
Abstract
The electronic properties of the graphene nanoribbons (GNR) with armchair chirality were studied using the density functional theory (DFT) combined with non-equilibrium green’s function method (NEGF) formalism. The role of donor and acceptor dopants of nitrogen and boron was studied separately and also in the situation of co-doping. The charge density, electronic density of states (DOS), and transmission coefficient at different bias voltages are presented for comparison between pure and doped states. It was found that this doping plays the main role in the distortion of the GNR lattices for cases of B and N as it affects straightly on the DOS and transmission coefficient of the systems under study. The band structure of edge was engineered by differently selecting the doping positions of B, N, and B-N hexagonal rings and it was found that there are significant changes in the electronic properties of these systems due to doping. This study can be used for developing GNR device based on doping B and N atoms.








Similar content being viewed by others

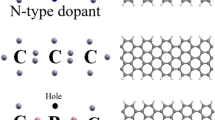
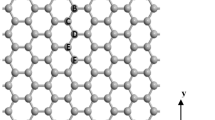
References
Yan Q, Huang B, Yu J, Zheng F, Zang J, Wu J, Gu BL, Liu F, Duan W (2007) Intrinsic current−voltage characteristics of graphene nanoribbon transistors and effect of edge doping. Nano Lett 7(6):1469–1473
Hermanson GT (2013) Bioconjugate techniques. Academic Press, pp. 627–648
Şahin H, Senger RT (2008) First-principles calculations of spin-dependent conductance of graphene flakes. Phys Rev B 78(20):205423
Chen J, Hu Y, Guo H (2012) First-principles analysis of photocurrent in graphene P N junctions. Phys Rev B 85(15):155441
Raza H, Kan EC (2008) Armchair graphene nanoribbons: electronic structure and electric-field modulation. Phys Rev B 77(24):245434
Biel B, Blase X, Triozon F, Roche S (2009) Anomalous doping effects on charge transport in graphene nanoribbons. Phys Rev Lett 102(9):096803
Brandbyge M, Mozos JL, Ordejón P, Taylor J, Stokbro K (2002) Density-functional method for nonequilibrium electron transport. Phys Rev B 65(16):165401
Xue Y, Datta S, Ratner MA (2002) First-principles based matrix Green’s function approach to molecular electronic devices: general formalism. Chem Phys 281(2–3):151–170
Treske U, Ortmann F, Oetzel B, Hannewald K, Bechstedt F (2010) Electronic and transport properties of graphene nanoribbons. Phys Status Solidi A 207(2):304–308
Taylor J, Guo H, Wang J (2001) Ab initio modeling of quantum transport properties of molecular electronic devices. Phys Rev B 63(24):245407
Wakabayashi K, Fujita M, Ajiki H, Sigrist M (1999) Electronic and magnetic properties of nanographite ribbons. Phys Rev B 59(12):8271
Kobayashi K (1993) Electronic structure of a stepped graphite surface. Phys Rev B 48(3):1757
Lv R, Terrones M (2012) Towards new graphene materials: doped graphene sheets and nanoribbons. Mater Lett 78:209–218
Rigo VA, Martins TB, da Silva AJ, Fazzio A, Miwa RH (2009) Electronic, structural, and transport properties of Ni-doped graphene nanoribbons. Phys Rev B 79(7):075435
Sevinçli H, Topsakal M, Durgun E, Ciraci S (2008) Electronic and magnetic properties of 3d transition-metal atom adsorbed graphene and graphene nanoribbons. Phys Rev B 77(19):195434
Mukherjee S, Kaloni TP (2012) Electronic properties of boron-and nitrogen-doped graphene: a first principles study. J Nanopart Res 14(8):1059
Topsakal M, Bagci VM, Ciraci S (2010) Current-voltage (I−V) characteristics of armchair graphene nanoribbons under uniaxial strain. Phys Rev B 81(20):205437
Javan MB (2015) Electronic transport properties of linear nC20 (n≤ 5) oligomers: theoretical investigation. Physica E: Low-Dimension Syst Nanostruct 67:135–142
Huang B (2011) Electronic properties of boron and nitrogen doped graphene nanoribbons and its application for graphene electronics. Phys Lett A 375(4):845–848
Varghese S, Swaminathan S, Singh K, Mittal V (2016) Energetic stabilities, structural and electronic properties of monolayer graphene doped with boron and nitrogen atoms. Electronics. 5(4):91
Soler JM, Artacho E, Gale JD, García A, Junquera J, Ordejón P, Sánchez-Portal D (2002) The SIESTA method for ab initio order-N materials simulation. J Phys Condens Matter 14(11):2745
Javan MB, Orimi RL (2015) Electric field and phosphorus doping roles on the electronic and optical properties of SiCNanocrystals: first principles study. J Comput Theor Nanosci 12(6):1023–1029
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77(18):3865
Dresselhaus MS, Dresselhaus G, Saito R, Jorio A (2005) Raman spectroscopy of carbon nanotubes. Phys Rep 409(2):47–99
Endo M, Hayashi T, Hong SH, Enoki T, Dresselhaus MS (2001) Scanning tunneling microscope study of boron-doped highly oriented pyrolytic graphite. J Appl Phys 90(11):5670–5674
Bylander DM, Kleinman L, Lee S (1990) Self-consistent calculations of the energy bands and bonding properties of B12 C3. Phys Rev B 42(2):1394
Bylander DM, Kleinman L (1991) Structure of B13 C2. Phys Rev B 43(2):1487
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Javan, M., Jorjani, R. & Soltani, A.R. Theoretical study of nitrogen, boron, and co-doped (B, N) armchair graphene nanoribbons. J Mol Model 26, 64 (2020). https://doi.org/10.1007/s00894-020-4307-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-4307-x