
Abstract
The Guide to the Expression of Uncertainty in Measurement (GUM) provides a framework for evaluating analytical data and characterizing their dispersion in a consistent manner. This is of eminent importance in the case of reference materials and their recommended values that are used for calibration of further measurements. The proper propagation of uncertainties for those data is essential. Guidance is provided in the GUM on how to calculate the combined standard uncertainty for a mean value or central value based on multiple individual measurements including their calibration uncertainty. However, according to published data, the guidance provided by GUM is not always properly applied in practice. Several published studies show calculated uncertainties much lower than those of input quantities. This may be caused by improper handling of the propagation of uncertainty components, thereby breaking the traceability chain for reported values. A simple check method using conventional statistical means is proposed to detect and to avoid such mistakes related to calibration uncertainties. It is intended to help to ensure a proper uncertainty propagation, to maintain a system of reliable and traceable reference materials. A spreadsheet is provided for the implementation of such a check. Example calculations for published data are presented.
Similar content being viewed by others


Introduction
Over the last decades, the consistency of scientific data reporting has increased considerably with the availability of accepted international guidelines. The Guide to the Expression of Uncertainty in Measurement (GUM) [1] and its supplements (see [2]) play a vital role as they provide a common set of procedures to ensure a consistent reporting of measurement data, accompanied by well-founded associated uncertainties. The term ‘combined standard uncertainty’ denotes the use of accepted principles in a measurement model to combine different components of uncertainty for a measurement result (law of propagation of uncertainty, clause 5 in [1]). However, its successful application is not always straightforward and deserves some discussion. It has been reported that even metrological institutions reporting data in compliance with GUM principles sometimes seem to underestimate data uncertainties, attributed to ‘dark uncertainty’ [3].
Proper uncertainty assessment is particularly important in addressing the properties of reference materials which are themselves used to calibrate further measurements worldwide. They have to ensure both the traceability of measurements [4] and their comparability and the use of a proper referencing strategy [5, 6]. The traceability of measurements is the ability to demonstrate a result of a measurement and its uncertainty in terms of the relevant SI unit. As δ-value isotope ratio measurements cannot presently be taken traceable to the SI system, they have to be made traceable to certified reference materials recognised as international standards (BIPM Traceability Exception). Those international standards like VSMOW2 (for hydrogen and oxygen) define the respective δ-scales (e.g. δ2H, δ18O). The traceability of measurements is then achieved by the use of reference materials (as similar as possible to the matrix and measurands) in an unbroken chain of comparisons back to the scale-defining international standards.
The important aspect is that the combined standard measurement uncertainty of the quantity value of a reference material in a calibration hierarchy has to incorporate the combined standard measurement uncertainty associated with the measured quantity value of the previous calibrator and must be evaluated and stated (see section 2.7 in [4]).
Therefore, for each generation of newly characterized reference materials, the combined standard uncertainties of their property values tend to gradually increase due to the calibration chain of existing reference materials for their establishment.
Unfortunately, this principle seems not to be fully implemented in a number of published studies on new reference materials and therefore breaks the traceability chain with possibly significant consequences.
This article aims to critically assess the published values and uncertainties of new stable isotope reference materials in various publications over the last decades and to check them for plausibility. It is motivated by the fact that calculated combined standard uncertainties for such new materials were in some cases by a factor of two or three lower than the assigned uncertainty of reference materials used for their calibration. This is in breach of principles of error propagation of the GUM.
A method will be presented for adequate conservative uncertainty propagation in case of multi-laboratory data evaluation. In order to keep focus on the main problem, only the most basic and relevant uncertainty components for this purpose are considered.
In the following, the term ‘sample’ is used for the new characterized reference material in a publication, and the term ‘calibration material’ is used for all those existing reference materials used for calibration of that sample.
First, the basic equation for two-point calibration will be presented; then, the uncertainty propagation in case of data from several laboratories will be discussed, and then, the proposed check method will be explained. Its application will be discussed in Annex A in a detailed example, and a large number of further examples using data from several publications are provided in a spreadsheet prepared for this purpose.
Calibration formula for stable isotope δ-scale measurements
The mostly used 2-point calibration formula for calibration of stable isotope ratio data for light elements in the δ-scale notation is applicable for mass spectrometric or laser spectrometric analyses. The example is given for carbon isotopes and their δ-definition [7]: δ13C = (Rsample − Rreference)/Rreference with R = r13/r12 as ratio R of isotope abundances ri of isotope i (atomic mass number i) of the given element carbon, and ‘reference’ referring to VPDB for definition of the zero-point of the δ13C scale (δ13Creference = 0). The δ-scale notation reports the dimensionless data commonly in per mill (‰), and it was suggested [8] to use equivalently the term mUr (a new notation which is followed by some cited publications).
The following basic two-point calibration formula applies:
with the following notation: δw13C denotes measured uncalibrated raw data (measured on machine working scale), δ13C denotes calibrated data on the VPDB/LSVEC δ13C-scale [9]. The subscripts ‘cal1’ and ‘cal2’ denote the two calibration standards used, in this case study the two reference materials NBS19 and LSVEC.Footnote 1
The above calibration Eq. (1) includes five input variables. Three of them describe measured raw data of the sample and the two standards (calibration materials) measured for the daily calibration (δw13Csample, δw13Ccal1, δw13Ccal2). These describe measurement data with statistical uncertainties of Type A [1]. The two other variables are the assigned uncertainties (Type B) of reference values of the two used standards ‘cal1’ and ‘cal2’ (δ13Ccal1, δ13Ccal2), as taken from the reference material certificate [1, 12]. The latter category could include contributions by the assessed inhomogeneity between individual bottles of the reference materials, or any remaining bias between laboratories which could not be directly corrected. Further details on this calibration formula can be found, for example, in [13, 14].
Uncertainty Propagation
The GUM [1] discusses in detail the use of variances for the calculation of uncertainties for measurements by use of standard deviations of measured data (type A) and by systematic effects like existing biases or the assigned uncertainty of reference materials (type B). Few aspects will be briefly repeated here to illustrate the proposed method of back-calculation of individual uncertainty components via variances from the published data and the combined uncertainty, when not all detailed information on measurements is available to the reader.
Two cases will be briefly discussed below: (a) several measurements of a sample taken in a single laboratory (possibly using different instruments, but using a joint calibration process) aggregated to a mean value with its combined standard uncertainty; (b) evaluation of sample measurements performed in different laboratories (possibly achieved by different methods, various number of measurements and different calibration means). The results are sometimes only available as summary information per laboratory (mean value, standard deviation, number of measurements) to derive a valid gross mean value with meaningful combined standard uncertainty. The main problems occur in the latter case b).
In all cases, a complete uncertainty budget for reference materials will include additional components addressing a potential inhomogeneity of the material, its storage stability and eventually further components. This aspect, however, will not further be discussed here as it basically is the addition of further static variance terms (of type B) to Eq. (2).
-
(a)
Single laboratory data set:
A single-laboratory stable isotope measurement data set typically consists of a series of individual measurements of the unknown sample and of normally two standards in case of a two-point calibration (and inclusion of quality assurance materials, further samples, etc.), performed multiple times possibly using various instruments, each one performed under repeatability conditions. Without restricting the possible complexity of settings, in this case the same five basic sources of uncertainty are to be considered and contributing as discussed in the previous section. These are three sources of Type A (statistical) measurement uncertainties associated with the measurements of the three materials, considered to be uncorrelated. In addition, the assigned uncertainties of the two reference materials used for calibration have to be included, which are fully correlated. Equation (2) applies.
For each single δ-value produced, its associated combined standard uncertainty u(δsample) can be derived as square root of its variance, from the calculation of the five individual variances as uncertainty components according to Eq. (2), being equivalent to equation 10 in the GUM section 5.1.2 [1] (and for simplicity omitting in the following formulas the ‘13C’ part at all δ-values):
with the following notation: f being the applicable calibration formula (here Eq. 1), and the u() terms indicating the respective uncertainty component (whether standard deviation or standard error of the mean), and each first term being in brackets being the partial derivative of the calibration formula for the indicated variable (its square is also called sensitivity factor). More on sensitivity factors can be found, for example, in [15].
The five partial derivatives of the calibration formula f (Eq. 1) in Eq. (2) are:
and they are used to calculate the combined uncertainty of the sample value.
Equation (2) is applicable strictly only for uncorrelated parameters (otherwise correlation terms have to be added); however, this condition can be achieved by an appropriate modification (see in clause 5.2.4 of [1]) by first treating the three uncorrelated terms of Eq. (2), and then adding only at a later stage the two last correlated terms of Eq. (2) separately.
In all practical cases even for a single laboratory, the reference value and uncertainty of a reference material will be calculated from a number of measurements. Then, the three uncertainty terms u(δw…) for measured data (of sample, cal1 and cal2) will represent effectively the contribution of these three standard deviations for those measurements.
To derive the uncertainty of the mean value, those three standard deviations are replaced by their respective standard-error-of-the-means (division of each standard deviation by the square root of the number of measurements). However, the two last terms in Eq. (2) stay unmodified, as the uncertainty assigned to the used calibration reference materials is independent of any number of performed measurements.
This results in the following modified Eq. (2a) (denoting each term in Eq. 2 only in an abbreviated form), e.g. for the first term: “(term1w-cal1)” = \({\left(\frac{\partial f}{\partial {{\delta }_{w}}_{\text{ cal1}}}\right)}^{2}\cdot u({{\delta }_{w}}_{\text{ cal1}}{)}^{2}\):
For simplicity, it is assumed that n=n1=n2=n3. The magnitude of the three measurement variance terms can be reduced by increasing the number n of repeated sample and standards measurements with dividing the individual respective variances by n (assuming for simplicity the same n for all measurements). However, the last two terms in Eq. (2) stem from the uncertainties assigned to the reference materials used stay constant, without any reduction, regardless of the number of repetitive measurements. They constitute completely correlated terms for all measurements. Therefore with increasing number of measurements n the final uncertainty will approximate the remaining calibration uncertainty from the two remaining terms [16].
The same principle applies in combining results from different instruments used in a single laboratory with common calibration principle and standards. In such case, instead of single measurements, the different mean values with standards errors of the means obtained by each instrument are combined to calculate a gross mean and its uncertainty. This is straightforward only if each instrument uses the same calibration reference materials, as then the calibration variance terms are all the same.
The combined standard uncertainty for the gross mean can be calculated in any of the cases above.
-
(b)
Combining multiple datasets as produced in different laboratories
In most cases—and for good reasons—the isotopic characterization of a new reference material is not performed at a single laboratory only but involves a group of selected expert laboratories. The merging of data from different laboratories follows the same GUM principles as in case (a) but needs to take into account the possible use of different instrumentation and of different calibration procedures by these laboratories. In addition, possible laboratory biases due to either applied analytical methods or to variable environmental conditions have to be considered.
The main difference in the process is the fact that all individual laboratories may have performed data following the process of Eq. (2), and therefore, all include their individual calibration uncertainty components. Thus, these data cannot be easily merged into a gross mean and gross uncertainty, as they are partially correlated due to the common calibration component.
For most experimentalists interested in proper data handling, but not being mathematicians, the requirements for handling correlated measurements can be a challenging experience [17, 18]. For stable isotopes, this is the case for even the easiest calibration formula with just five input quantities, where the creation of the correlation matrix requires the calculation of up to twenty double partial derivatives.
Two alternatives exist to deal with the correlations: (a) using Monte Carlo simulations to derive the effects of correlations—this approach is not discussed here further as it still needs some programming skills for users; (b) removing the data correlation caused by the unavoidable use of common calibration standards (see the last two variances in Eq. 2). The related suggestion to use different independent calibration standards for each laboratory [19] and thus being able to reduce even further the resulting uncertainty is no solution, as all stable isotope reference materials are linked to each other due to their calibration hierarchy and are thus all correlated to the scale-defining primary calibrants. Fortunately, for this second scenario (b) a real implementation solution is possible with low calculation efforts.
In the easiest case of a strict protocol applied by all laboratories, exactly the same calibration uncertainty (variances term4 and term5) applies to all individual data. These terms can therefore be temporarily subtracted from the variances in Eq. (2), and for the remaining measurement terms the same calculation procedure can be applied as above for use of different instruments in a single laboratory. Only then, when the gross measurement uncertainty is calculated, in a last step the variance of the calibration uncertainty (term4 and term5) is added again, and the combined standard uncertainty of the gross mean is calculated. This avoids the need to include covariance terms in the calculations.
In order to minimize potential complications in the calculation process, the careful design of a study limits the complexity of the evaluation, best done with a priori fixed rules for analytical sequences. This may include a fixed number of measurements for samples and standards, the mandatory use of the same standards for calibration in each laboratory and performing additional adequate quality checks to detect a possible laboratory bias. Otherwise, considerable approximations have to be applied, especially if the reference materials used differ from laboratory to laboratory, thus resulting in considerably varying calibration components (term4 and term5) for each laboratory. Fur such a case, exact mathematical formulas for solutions cannot be applied, necessitating other evaluation methods like Monte Carlo techniques. Real cases with such complications will be discussed shortly and appropriate calculations suggested (see example in Annex A).
In practice, this approach is a mathematically solid solution for a stringent measurement scheme as applied in all laboratories, using all the same number of measurements and same calibration standards. In other cases, approximations are to be used.
Description of the used check method
The variance of the overall mean value contains the variance contributions attributed to the necessary measurements (both of the sample and of all the reference materials used), the variance from the assigned uncertainty of the reference materials (as stated in the reference material certificate), plus several other possible variances related to other relevant uncertainty contributions (inhomogeneity assessment of the material, its long-term stability, any other relevant factor as stated in the publication). Fortunately, both the terms related to measurements and related to calibration can be re-calculated, and other terms can be easily incorporated when being stated in a publication. Uncorrelated variances are additive.
For a given publication, a comparison of the stated overall uncertainty with its re-evaluated major components (measurements, calibration) allows a statement on the compliance with the necessary uncertainty propagation. In case that the publication does not provide all details to exactly reprocess the data, still a basic re-evaluation is possible. This possibility may be especially useful if recommended values for reference materials would change at a later date, and a necessary retroactive adjustment of data in this publication is not directly possible anymore.
The suggested re-evaluation method requires the existence of the following basic information in the publication:
-
(a)
the overall mean value of the sample and its combined standard uncertainty (or its expanded uncertainty with stated k-factor);
-
(b)
in case of use of several instruments or laboratories, the individual data sets each consisting of the respective mean value, the standard uncertainty and the number of measurements, used in the publication to derive the overall mean value;
-
(c)
for each individual laboratory or instrument having performed measurements, statements on the used reference materials with their reference values and assigned uncertainties; and the measured mean values and measured uncertainties of these reference materials;
-
(d)
optional information on further uncertainty components included in the overall mean value uncertainty, like data on inhomogeneity level or on long term stability.
The square of the combined standard uncertainty of the overall mean value is its overall variance. According to Eq. (2), it equals the sum of all relevant variances in the evaluation process.
For any publication characterizing a new reference material, at least all data for the categories (a) to (c) have to be available. From the reported data on measured values for sample and standards, the variance for the measurements in each laboratory can be fully reconstructed using Eq. (4), even if individual measurement data are not published (see, e.g. [20]). In the supplementary Excel file, the same calculation is realised by use of a user-defined function called ‘sdAoM’ (‘standard deviation for Average of Means’).
In the following, a brief description is given on the process to apply the check method and to use the supplied Excel file. In Annex A, a full example for the numerical re-evaluation of a reference material is provided to illustrate the following description.
-
(a)
Recalculation of overall data (mean and Type A and Type B uncertainties) from published data
The data in a given publication can be used to recalculate the means and uncertainty components from measurements (Type A uncertainty), by using the available gross data for each laboratory or instrument, which consist for each data set at least of the individual mean, its standard deviation and the number of measurements.
The formula to derive the overall arithmetic mean value X from k individual mean values xi and number of related individual measurements ni with N= \(\sum_{i}^{k}{n}_{i}\) is:
The corresponding formula to calculate for X its related variance S2 from the given data (see, e.g. page 124 in [20]) is given by:
The square root of this variance S2 is the standard deviation S for all data used to derive the overall mean value X.
The data provided in such publication will also state the used calibration materials with their assigned mean values and assigned uncertainties. From this information, the uncertainty of the calibration process (uncertainty of Type B) can be calculated easily when the same calibration process is used by all laboratories. If the calibration process varies between laboratories, the calculation gets a bit more complicated as then approximations have to be applied.
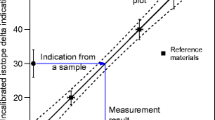
For a one-point calibration, the resulting calibration variance is calculated as square of the respective assigned uncertainty of the used reference material. As shown in Fig. 1 for the case of a two-point calibration with two reference materials of same uncertainty level, the theoretical lower uncertainty limit for a sample (with very large number of analyses and therefore vanishing influence of the measurement variances) follows the curved line. Approximately the same behaviour is expected from a multi-point calibration using several reference materials. In a first-order approximation for Fig. 1, taking the average value of the uncertainties of the reference materials to calculate the corresponding variance, it will result in a maximal 30 % overestimation of this component. It is considered as a conservative approach in the check process.

The theoretical lower calibration standard uncertainty limits are shown by the curved line for δ18O of samples intermediate to two calibration materials. Here used are VSMOW2 and SLAP2 (filled circles) with assigned combined standard uncertainties of 0.02 ‰). For GRESP [21], a theoretical minimal calibration uncertainty of 0.015 ‰ could be possible (open circle, neglecting all other uncertainty components, for example, due to measurement uncertainties). In practice, the achieved combined standard uncertainty for GRESP is much larger at 0.04 ‰ (filled circle)
All those data necessary for both calculations above will be present in any publication on the characterization of new reference materials. Basic statistical methods are sufficient for their calculation in usual cases.
An Excel template is provided as supplementary file for these calculations on any published reference materials data. Further input is needed for such calculation; this is first the overall gross mean and uncertainty of the sample as published, and secondly a decision what kind of uncertainty is stated in the publication at the individual laboratory level (whether these are just standard deviations of measured data, or they constitute combined standard uncertainties including the calibration process), as different calculations have to be performed depending on these two cases.
-
(b)
Comparison of means and uncertainties
After the recalculation of variances, the original published data may be easily compared to the re-evaluated data. The supplementary Excel file provides some standardized comparison results to facilitate the process, and feedback is given therein in case of potential incompatibilities.
The re-evaluated arithmetic sample mean should be generally comparable with the published mean value. Deviations for these two mean values may exist if in the publication either weighted means had been used or laboratory offsets across samples had been considered, as often the case in Bayesian evaluation methods.
The re-evaluated combined uncertainty from both measurement and calibration uncertainty components should be generally comparable with the published combined standard uncertainty of the sample, if GUM principles are followed.
Of particular interest is the comparison of the published overall uncertainty as claimed for the sample with one input uncertainty component, the assigned uncertainty from reference materials used. In case the combined standard uncertainty variance of the sample is lower than one of its input components, obviously a major problem exists.
As the re-evaluation with its uncertainty calculation considers only the five variance components of Eq. (2), the original published combined standard uncertainty is expected to be in general even larger (due to further uncertainty components included there like the material inhomogeneity).
In case of a publication stating significantly lower combined standard uncertainties for samples than those of the re-evaluation, a lot of caution is necessary to carefully examine statements to come up with a robust conclusion on the suitability of the sample data for their intended purpose.
Results from the re-evaluation of some published reference material data
In this section, several publications will be discussed, in which new reference materials were characterized (see Table 1). Some of their published results were checked with the method suggested in this publication, with the results indicated in the last two columns (‘Yes’ indicate comparable results, ‘No’ indicate significantly deviating results, brackets around statements denote mixed results). Those evaluated data are stored and made available in the supplementary Excel file.
IAEA, 2021 (GRESP reference material, water) [24]
Result in short: Published data and re-evaluation data are fully comparable, both for the mean and the uncertainty. All instrument used the same calibration approach (2-point, same calibration materials). The bias among different measurement instruments during the study was fully included by increasing the combined uncertainty accordingly.
The published report (IAEA 2021)[24] describes the calibration of the new water reference material GRESP for δ2H and δ18O directly versus two primary reference materials, by using more than ten different instruments/methods over a period of nearly ten years (over 5000 analyses in total). Only the analyte δ18O is discussed here. The original purpose was to try to reduce the combined uncertainty of the new material GRESP by applying a large number of high-precision measurements to an uncertainty level below that of the used calibration standards (Fig. 1). It was, however, realised that biases between individual instruments seriously increased the achievable uncertainty to a level much above those of the calibration standards.
Verkouteren 2004 (calibration of three NIST CO2 RMs, carbonates) [22]
Result in short: Published data and re-evaluation data are comparable, both for means and for uncertainties. All laboratories used the same strict measurement protocol with a defined number of measurements and sequence and using the same calibration approach (1-point, same calibration material).
A classical calibration study (Verkouteren 2004) [22] compiles data from six carbonate and carbon dioxide reference materials for δ13C and δ18O as analysed by seven laboratories using dual-inlet mass spectrometry. All laboratories followed a given strict analytical protocol, and calibrated data versus the NBS19 primary reference material. This study established a benchmark for further calibrations.
Coplen 2006 (re-calibration of carbonates, CO2 and organic RMs) [9]
Result in short: Published data and re-evaluation data show deviations for mean values (Bayesian approach versus single sample basic statistics). Uncertainties are mostly comparable with few exceptions. All laboratories used the same measurement protocol and used the same calibration approach (1-point, led to the suggestion of the 2-point normalization for the δ13C-scale introduced after this study).
A further publication Coplen et al. [9] extended the scope of the Verkouteren and Klinedinst [22] study to add further carbon reference materials, especially some of organic origin. The evaluation was done using a Bayesian model to include laboratory biases, which led to the recommendation of the second anchor LSVEC for the δ13C VPDB-scale so far realised only by one primary reference material NBS19. Four laboratories provided data following a given protocol and all calibrating data versus NBS19 (and LSVEC). At the time of that study, the problem of varying isotopic shifts in different LSVEC vials was not known, as discovered only in 2016 [25]. Therefore, one laboratory having reported a LSVEC value deviating a lot from those of the other laboratories was considered as being biased. This caused a large (wrong) bias correction when normalizing all mean data to a fixed LSVEC value (which was an understandable approach for normalization, but based on wrong assumptions as it is known today). Consequently, that published normalized mean value was significantly shifted.
Schimmelmann 2016 (USGS61-79 organic materials) [23]
Result in short: Published data and re-evaluation data are comparable for the mean values. However large differences occur for uncertainties for carbon and nitrogen, with the published combined standard uncertainties being by a factor of 2–3 lower than those of the re-evaluation. Each of the nine participating laboratories used its own set of different reference materials, even sometimes changing RMs between single runs. There was not common measurement protocol applied. As no related measurement data for the reference materials had been published, their respective uncertainties were estimated using related sample data from the publication.
In the publication of Schimmelmann et al. [23], 19 new organic reference materials for analysis of hydrogen, carbon and nitrogen were announced following an international calibration effort. This was the result of an immense preparation effort to create—over a period of some years—several sets of organic materials with distinct isotopic differences within each set achieved by use of spiked materials. Eleven laboratories were involved with a large number of measurements, and a considerable final evaluation effort was done. It culminated in the isotopic characterization of 19 new organic reference materials. However, the stated combined standard uncertainties for carbon and nitrogen δ-values of most of these new materials revealed a problem; they were even lower than the assigned uncertainties of reference materials used for their calibration (much below the possible theoretical limit as shown in Fig. 1). This seems to violate the principle of proper uncertainty propagation and unfortunately in a strict sense may leave the recommended values for these materials as being unsuitable according to clause 10 of the ISO Guide 35 [26] for use as secondary reference materials. This needs a corrective action, with a temporary solution by the recent re-evaluation as described below. Possibly a new evaluation of the data set would clarify root causes for this discrepancy, to make these valuable materials fully suitable for their intended purpose.
In the study [23], eleven laboratories participated with individually variable numbers of measurements for each sample. A Bayesian statistics approach was used in the publication. Most laboratories used different calibration materials. Some used a 2-point calibration, others a multi-point calibration. Without access to the raw data, it could not be fully clarified how individual measurements were calibrated in each laboratory.
In an attempt by the author to better understand supposed inconsistencies of the original data evaluation, a conventional statistical approach was developed and applied to the original data of that publication (and being briefly described in a supplementary file to [23]). Real problems for the original data evaluation became evident as the low uncertainties in the publication could not be confirmed or validated.
Qi et al. 2016 (USGS54 – USGS56 reference materials, wood) [27]
Result in short: Published δ13C data and re-evaluation data are comparable for the mean values. However, published combined standard uncertainties are unreasonable low, especially in view of a total of only 18 replicate measurements in three runs using an EA technique, resulting in an uncertainty for δ13C of only 0.01 ‰? Published δ18O and δ15N data and re-evaluation data are comparable for means and uncertainties.
The publication [27] characterized three wood materials for stable isotopes of hydrogen, carbon, nitrogen and oxygen. Here δ13C, δ15N and δ18O data were considered. Data for nitrogen and oxygen were comparable both for means and uncertainties. It is to be noted that the δ13C uncertainties as stated in the abstract imply that these materials would belong to the most accurately determined carbon stable isotope reference materials ever. In the performed 2-point calibration, the assigned uncertainty value for the normalization material LSVEC had been set to 0. In the same year in another publication the LSVEC uncertainty had been set to 0.15 ‰ ([23], including two joint co-authors), in view of previously discovered isotopic drift problems of that material. Considering this fact of a neglected significant uncertainty contribution in [27], the properly evaluated δ13C uncertainty for these three materials should be rather close to the 0.10 ‰ level.
Chartrand et al. 2019 (NRC sugar RMs) [28]
Result in short: Published δ13C data and re-evaluation data are comparable for the mean values. Published combined standard uncertainties seem to be consistently lower than the assigned uncertainties of the three reference materials used for calibration, however reviewing the isotopic compositions of reference materials and samples according to Fig. 1, the three samples could have been assessed effectively like in a single point calibration, then reflecting the uncertainty of the isotopically closest reference material being in that uncertainty range.
The study was using a random laboratory effects statistical model accounting for correlations. It considered and included uncertainties from the characterization as well as homogeneity and stability. The good performance of laboratories with a narrow data range obviously did not require many corrections, so mean values coincide with the basic statistics of the re-evaluation. The original uncertainty could be de facto interpreted as being close to that from a one-point calibration with the calibration material closest to the respective sugar δ13C isotopic composition.
Schimmelmann et al. 2020 (USGS82-USGS91, food matrix RMs) [29]
Result in short: Published δ13C data and re-evaluation data are comparable for the mean values. However large differences occur for δ13C uncertainties, the published combined standard uncertainties are lower by a factor of 2–3 compared to the re-evaluation uncertainties. Note that several reference materials used in this study for calibration had been characterized by Schimmelmann et al. [23] and are also subject to doubts on their uncertainties (this work).
The publication [29] characterized 10 food matrix related materials for stable isotopes of hydrogen, carbon, nitrogen, oxygen and sulphur. It is noted that very large k-factors were used in the reported data (k-factors between 4 and 9), which is an unusual practice and may cause problems when not appropriately recognized by readers. While the re-evaluation was done only for carbon, it is expected that also for nitrogen the uncertainty values could be also low. As the publication uses several reference materials characterized in Schimmelmann et al. [23] for calibration, which are also subject to possible underestimation of their uncertainties, this effect would be even larger when fully applied to the re-evaluation.
Results of the data re-evaluation
Table 1 provides an overview of the studies selected for re-evaluation and provides a general overview on the comparability of results obtained.
As an example of this evaluation approach based on the studies above, the original recommended values and uncertainties of reference materials are listed in Tables 2, 3 and 4 for carbon, nitrogen and oxygen: both the original published data and the re-evaluated data (this study, marked in bold) are listed.
For each stated material and evaluation line in Tables 2, 3 and 4, the respective full calculation can be found in the supplementary spreadsheet.
Discussion
Seven relevant publications on the characterization of stable isotope reference materials published during the last twenty years were selected and ten separate data sets on reference materials for one analyte each were extracted and re-evaluated. In regard to recommended mean values, in nine out of ten studies the comparability of mean values was confirmed. In one case, the used Bayesian evaluation method triggered a large correction of mean values due to the detection of supposed laboratory biases, which were indeed most probably caused by the isotopic variability of LSVEC not known at that time. In this case, a deviation of mean values was to be expected due to the (wrongly) applied laboratory bias correction versus a re-evaluation of individual samples only. With regard to reported uncertainties in the seven publications, their consistency check by the re-evaluation provided a scattered outcome. The re-evaluations of three datasets were in full conformity with the original reported uncertainties, and three more data sets were in partial conformance. However, the re-evaluation of uncertainties for four data sets resulted in significant discrepancies, with the original reported uncertainties found to be much lower than to be expected in view of the assigned uncertainties of used calibration materials.
The author has slightly changed the evaluation approach first discussed in supplementary material of [23] from the use of weighted means to arithmetic means (this work) for simplified calculation.
A few further remarks are provided related to the original published study [23]:
To the best knowledge of the author, in one of the major studies [23] no distinct heterogeneity study of the individual materials by one laboratory was performed or published on most of the new materials (with the exception of USGS61-USGS63), as it is required by ISO 17034 [30] and ISO Guide 35, clause 7 [26]. Thus, a mandatory and potentially significant uncertainty component for the assigned value was not included in the assessment of these materials. It is not possible to conclude whether the bias between laboratories is based on local measurement offset or on existing heterogeneities in the bottled material; therefore, an effect on the overall uncertainty for those materials cannot be excluded. This could possibly be resolved by a further study of the original measured data.
The use of the primary calibration material NBS19 for calibration of δ13C data in several of the example studies was mistakenly taken to imply—beside a zero-uncertainty for the scale definition—also a zero-uncertainty for its measurement by use of single units of this material. However, a zero-uncertainty definition does not apply for use of single units of any physically existing solid material at least due to the possible presence of bottle to bottle heterogeneity. This is an uncertainty component which needs to be included for consistency. The same applies for the use of the very old reference material VSMOW for δ18O.
Another serious complication for proper calibration of δ13C in the last ten years was the discovery around the years 2014–2016 of a significant isotopic variability of two international reference materials used regularly as secondary anchor in a two-point calibration process for δ13C data normalization. The two affected δ13C reference materials are LSVEC and USGS41, which resulted in a considerable increase of their assigned uncertainties (by a factor of about four) and the discontinuation of their distribution as δ13C reference materials. As the problem was detected only after the measurements of one major study [23] had already been performed, a retroactive correction of the measurement had to be carried out, increasing further the overall uncertainty for all laboratories having used those standards for calibration. For LSVEC, the observed range of 0.25 ‰ drifts in individual bottles towards more positive δ13C values is not fully covered by the stated increased assigned uncertainty of 0.15 ‰ around the formerly fixed value. All these effects could further increase calculated uncertainties.
Conclusion
The re-evaluated uncertainties for a large number of reference materials in this study are considered as a conservative estimate for newly assigned uncertainties of these reference materials, and they are suggested to be used until a thorough investigation using raw data is made available. The re-evaluation took into account a proper propagation of uncertainty, now in compliance with international recommendations on the reporting of uncertainties [26]. With the proposed revised uncertainty data and the few slightly changed reference values, those materials are now believed to be ready for use as calibrants for hydrogen, carbon and nitrogen δ-scale measurements. It is proposed to include the revised values in the forthcoming update of the Brand et al. [6] publication on stable isotope reference materials.
Similar basic re-evaluations could be applied to other recent studies on new reference materials. It is proposed to place much care in the design of future reference material assessment studies to avoid the underlying problems. Only then can the full potential of more sophisticated statistical approaches like the Bayesian method be fully utilized.
One root cause of unreliable δ13C uncertainty statements relates to the unfortunate long-term isotopic shifts in the two reference materials LSVEC and USGS41 discovered only in 2014. Previous data can hardly be corrected, as the isotopic shifts even varied significantly between individual bottles of these two materials. It is therefore recommended to completely abolish the use of these materials in laboratories and to establish a larger set of suitable reference materials as replacements. This would allow an easier detection of any such future potential isotopic drift in a single material. The proposed establishment of the VPDB2020 scale [11] is addressing this problem.
The assignment of a zero-uncertainty to physical available reference materials needs to be ceased, as it resulted in severe misconceptions when using calibration measurements of such materials by ignoring their measurement uncertainty.
Supplementary file
An Excel file is supplied, which provides altogether 69 performed data re-evaluations of reference material values from the discussed seven publications using the original data as published, provides an empty template for further calculations, and provides the re-evaluation formulas, including several functions in VBA macro language to facilitate such evaluation.
A Word file Annex A is providing the stepwise numerical calculations as performed by the method and used in the electronic Excel spreadsheet for the reference material USGS63 taken as example.
References
Joint Committee for Guides in Metrology (2008) Evaluation of measurement data—guide to the expression of uncertainty in measurement (GUM) (JCGM 100:2008)
Joint Committee for Guides in Metrology (2009), Evaluation of measurement data—an introduction to the “Guide to the expression of uncertainty in measurement” and related documents (JCGM 104:2009)
Thompson M (2011) Uncertainty functions, a compact way of summarising or specifying the behaviour of analytical systems. Trends Anal Chem 30:1168–1175. https://doi.org/10.1016/j.trac.2011.03.012
De Bievre P, Dybkaer R, Fajgelj A, Hibbert DB (2011) Metrological traceability of measurement results in chemistry: concepts and implementation (IUPAC Technical Report). Pure Appl Chem 83:1873–1935. https://doi.org/10.1351/PAC-REP-07-09-39
Werner RA, Brand WA (2001) Referencing strategies and techniques in stable isotope ratio analysis. Rapid Commun Mass Spectrom 15:501–519
Brand WA, Coplen TB, Vogl J et al (2014) Assessment of international reference materials for isotope-ratio analysis (IUPAC Technical Report). Pure Appl Chem 86:425–467. https://doi.org/10.1515/pac-2013-1023
Gonfiantini R (1981) The δ-notation and the mass-spectrometric measurement techniques. In: Gat JR, Gonfiantini R (eds) Stable Isotope Hydrology, deuterium and oxygen-18 in the water cycle. International Atomic Energy Agency, Vienna, pp 35–84
Brand WA, Coplen TB (2012) Stable isotope deltas: tiny, yet robust signatures in nature. Isotopes Environ Health Stud 48:393–410
Coplen TB, Brand WA, Gehre M et al (2006) New guidelines for δ13C measurements. Anal Chem 78:2439–2441
Assonov S, Gröning M, Fajgelj A et al (2020) Preparation and characterisation of IAEA-603, a new primary reference material aimed at the VPDB scale realisation for δ13C and δ18O determination. Rapid Commun Mass Spectrom 34:1–16. https://doi.org/10.1002/rcm.8867
Assonov S, Fajgelj A, Allison C, Gröning M (2021) On the metrological traceability and hierarchy of stable isotope reference materials aimed at realisation of the VPDB scale: revision of the VPDB δ13C scale based on multipoint scale-anchoring RMs. Rapid Commun Mass Spectrom 35:e9018. https://doi.org/10.1002/rcm.9018
ISO (2015) Reference materials—contents of certificates, labels and accompanying documentation (ISO Guide 31:2015). International Organization for Standardization, Geneva, Switzerland
Gröning M (2011) Improved water δ2H and δ18O calibration and calculation of measurement uncertainty using a simple software tool. Rapid Commun Mass Spectrom 25:2711–2720. https://doi.org/10.1002/rcm.5074
Gröning M (2021) SICalib216.zip file as link
Gröning M, Rozanski K (2003) Uncertainty assessment of environmental tritium measurements in water. Accred Qual Assur 8:359–366
van der Veen AMH, Pauwels J (2000) Uncertainty calculations in the certification of reference materials. 1. Principles of analysis of variance. Accred Qual Assur 5:464–469
Priel M, Désenfant M (2015) Implementation of the calibration’s VIM3 definition using the matrix of variance–covariance of input data. Accred Qual Assur 20:107–114. https://doi.org/10.1007/s00769-015-1107-6
Wiora J (2016) Problems and risks occurred during uncertainty evaluation of a quantity calculated from correlated parameters: a case study of pH measurement. Accred Qual Assur 21:33–39. https://doi.org/10.1007/s00769-015-1183-7
Hässelbarth W, Bremser W (1998) Derived measurement standards of reduced uncertainty—a contradiction? Accred Qual Assur 3:337–339. https://doi.org/10.1007/s00769-004-0782-5
Brandt S (2014) Data analysis. Statistical and computational methods for scientists and engineers, 4th edn. Springer, Heidelberg
IAEA (2021) Certification report on value assignment for the δ2H and δ18O stable isotopic composition in the water reference material GRESP (Greenland Summit Precipitation). International Atomic Energy Agency, Vienna
Verkouteren RM, Klinedinst DB (2004) Value assignment and uncertainty estimation of selected light stable isotope reference materials: RMs 8543–8545, RMs 8562–8564, and RM 8566. National Institute of Standards and Technology, Gaithersburg
Schimmelmann A, Qi H, Coplen TB et al (2016) Organic reference materials for hydrogen, carbon, and nitrogen stable isotope-ratio measurements: caffeines, n-alkanes, fatty acid methyl esters, glycines, l-valines, polyethylenes, and oils. Anal Chem 88:4294–4302. https://doi.org/10.1021/acs.analchem.5b04392
IAEA (2021) GRESP webpage (Excel data files). In: IAEA Ref. Prod. Environ. Trade. https://nucleus.iaea.org/sites/ReferenceMaterials/SitePages/Home.aspx. Accessed 26 Jan 2021
Assonov S, Gröning M, Fajgelj A (2016) IAEA stable isotope reference materials: addressing the needs of atmospheric greenhouse gas monitoring. In: 18th WMO/IAEA meeting on Carbon Dioxide, Other Greenhouse Gases and Related Measurement Techniques (GGMT-2015)—GAW Report 229. World Meteorological Organization Global Atmospheric Watch, Geneva, Switzerland
ISO (2017) Reference materials—guidance for characterization and assessment of homogeneity and stability (ISO Guide 35:2017). International Organization for Standardization, Geneva, Switzerland
Qi HP, Coplen TB, Jordan JA (2016) Three whole-wood isotopic reference materials, USGS54, USGS55, and USGS56, for δ2H, δ18O, δ13C, and δ15N measurements. Chem Geol 442:47–53. https://doi.org/10.1016/j.chemgeo.2016.07.017
Chartrand MMG, Meija J, Kumkrong P, Mester Z (2019) Three certified sugar reference materials for carbon isotope delta measurements. Rapid Commun Mass Spectrom 33:272–280. https://doi.org/10.1002/rcm.8357
Schimmelmann A, Qi H, Dunn PJH et al (2020) Food matrix reference materials for hydrogen, carbon, nitrogen, oxygen, and sulfur stable isotope-ratio measurements: collagens, flours, honeys, and vegetable oils. J Agric Food Chem 68:10852–10864. https://doi.org/10.1021/acs.jafc.0c02610
ISO (2016) General requirements for the competence of reference material producers (ISO 17034:2016). International Organization for Standardization, Geneva, Switzerland
Acknowledgements
This work is the result of intense discussions on the principles of evaluation of combined standard uncertainties with several colleagues at the International Atomic Energy Agency and the members of the Commission of Isotopic Abundances and Atomic Weights of the International Union of Pure and Applied Chemistry. The manuscript has greatly benefited from careful reviews by two anonymous reviewers.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has no relevant financial or non-financial interest to disclose. The author is a co-author of several of the studies taken as examples and discussed in this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gröning, M. Some pitfalls in the uncertainty evaluation of isotope delta reference materials. Accred Qual Assur 28, 101–114 (2023). https://doi.org/10.1007/s00769-022-01527-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00769-022-01527-6