
Abstract
This work presents a systematic investigation of the spectroscopic properties at anharmonic force fields of ground electronic state (\( {\tilde{X}}^1{A}_1 \)) of LiNH2, which are calculated using second-order Møller-Plesset perturbation theory (MP2) and density functional theory (DFT) with hybrid GGA and meta-hybrid GGA (M06-2X) exchange-correlation functional. Two high angular momentum basis sets of 6-311+g (2d, p) and 6-311++g (3df, 2pd) are used. The equilibrium geometries, ground-state rotational constants, harmonic frequencies, and quartic and sextic centrifugal distortion constants of LiNH2 are calculated and compared with corresponding experimental or theoretical data. The predicted accuracy of the calculated constants has been confirmed by analyzing the deviations with respect to experiment. In addition, the anharmonic constants, vibration-rotation interaction constants, force constants, and Coriolis coupling constants of LiNH2 are firstly obtained. The infrared spectrum is predicted and together with the first prediction on the higher-order anharmonic constants contributes to a better understanding of the vibrational and rotational characteristics of LiNH2, thus revealing its internal structure.

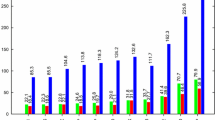
The IR spectra and the magnified IR spectra at 3500 cm−1 in harmonic approximations of LiNH2 using B3P86, M06-2X and MP2 methods combining with 6-311++g(3df,2pd)




Similar content being viewed by others

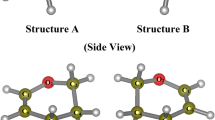
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Fieser L, Fieser M (1967) Reagents for organic synthesis, vol 1. Reagents for organic synthesis. Wiley, New York
Gray M, Tinkl M, Snieckus V (1995) Lithium. In: Abel EW, Stone FGA, Wilkinson G (eds) Comprehensive organometallic chemistry II, vol 11. Pergamon, New York, pp 1–92
Lange L, Triebel W (2000) “Sodium amide” in Ullmann’s encyclopedia of industrial chemistry. Wiley-VCH, Weinheim
Kaye IA, Kogon IC (1951) N-Monosubstituted 2-aminopyridines, 2-aminopyrimidines and 2-aminolepidines. J Am Chem Soc 73(12):5891–5893. https://doi.org/10.1021/ja01156a534
Hauser CR, Lindsay JK (1957) Some typical aldehyde addition and condensation reactions of formylferrocene1. J Org Chem 22(8):906–908. https://doi.org/10.1021/jo01359a013
Orimo S, Nakamori Y, Eliseo JR, Züttel A, Jensen CM (2007) Complex hydrides for hydrogen storage. Chem Rev 107(10):4111–4132. https://doi.org/10.1021/cr0501846
Hu YH, Ruckenstein E (2003) Ultrafast reaction between LiH and NH3 during H2 storage in Li3N. J Phys Chem A 107(46):9737–9739. https://doi.org/10.1021/jp036257b
Pinkerton FE (2004) Decomposition kinetics of lithium amide for hydrogen storage materials. J Alloys Compd 837(1–2):76–82. https://doi.org/10.1557/PROC-837-N5.3
Chen P, Xiong Z, Luo J, Lin J, Tan KL (2002) Interaction of hydrogen with metal nitrides and imides. Nat 420(6913):302–304. https://doi.org/10.1038/nature01210
Prasad DL, Ashcroft NW, Hoffmann R (2012) Lithium amide (LiNH2) under pressure. J Phys Chem A 116(40):10027–10036. https://doi.org/10.1021/jp3078387
Gregory K, Schleyer PVR, Snaith R (1991) Structures of organonitrogen-lithium compounds: recent patterns and perspectives in organolithium chemistry. Adv Inorg Chem 37:47–142. https://doi.org/10.1016/s0898-8838(08)60005-7
Collum DB (1993) Solution structures of lithium dialkylamides and related N-lithiated species: results from lithium-6-nitrogen-15 double labeling experiments. Acc Chem Res 26(5):227–234. https://doi.org/10.1002/chin.199337340
Clegg W, Liddle ST, Mulvey R, Robertson A (1999) Dis-assembling lithium amide ladder structures: new insight through the structure of [{[PhCH2N(H)Li]2·thf}∞], a polymer of (NLi)2 planar rings connected by thf-bridged (NLi)2 butterfly junctions. Chem Commun 1999(6):511–512. https://doi.org/10.1039/A900392D
Juza R, Opp K (1951) Metallamide und metallnitride, 24. Mitteilung. Die Kristallstruktur des Lithiumamides. Z Anorg Allg Chem 266(6):313–324. https://doi.org/10.1002/zaac.19512660606
Jacobs H, Juza R (1972) Neubestimmung der kristallstruktur des lithiumamids. Z Anorg Allg Chem 391(3):271–279. https://doi.org/10.1002/zaac.19723910308
Bohger JPO, EßMann RR, Jacobs H (1995) Infrared and Raman studies on the internal modes of lithium amide. J Mol Struct 348(none):325–328. https://doi.org/10.1016/0022-2860(95)08654-e
Grotjahn DB, Sheridan P, Al Jihad I, Ziurys LM (2001) First synthesis and structural determination of a monomeric, unsolvated lithium amide, LiNH2. J Am Chem Soc 123(23):5489–5494. https://doi.org/10.1021/ja003422h
Kojima Y, Kawai Y (2005) IR characterizations of lithium imide and amide. J Alloys Compd 395(1–2):236–239. https://doi.org/10.1016/j.jallcom.2004.10.063
Miceli G, Cucinotta CS, Bernasconi M, Parrinello M (2010) First principles study of the LiNH2 /Li2NH transformation. J Phys Chem C 114(35):15174–15183. https://doi.org/10.1021/jp100723p
Zhou SQ, Zhou SM, Hu T, Ju XH (2011) Density functional theory study on (LiNH2)n (n=1-5) clusters. J Mol Model 17(2):235–242. https://doi.org/10.1007/s00894-010-0717-5
Burton MA, Russ BT, Bucchino MP, Sheridan PM, Ziurys LM (2019) Quadrupole coupling in alkali metal amides MNH2 (\( {\tilde{X}}^1{A}_1 \)): an experimental and computational study. J Mol Spectrosc 365. https://doi.org/10.1016/j.jms.2019.111211
Becke A (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98(7):5648–5652. https://doi.org/10.1063/1.464913
Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33(12):8822–8824. https://doi.org/10.1103/PhysRevB.33.8822
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785–789. https://doi.org/10.1103/physrevb.37.785
Perdew JP, Wang Y (1992) Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B 45(23):13244–13249. https://doi.org/10.1103/PhysRevB.45.13244
Zhao Y, Truhlar DG (2007) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Accounts 120(1–3):215–241. https://doi.org/10.1007/s00214-007-0310-x
Møller C, Plesset MS (1934) Note on an approximation treatment for many-electron systems. Phys Rev 46(7):618–622. https://doi.org/10.1103/PhysRev.46.618
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2019) Gaussian 16, Revision C.01. Gaussian, Inc, Wallingford
Clabo DA, Allen WD, Remington RB, Yamaguchi Y, Schaefer HF (1988) A systematic study of molecular vibrational anharmonicity and vibration—rotation interaction by self-consistent-field higher-derivative methods. Asymmetric top molecules. Chem Phys 123(2):187–239. https://doi.org/10.1016/0301-0104(88)87271-9
Barone V (2005) Anharmonic vibrational properties by a fully automated second-order perturbative approach. J Chem Phys 122(1). https://doi.org/10.1063/1.1824881
Krishnan R, Binkley JS, Seeger R, Pople JA (1980) Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys 72(1):650–654. https://doi.org/10.1063/1.438955
Chi LH, Wang MS, Yang CL, Li X, Ma XG (2019) Spectroscopic constants and anharmonic force fields of F2SO: an ab initio study. Chem Phys Lett 736. https://doi.org/10.1016/j.cplett.2019.136814
Xu QS, Wang MS, Zhao YL, Yang CL, Ma XG (2017) Ab initio studies on spectroscopic constants for the HAsO molecule. J Phys Chem A 121(37):7009–7015. https://doi.org/10.1021/acs.jpca.7b01665
Zhao YL, Wang MS, Yang CL, Ma XG, Li J (2018) DFT calculations for anharmonic force field and spectroscopic constants of YC2 and its 13C isotopologues. Spectrochim Acta A 191:382–388. https://doi.org/10.1016/j.saa.2017.10.016
Boussessi R, Tasinato N, Pietropolli Charmet A, Stoppa P, Barone V (2020) Sextic centrifugal distortion constants: interplay of density functional and basis set for accurate yet feasible computations. Mol Phys 118:1734678–1734692. https://doi.org/10.1080/00268976.2020.1734678
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592. https://doi.org/10.1002/jcc.22885
Mills IM, Robiette AG (2007) On the relationship of normal modes to local modes in molecular vibrations. Mol Phys 56(4):743–765. https://doi.org/10.1080/00268978500102691
Guido Della Valle R (1988) Local-mode to normal-mode hamiltonian transformation for X-H stretchings. Mol Phys 63(4):611–621. https://doi.org/10.1080/00268978800100421
Mills IM (1972) Vibration-rotation structure in asymmetric and symmetric top molecules. In: KN RAO (ed) Molecular spectroscopy. Modern research academic press, pp 115–140. https://doi.org/10.1016/B978-0-12-580640-4.50013-3
Fogarasi G, Pulay P (1985) In: Durig JR (ed) Vibrational spectra and structure, vol 14. Elsevier, Amsterdam, pp 1–89
Pang WX, Wang MS, Yang CL, Zhang YF (2007) Ab initio study of spectroscopic constants and anharmonic force field of 74GeCl2. J Chem Phys 126(19):194301. https://doi.org/10.1063/1.2733654
Funding
This work was supported by the National Natural Science Foundation of China (Grant No.11474142) as well as the Taishan Scholars Project of Shandong Province (ts2015 11055). All calculations were carried out at the Langchao Supercomputer Center (LCSCC) of Ludong University.
Author information
Authors and Affiliations
Contributions
Shanshan Ma: Writing – original draft preparation, Methodology, Data curation.
Meishan Wang: Software, Supervision, and Writing – reviewing and editing.
Yanli Liu: Writing – reviewing and editing.
Chuanlu Yang: Project administration.
Lihan Chi: Formal analysis.
Qiushuang Xu: Formal analysis.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
Compliance with ethical standards.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, S., Wang, M., Liu, Y. et al. Ab initio study of spectroscopic properties at anharmonic force fields of LiNH2. J Mol Model 27, 33 (2021). https://doi.org/10.1007/s00894-020-04641-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04641-9