
Abstract
It is now widely accepted that the measurement process usually begins when the primary sample is taken. The uncertainty of measurement (MU) must therefore include contributions that arise from the primary sampling, and also from any physical preparation of the sample which often occurs before the sample reaches the laboratory. Guidance on how to estimate MU that includes that arising from sampling (UfS) has been widely applied to a wide range of application sectors (e.g. food, feed, water, sediment, soil, gases). Recent revision of ISO/IEC 17025:2017 (https://www.iso.org/standard/66912.html) has also recognised the inclusion of sampling within the measurement process. This recognition has implications for the validation of measurement procedures that include sampling (VaMPIS). The scope of method (or procedure) validation has therefore to be expanded and reassessed, in order to include all of these components. The uncertainty of the measurement value (MU) is a key parameter that encompasses the effects of all the other operating characteristics of the analytical procedure that is traditionally considered during its validation. It has the further advantage that it can also incorporate the uncertainty due to sampling and physical sample preparation, thus providing a single value of uncertainty that derives from the entire measurement procedure. The fitness for purpose (FnFP) of the whole measurement procedure, which is required for validation, can be judged by comparing the estimated MU (including UfS), against a Target MU, however that is set. A case study for the determination of nitrate in glasshouse lettuce shows how this VaMPIS approach can be applied to a whole measurement procedure. The experimental MU is estimated using the Duplicate Method and compared against a Target MU set using the Optimised Uncertainty (OU) method. The measurement procedure published in EU guidance is shown not to be fit for purpose (FFP). However, this approach identifies how that sampling procedure can be modified to achieve FnFP for the whole procedure, by increasing the number of sample increments per batch from 10 to 40.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is now widely accepted that primary sampling is the first step in the measurement process. This realisation is particularly clear in the case of in situ measurement techniques, such as PXRF (Portable X-ray Fluorescence) or other techniques using sensors in which no physical sample is removed. For example, once a PXRF analyser or spectrometer is placed on an area of topsoil, prior to making a measurement, a ‘sample’ has been selected. Replacing the PXRF in nominally the same position, as specified by the measurement procedure, will usually result in a slightly different ‘sample’ being interrogated and measured. The resultant uncertainty arising from the sampling procedure produces uncertainty in the resultant measurement values (i.e. MU), in addition to that arising purely from the analytical contribution. A similar argument can be made for all of the potential actions of physical sample preparation of physical primary samples that are often applied prior to ex situ measurement (e.g. filter, acidify, dry, store, sieve, grind, split), which can happen either in the field or in the laboratory. Primary sampling and physical sample preparation are both parts of the measurement procedure, and potentially important sources of MU, and therefore need to be included not only in the validation, but also in subsequent quality control (QC) processes [2]. A new approach to validation is required, therefore, to enable developers of new measurement procedures that include sampling (and physical sample preparation) to judge their FnFP.
Measurement uncertainty, and its role in validation
Measurement uncertainty (MU) has had several definitions, but one historic definition that is useful for this discussion is ‘an estimate attached to a test result (x), which characterises the range of values within which the true value is asserted to lie’ (ISO 3534 to 1: 1993 Statistics–Vocabulary and Symbols, now withdrawn). The ‘true value’ referred to is equivalent to ‘value of the measurand’ in more recent definitions of MU, such as a ‘parameter, associated with the result of a measurement that characterises the dispersion of the values that could reasonably be attributed to the measurand’ [3].
MU includes both random effects (e.g. precision estimates) and systematic effects (e.g. trueness, estimated as bias), and arises from all steps in measurement, including sampling and physical sample preparation. The MU relates to a particular ‘sampling target’, which is defined as a ‘portion of material, at a particular time, that the sample is intended to represent’ [2] and is typically a lot or batch of material or product.
MU can be used as the key parameter for expressing the quality of measurements, and therefore also of the sampling procedures used to generate them. This approach is more realistic, and metrologically sound, than the traditional approach to sampling quality, which assumes that samples are ‘correct’, and hence ‘representative’, if they have been taken ‘correctly’ by a ‘correct’ sampling procedure [4, 5].
MU can be expressed in five general ways, which are: (1) the standard uncertainty (u, usually a value of standard deviation s), (2) the expanded uncertainty (U, e.g. 2 s for a 95 % confidence interval), (3) the expanded relative uncertainty (U’, relative to the concentration value, x, usually the mean), and (4) the uncertainty factor (FU) (See Sect. 9.5.3 in Ramsey et al. [2]). The further and perhaps the simplest expression of MU is (5) to quote the uncertainty interval (or Confidence Interval CI) between a stated Lower Confidence Limit (LCL) and Upper Confidence Limit (UCL), however derived. The LCL can be calculated, for example, as x—U, or x /FU, and the UCL as x + U, or x×FU.
One advantage of the 1993 definition of MU, which includes the concept of ‘true value’, is that it can be used to define the statistical model that explains the relationship between the measured value (x) and the true value (Xtrue) of the analyte concentration in one sampling target. The difference between the measured and true value is \({\upvarepsilon }_{{\text{sampling}}}+{\upvarepsilon }_{{\text{analytical}}}\), where these terms are effects on measured concentration value from sampling and analysis, respectively. This relationship can also be extended to include the effects of the multiple sampling targets that are generally encountered in practice and are also usually needed for MU estimation, giving:
where \({\upvarepsilon }_{{\text{target}}}\) represents the effect of variation of concentration between the targets.
When we consider the estimated variances (s2) association with each of these terms, we have:
where s is the standard deviation, s2 being the statistical estimate of variance. In this case, \({s}_{{\text{total}}}^{2}\) is the total variance, \({s}_{{\text{sampling}}}^{2}\) is the between-sample variance on one target (largely due to analyte heterogeneity), \({s}_{{\text{analytical}}}^{2}\) is the between-analysis variance on one sample (usually expressed as repeatability), and \({s}_{{\text{between}}-{\text{target}}}^{2}\) is the variance between the multiple sampling targets.
MU is not traditionally considered as one of the seven performance criteria that are used to validate analytical procedures or methods (i.e. precision, trueness estimated as bias, LOD/LOQ, working range, selectivity, sensitivity, ruggedness) [6]. It has been argued, correctly, that MU is a quality of the measurement value and not of the procedure. However, this overlooks the fact that the only useable outcome of a measurement procedure is the measurement result, which should include the value of the MU [3]. MU is the only metric that enables a user to directly assess the quality of a measurement procedure in fulfilling the objectives of the investigation. In the case of a compliance decision, for example, the inclusion of a reliable estimate of MU is integral to the making of a correct decision [7]. Eurachem is preparing Supplementary Guidance (SG) on how to achieve VaMPIS, using MU as the main parameter by which to judge FnFP [8].
Duplicate method for estimating MU including UfS
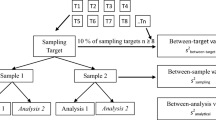
Duplication is the most cost-effective form of replication, because it allows the maximum number (and therefore greatest diversity) of sampling targets to have replicate samples taken from them. The Duplicate Method only requires the participation of one ‘sampler’, or measurement scientist. However, the estimate of MU can be made even more realistic by including between-sampler bias revealed by using multiple ‘samplers’, using results from either a Collaborative Trial in Sampling (CTS) or a Sampling Proficiency Test (SPT) (See Sect. 9.4.1, in Ramsey et al. [2], [9]).
The Duplicate Method often uses a fully balanced two-stage nested experimental design (Fig. 1) that typically requires the taking of two duplicate samples, both with duplicate chemical analyses, on around 5 to 10 %, but at least 8, of the sampling targets. An unbalanced experimental design is also possible to reduce costs (See Annex D in Ramsey et al. [2]), but this has a larger confidence interval on the resulting estimates of MU.

Realistic taking of the duplicate samples is crucial and should never be made by just the splitting of a single sample. Duplicate samples need to be taken independently by a fresh interpretation of the sampling procedure. The distance between where the duplicate samples are taken (in space or time) needs to reflect the ambiguity in sampling procedure and the spatial uncertainty in the surveying device that is used. These issues are best explained by use of an example, such as that discussed below.
Validation of measurement procedures that include sampling (VaMPIS).
The proposed approach (VaMPIS) is to judge the fitness for purpose (FnFP) of the measurement results in terms of their MU. In compliance (or conformity) assessment, it is the uncertainty (e.g. MU = Umeas) of the measurement value (x) that controls the decision of either acceptance or rejection that is made on the sampling target. All seven of the performance characteristics that are traditionally used for validation of analytical procedures in isolation (listed above) ultimately affect the MU within the measurement result (x ± U) and hence the decision that is made. The FnFP of the whole measurement procedure can be judged by deciding whether Umeas is less than the target value (UTarget Value), however that is set. If the Umeas is greater than UTarget Value, then, the contributions from both of the main components of the measurement procedure (i.e. sampling Usamp and analysis Uanal) need to be considered individually (possibly in terms of their cost–benefit). If the analytical component of Umeas requires reduction, then, each of the seven performance characteristics needs to be considered, in order to decide which needs to be improved in order for Uanal to be reduced to the point where Umeas is below (or sufficiently close to) UTarget Value and FnFP can thereby be achieved. Alternatively, if the sampling component of MU (Usamp) requires reduction, then, the sampling procedure will need to be modified to achieve that reduction (e.g. by increasing the number of increments in each composite sample).
In the simultaneous approach to VaMPIS, all of the components of the measurement procedures are validated within the same experimental design. However, it is also possible to use a sequential approach in which a previously validated analytical procedure is used to validate a sampling procedure as part of the whole measurement procedure. The validity of the analytical procedure considered in isolation can then subsequently be reviewed in the context of the new combined measurement process.
The general flow-chart for VaMPIS [8], applicable to both ex situ and in situ measurement procedures, is shown in Fig. 2.

Validation of Measurement Procedures Including Sampling (VaMPIS)—Flow-chart shown 11 main steps, in which the measurement procedure (MP) being assessed has a component sampling procedure (SP) and an analytical procedure (AP). (MU is measurement uncertainty, FnFP is fitness for purpose, AQC is analytical quality control) [8]
Case study for sequential VaMPIS of ex situ measurement of nitrate in lettuce
The mean nitrate concentration in a batch of glasshouse lettuce just before picking (i.e. the sampling target) had an upper threshold limit set by the European Union at the time of the study at 4500 mg kg−1 [10]. In a study of eight such sampling targets, each batch/target consisted of around 12,000 to 20,000 lettuce heads (See Example A1 in Ramsey et al. [2]) (Step 1, Fig. 2). The EU sampling procedureFootnote 1 that was applied specified the taking of a single composite sample from each sampling target made up of 10 heads (increments), taken in the pattern of a ‘W’ shape. The analytical procedure using HPLC,Footnote 2 which had previously been validated using a Collaborative Trial [11], was applied to suitably prepared laboratory samples (Step 2, Fig. 2). That validation gave the estimate of analytical reproducibility as ~ 3 % relative standard deviation, suggesting an estimated U’analysis of around 6 %. This case study aims to validate the whole measurement procedure, including the steps of primary sampling and physical sample preparation, using the sequential approach. To this end, the FnFP of the whole measurement procedure is judged by comparing the estimated MU against a Target MU (U’Target Value, Step 3, Fig. 2). The MU (U’meas) and its components (U’anal and U’samp) were estimated using the Duplicate Method (Fig. 1) applied to these eight randomly selected sampling targets (i.e. bays of lettuce heads). The duplicate samples for each bay were taken using an independent interpretation of the sampling procedure. This used an “M’ pattern that is the inverted orientation of the ‘W’ pattern, but an equally likely interpretation (Step 4, Fig. 2).
The four measurement values of nitrate concentration by HPLC for each of the eight sampling targets (A–H) are given in Table 1 (Step 5, Fig. 2). Visual inspection of the analytical duplicates (e.g. S1A1 and S1A2) generally show quite good agreement, suggesting a repeatability precision of around 10 %. The sampling duplicates (S1 and S2) generally differ by a larger margin, suggesting a sampling precision of closer to 20 %. However, there are some duplicate pairs, both sampling (target C) and analytical (S2 for target H), that show markedly greater differences, suggesting the presence of outlying values not consistent with an assumed normal (i.e. Gaussian) frequency distribution. A histogram of the 32 measurement values (Fig. 3) confirms that the frequency distribution is generally close to normal, but that there is evidence of a small proportion of outlying values (e.g. from target G at around 3000 mg kg−1) (part of Step 6, Fig. 2).

The quantitative estimation of MU was made using Analysis of Variance (ANOVA) applied to the 32 measurement values (Table 1). Robust ANOVA (using RANOVA3 [12]) was selected, because it can accommodate up to 10 % of suspected outlying values, whether they are analytical, sampling, or between target in origin [13] (Step 7, Fig. 2).
The robust estimate of U’meas (Table 2) was 16.4 %, with umeas in the original concentration units being 360 mg kg−1. The analytical component U’anal, estimated as repeatability was 7.6 %, which is a little larger, but very similar to the value of 6 % reported from the between laboratory validation of analytical procedure in isolation (part of Step 11, Fig. 2). Analytical recovery for nitrate was tested, but found not to be statistically different from 100 % (part of Step 6, Fig. 2). Analytical bias was therefore not detected and does not need to be combined into the MU estimate in this case.
Validation of measurement procedure—judging FnFP against target MU
In order to judge FnFP (Step 8, Fig. 2), the measured MU estimate (Umeas) can be compared against a value of Target MU (UTarget Value). One possible source for a value of UTarget Value can be from an external body, such as a regulator that has decided upon such a value. If, for the sake of argument, U’Target Value had been set externally at an arbitrary value of 20 % for this situation, then, the estimated U’meas of 16 % would be less than U’Target Value and this measurement procedure would therefore be considered as FFP. In this case, however, no value for UTarget Value has been set for the mean nitrate concentration in lettuce batches in this context. A second option for setting UTarget Value is to calculate the value required for the particular purpose for which the measurement values are to be used (Step 8b, Fig. 2). For the particular purpose of geochemical mapping of contaminant concentration between targets, for example, the FnFP criteria suggested is that MU should contribute less than 20 % to the total variance (using Eq. 2) (See Sect. 16.2 in Ramsey et al. [2]). For the current measurement procedure, the MU contributes an estimated 29 % to the total variance (Table 2), so it would not be fit, or hence validated, for this purpose.
A second way of calculating, a UTarget Value, is to use the Optimal Uncertainty (OU) Method that minimises the overall cost, including the consequences of incorrect decisions (See Sect. 16.3 in Ramsey et al. [2]). This approach is shown in Fig. 4 and based upon the original general methodology of Thompson and Fearn [14].

Generalised approach to setting of UTarget Value at the value of MU that minimises the total cost, using the Optimal Uncertainty (OU) method. Measurement cost (sampling + analysis) decreases as the required MU increases. However, the cost of incorrect decisions (e.g. false positive or false negative classification of batches) increases with increasing MU. The solid line shows the sum of these two costs, with a minimum total cost at an optimal level of MU (thick dotted line), that can be used as the Target MU to judge FFP for the whole measurement procedure. Adapted from [2]
In the OU method, the UTarget Value is set at a value of MU at which the total cost arising from the measurement (or more accurately the ‘Expectation of Loss’ [14]) is broadly at its minimum. This total cost is not just the cost of the sampling and chemical analysis but also includes the potential cost of incorrect decisions caused by a given level of MU, also called the consequence cost (Fig. 4).
The second part of the OU calculation aims to decide how a Target MU, however set, can be achieved most cost-effectively for a particular case study. This is achieved by knowing the two components of the MU (i.e. Uanal and Usamp) and also their corresponding costs. The OU method calculates whether it is more cost-effective to achieve UTarget Value by spending more on either chemical analysis (e.g. by using a more precise analytical technique), or on sampling (e.g. by taking more increments within each composite sample). It is also possible that the estimated MU is less than the Target MU, and, in this case, money can be saved by spending less on either sampling or analysis.
Application of the OU method to set the target MU for the lettuce case study (Step 8b, Fig. 2)
The OU method was applied to the case study, using the input data shown in Table 3.
The costs are the commercial unit costs of the sampling and the chemical analysis. When estimating the consequence cost, there are two general options. When a product is erroneously passed as being compliant with the regulations, a false compliance occurs (e.g. a false negative). If this is subsequently discovered, then, the producer may be fined and a product recall may be required, and the producer may not retain public support and so experience a drop in sales or a sharp deduction in share price. Past examples can be utilised when evaluating this parameter. Alternatively, when a product is wrongly classed as being non-compliant with a regulatory threshold, a false non-compliance scenario occurs (e.g. a false positive). This typically results in the unnecessary rejection of a batch of product. The cost here is typically evaluated as the cost of the batch. In this example, the consequence cost (i.e. €5280) is calculated for a false non-compliance (i.e. false positive) decision based upon the value of an entire batch of 12,000 heads of lettuce at €0.44 (all prices applied at time of validation). The theory of the OU method is outlined in the Annex to this publication and the calculation for this case study explained in more detail elsewhere [15].
The uncertainty values are derived from the robust ANOVA output (Table 2). The threshold concentration (T) is that specified in EU regulations at the time of the study [10].
The concentration at which the system is to be optimised (cm) is generally selected so that there is an appreciable probability of misclassification. Previous applications of the OU method have utilised several slightly different criteria for the setting of cm (e.g. 1.1 T [16]). For this investigation, the level of cm was set at a hypothetical enforcement limit of nitrate in lettuce, as follows. [16].
The value cm was chosen to be the minimum measurement that would indicate that the nitrate concentration was greater than the threshold T at 95 % confidence. This is where cm–Uanal = T, (where Uanal is calculated for a measured concentration cm). The relative expanded analytical uncertainty (U’anal) was estimated to be 7.62 % (from Table 2), and T = 4500 mg kg−1 (Table 3). Using this method, the value for cm can be derived as cm = 4500/(1 – 0.0762) = 4871 mg kg−1.
The graphical output of the OU method applied to this case study (Fig. 5) shows that the existing measurement procedure is not FFP (Step 9, Fig. 2). The actual estimated umeas is 360 mg kg−1 (U’ = 16.4 %), which predicts total cost (including consequences) of around €800 per target. This is much higher than the calculated optimal MU value of 184 mg kg−1 (U’ = 8.3 %) at the minimum total cost of around €400. The optimal value is calculated by minimising the expectation of loss (E(L)) from Equation A1 (in Annex) over the whole range of MU [17]. This large excess of the actual MU over the optimal value of MU indicates that the whole measurement procedure is NOT fit for the purpose of classifying these batches of lettuce (i.e. not FFP).

Application of the OU method in general (See Fig. 4) to the case study shows that this measurement procedure is NOT FFP. This is because the actual estimated MU (smeas = 360 mg kg−1) is far above (roughly twice) the value of the optimal MU (uTarget Value = 184 mg kg−1)
In order to achieve FnFP, we need to reduce the MU by a factor of approximately two. The ANOVA has shown that sampling accounts for 78 % of MU (100×22.64/28.91, from Table 2). This indicates that reducing UfS is the most cost-effective way to reduce overall MU, given that the costs of sampling and analysis are identical.
Sampling theory (See Sect. 10.2.8 in Ramsey et al. [2]) can be used to predict that we can reduce UfS by a factor of 2 by increasing sample mass by factor of 4 (i.e. 22). This calculation predicts that the taking of composite samples with 40 lettuce heads, instead of 10 heads, should make the measurement procedure FFP (Step 10, Fig. 2). The second part of the OU method [17] (Equation A5 in Annex) gives the optimal sampling uncertainty (usamp) as 149 mg kg−1. This suggests that usamp should be reduced by a factor of 2.14 (319/149 mg kg−1), which is also effectively achieved by this proposed four-fold mass increase in sample mass.
Second experiment to reduce MU in order to achieve FnFP
The design of the previous experiment was repeated on eight new sampling targets, but with an increased number of increments to 40 heads (rather than the previous 10) taken for the composite sample from each target. The detailed calculations are reported elsewhere [15], and the results adjusted to account for seasonal changes in the typical nitrate concentration in the lettuces between the two experiments. The results broadly showed that usamp was reduced from 319 to 177 mg kg−1, i.e. by a factor of 1.8, which is similar to the sampling theory model prediction of a factor of 2.0.
The overall MU (umeas) was reduced from 360 to 244 mg kg−1 (U’ from 16.4 to 11.1 %). This new umeas of 244 mg kg−1 was much closer to the optimal value (utarget = 184 mg kg−1) and at a similar total cost of ~ €500, which is substantially reduced from €800 per target originally.
Although the values do not exactly match the uTarget Value of 184 mg kg−1 at a predicted total cost of €400, they do reduce the predicted total cost by 75 %. This modified measurement procedure can therefore be considered to effectively achieve fitness for the purpose of classifying the batches of lettuce (i.e. it is FFP), by giving an MU that substantially minimises the overall financial loss. The ongoing verification of this modified measurement procedure will require implementation of a subsequent QC process that ideally uses a small proportion of duplicated samples (See Sect. 13 in Ramsey et al. [2]).
It is useful to review the FnFP of the previously validated analytical procedure in the context of this whole measurement procedure (Step 11, Fig. 2). As noted above, the separate validation from an inter-laboratory CT gave U’anal as 6 %, which is arithmetically smaller than the within laboratory repeatability U’anal from this ANOVA (7.6 %, Table 2). The second stage of the OU method (Equation A6), [17] indicates that U’Target Value could be achieved by also slightly reducing U’anal to 4.9 % (uanal = 108 mg kg−1). This might be achieved by reviewing and improving some of the seven performance characteristics [6] already discussed. Interestingly, if this lower U’anal was achieved by improving the analytical method, the overall MU would be predicted to drop further to 207 mg kg−1 (U’meas = 9.4 %), which is even closer to the U’Target Value of 184 mg kg−1 (U’meas = 8.4 %). However, although that improvement would be beneficial, at a practical level there is no necessity to reduce U’anal to achieve overall FnFP. This is because the implemented modification to the sampling procedure has achieved the vast majority of the reduction in the MU required to effectively achieve FnFP.
This example has demonstrated VaMPIS for an example of an ex situ measurement procedure, but it is equally applicable to in situ measurement procedures, as demonstrated in a second example given in upcoming guidance from Eurachem [8].
More reliable compliance decision using UfS within MU
It is becoming accepted practice that MU is included in the compliance decision as to whether a batch with analyte concentration (x) exceeds some regulatory threshold value (T) [7]. However, most examples of this approach only use the value of MU estimated for the analytical part of the measurement procedure (i.e. Uanal). The case study already discussed will be used to show that reliable compliance decisions can only be made if the value of MU used in the compliance decision also includes the contribution from the sampling procedure (UfS, Usamp).
For example, let us consider the compliance decision for the first batch of lettuces (labelled Target A in Table 1). In routine practice, the target would be classified using just a single measurement value made on just one composite sample. We can, therefore, take the first single measurement value (x = 3898 mg kg−1). This is the first analysis (A1) on the first sample (S1) for Target A (S1A1)), which is the value upon which the compliance decision for that target would usually be made. If we only consider the analytical component of the MU, we have uanal = 168, (Table 2) which gives Uanal = 336 mg kg−1 (i.e. 168×2, for 95 % confidence). The UCL of the uncertainty interval is therefore 4234 (i.e. x + Uanal = 3898 + 336) mg kg−1. This UCL of 4234 is the upper limit of the estimated range within which the true value lies, so the true value cannot possibly exceed the threshold value for compliance of 4500 mg kg−1, at 95 % confidence. It appears that the true value cannot exceed the threshold and that the measurement value, therefore, indicates that Target A is compliant.
However, if we now use a value of MU that includes UfS we get umeas = 360, Umeas = 720 mg kg−1 (Table 2), the UCL of the uncertainty interval is now 4618 (i.e. 3898 + 720) mg kg−1, which indicates that the true value could possibly exceed the threshold value of 4500 mg kg−1. This same measurement value, but with the more realistic estimate of MU, now indicates that Target A is non-compliant.
Incidentally, it is interesting to speculate upon the outcome if these measurement values had been made using the modified sampling procedure with 40 heads. The reduced Umeas with 40 heads was 488 mg kg−1, which would give a UCL of 4386 that is less than the threshold of 4500 mg kg−1. Target A might therefore have been shown to be compliant, if that FFP measurement procedure had been used.
To summarise, the non-compliance of Target A (False positive) appears to be impossible when using an MU based only upon Uanal. Only by including UfS within MU can a non-compliant batch (e.g. Target, A) be rejected reliably.
Conclusions
It is now widely accepted that primary sampling is the essential first step in, and therefore an integral part of the whole measurement procedure. Measurement uncertainty (MU) is the key metric that expresses the reliability of a measurement value and should be used to inform the decisions made using that value, such as for compliance assessment. MU is also a unifying factor that is generated by all steps in the measurement procedure, including that arising from the sampling (UfS). MU can therefore be used to judge the FnFP of that whole measurement procedure and hence to validate that whole procedure. International guidance already exists to explain how to estimate MU that includes UfS, using procedures such as the Duplicate Method, and also Collaborative Trials in Sampling or Sampling Proficiency Testing [2]. Further guidance is being developed by Eurachem that explains how MU can be used to validate measurement procedures that include sampling (VaMPIS) [8]. A case study shows how a whole measurement procedure (including sampling) can be validated, by comparing the MU against a target uncertainty, in this case set using the Optimised Uncertainty (OU) method. The case study also demonstrates how a more realistic estimate of MU (that include UfS) generally gives a more reliable decision in compliance assessment.
Notes
European Directive 79/700/EEC. OJ L 207, 15.8.1979, in force at the time of the study, now replaced by Commission Directive 2002/63/EC of 11 July 2002.
BS EN 12014–2:1997 Foodstuffs-Determination of nitrate and/or nitrite content; Part 2; HPLC/IC method for the determination of nitrate content of vegetables and vegetable products. In force at the time of the study, withdrawn December 2017.
References
ISO/IEC 17025:2017 General requirements for the competence of testing and calibration laboratories. International Organization for Standardization, Geneva, Switzerland. https://www.iso.org/standard/66912.html
Ramsey MH, Ellison SLR, Rostron P (eds.) (2019) Eurachem/EUROLAB/ CITAC/Nordtest/ AMC Guide: Measurement uncertainty arising from sampling: a guide to methods and approach, Second Edition, Eurachem, ISBN 978–0–948926–35–8 http://www.eurachem.org/index.php/publications/guides/musamp
Joint Committee for Guides in Metrology, JCGM 100:2008 Evaluation of measurement data - Guide to the Expression of Uncertainty in Measurement (GUM). Sevres, (2008) https://www.bipm.org/documents/20126/2071204/JCGM_100_2008_E.pdf/cb0ef43f-baa5-11cf-3f85-4dcd86f77bd6
Gy PM (1979) Sampling of Particulate Materials – Theory and Practice. Elsevier, Amsterdam, p 431
Ramsey MH (2016) Appropriate sampling for optimised measurement (ASOM), rather than the theory of sampling (TOS) approach, to ensure suitable measurement quality: a refutation of Esbensen and Wagner (2014). Geostandards Geoanal Res 40(4):571–581. https://doi.org/10.1111/ggr.12121
Magnusson B, Örnemark U (eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods – A Laboratory Guide to Method Validation and Related Topics, (2nd ed. 2014). ISBN 978–91–87461–59–0. Available from www.eurachem.org.
Williams A, Magnusson B (eds.) Eurachem/CITAC Guide: Use of uncertainty information in compliance assessment (2nd ed. 2021). ISBN 978–0–948926–38–9 https://www.eurachem.org/images/stories/Guides/pdf/MUC2021_P1_EN.pdf
Validation of Measurement Procedures that Include Sampling SG-VaMPIS, supplementary guidance in preparation by Eurachem/Eurolab Joint Task Group
Ramsey MH, Geelhoed B, Damant AP, Wood R (2011) Improved evaluation of measurement uncertainty from sampling by inclusion of between-sampler bias using sampling proficiency testing. Analyst 136(7):1313–1321. https://doi.org/10.1039/C0AN00705F
Commission Regulation (EC) No 563/2002 of 2 April 2002 amending Regulation (EC) No 466/2001 setting maximum levels for certain contaminants in foodstuffs, Official Journal of the European Communities, L 86/5 to L 86/6. https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2002:086:0005:0006:EN:PDF
Farrington D, Damant AP, Powell K, Ridsdale J, Walker M, Wood R (2006) A comparison of the extraction methods used in the UK nitrate residues monitoring program. J Associat Public Anal 34:1–11
RANOVA3, software for estimation of measurement uncertainty including that arising from sampling, from Analytical Methods Committee of Royal Society of Chemistry, London https://www.rsc.org/membership-and-community/connect-with-others/join-scientific-networks/subject-communities/analytical-science-community/amc/software/
Analytical Methods Committee (2001). Robust statistics: a method of coping with outliers. Technical Brief No.6, Royal Society of Chemistry, London http://www.rsc.org/Membership/Networking/InterestGroups/Analytical/AMC/TechnicalBriefs.asp
Thompson M, Fearn T (1996) What exactly is fitness for purpose in analytical measurement? Analyst 121:275–278
Lyn JA, Palestra IM, Ramsey MH, Damant AP, Wood R (2007) Modifying uncertainty from sampling to achieve fitness for purpose: a case study on nitrate in lettuce. Accred Quality Assur: J Quality Comp Reliab Chem Meas 12:67–74. https://doi.org/10.1007/s00769-006-0239-0
Lyn JA, Ramsey MH, Wood R (2003) Multi-analyte optimization of uncertainty in infant food analysis. Analyst 128:379–388
Lyn JA, Ramsey MH, Wood R (2002) Optimised uncertainty in food analysis: application and comparison between four contrasting ‘analyte-commodity’ combinations. Analyst 127:1252–1260
Author information
Authors and Affiliations
Contributions
M.H.R. wrote the main manuscript text. P.R. prepared figures and tables for presentation. Both authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendices
Appendix 1
Theoretical basis of optimised uncertainty (OU) method
The Optimised Uncertainty (OU) Method is one approach to setting the Target MU for a measurement procedure that includes sampling as well as chemical analysis. The OU method enables the setting of one fitness for purpose (FnFP) criterion that can be used to validate the whole measurement procedure. It can also optimise the relative proportions of MU that is generated by both the sampling and analytical procedures.
The equation that gives the total cost (formally expectation of loss) as a function of the MU (solid line in Fig. 4), taken from [14], is:
where:-
E(L) = Expectation of Loss (the formal term for the total cost).
C = Consequence cost.
ε = error limit and \(\upvarepsilon =\left|T-{c}_{m}\right|\)
T = threshold value.
cm = analyte concentration at which the optimisation is made.
smeas = standard deviation of the measurement values (i.e. standard uncertainty).
D = cost for unit variance for total measurement process (explanation in Equation A4).
Φ = probability that the true concentration will be compliant with the threshold, in the case of a false compliance case. By subtracting this probability from 1, the probability that the true concentration is non-compliant is calculated, hence the probability of misclassification. (Φ is given by function NORMSDIST in Excel™).
Calculated input parameters
Using the main input parameters (e.g. outline above in Table 3), further parameters can be calculated.
The cost for unit variance (actual s2samp) for sampling
The cost for unit variance (actual s2anal) for analysis
where Lsamp and Lanal are the unit costs of sampling and analysis, respectively, s2samp and s2anal are the variances of sampling and analysis from ANOVA.
The cost for unit variance for the total measurement process
Optimal apportionment of expenditure
Once the optimal value of uncertainty has been determined the optimal values of sampling variance (\({\nu }_{{\text{samp}}}{\prime}\)) and analytical variance (\({\nu }_{{\text{anal}}}{\prime}\)) are evaluated from the measurement variance (\({\nu }_{{\text{meas}}}{\prime})\). By taking the square root of these variance estimates, optimal uncertainties for sampling (s′samp) and analysis (s′anal) can be derived.
The optimum variance of sampling
The optimum variance of analysis
Subsequently the optimal costs of sampling and chemical analysis, L′samp and L′anal, respectively, are also computed.
The optimum cost of sampling
The optimum cost of analysis
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ramsey, M.H., Rostron, P.D. Measurement uncertainty from sampling and its role in validation of measurement procedures. Accred Qual Assur 29, 153–162 (2024). https://doi.org/10.1007/s00769-024-01575-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00769-024-01575-0