Chem Chem Phys 22:4715–4730
Yu DH, Stuyver T, Rong CY, Alonso M, Lu T, De Proft F, Geerlings P, Liu SB (2019) Global and local aromaticity of acenes from the information-theoretic approach in density functional reactivity theory. Phys Chem Chem Phys 21:18195–18210
Zhao DB, He X, Li M, Wang B, Guo CN, Rong CY, Chattaraj PK, Liu SB (2021) Density functional theory studies of boron clusters with exotic properties in bonding, aromaticity and reactivity. Phys Chem Chem Phys 23:24118–24124
Stanger A (2010) Obtaining relative induced ring currents quantitatively from NICS. J Org Chem 75:2281–2288
Stanger A (2006) Nucleus-independent chemical shifts (NICS): distance dependence and revised criteria for aromaticity and antiaromaticity. J Org Chem 71:883–893
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang WT (2010) Revealing noncovalent interactions. J Am Chem Soc 132:6498–6506
Zhao DB, He X, Li M, Guo CN, Rong CY, Chattaraj PK, Liu SB (2021) A density functional theory study of H3+ and Li3+ clusters: similar structures with different bonding, aromaticity, and reactivity properties. In: Chattaraj - atomic clusters with unusual structure, bonding and reactivity. Elsevier
Havenith RWA, De Proft F, Fowler PW, Geerlings P (2005) σ-Aromaticity in H3+ and Li3+: insights from ring-current maps. Chem Phys Lett 407:391–396
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423
Fisher RA Theory of Statistical Estimation. (1925) Math Proc Cambridge Philos Soc 22:700–725
Zhao DB, Liu SY, Rong CY, Zhong AG, Liu SB (2018) Toward understanding the isomeric stability of fullerenes with density functional theory and the information-theoretic approach. ACS Omega 3:17986–17990
Liu SB (2016) Information-theoretic approach in density functional reactivity theory. Acta Phys-Chim Sin 32:98–118
Liu SB (2019) Identity for Kullback-Leibler divergence in density functional reactivity theory. J Chem Phys 151:141103
He X, Li M, Yu DH, Wang B, Zhao DB, Rong CY, Liu SB (2021) Conformational changes for porphyrinoid derivatives: an information-theoretic approach study. Theor Chem Acc 140:123
Li M, He X, Chen J, Wang B, Liu SB, Rong CY (2021) Density functional theory and information-theoretic approach study on the origin of homochirality in helical structures. J Phys Chem A 125:1269–1278
Liu SB (2021) Principle of chirality hierarchy in three-blade propeller systems. J Phys Chem Lett 12:8720–8725
Acknowledgements
Professor Shubin Liu from the University of North Carolina is gratefully acknowledged for helpful discussion.
Funding
SJZ and CYR received support from the Science and Technology Innovation Program of Hunan Province (2021RC1003). PKC received from DST, New Delhi, India, the J. C. Bose National Fellowship, grant number SR/S2/JCB-09/2009. DBZ was supported by the startup funding of Yunnan University.
Author information
Authors and Affiliations
Contributions
P. K. C., C. Y. R., and D. B. Z. conceived and designed the overall project. X. H., C. N. G., M. L., S. J. Z., and X. J. W. carried out the computational studies. X. H., P. K. C., and D. B. Z. analyzed the data and wrote the manuscript with the input of all authors. All authors edited and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
He, X., Guo, C., Li, M. et al. Revisiting the trapping of noble gases (He–Kr) by the triatomic H3+ and Li3+ species: a density functional reactivity theory study. J Mol Model 28, 122 (2022). https://doi.org/10.1007/s00894-022-05099-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-022-05099-7