
Abstract
As a catabolic process, autophagy through lysosomes degrades defective and damaged cellular materials to support homeostasis in stressful conditions. Therefore, autophagy dysregulation is associated with the induction of several human pathologies, including cancer. Although the role of autophagy in cancer progression has been extensively studied, many issues need to be addressed. The available evidence suggest that autophagy shows both cytoprotective and cytotoxic mechanisms. This dual role of autophagy in cancer has supplied a renewed interest in the development of novel and effective cancer therapies. Considering this, a deeper understanding of the molecular mechanisms of autophagy in cancer treatment is crucial. This article provides a summary of the recent advances regarding the dual and different mechanisms of autophagy-mediated therapeutic efficacy in cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
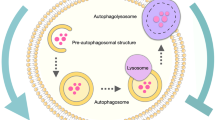
Autophagy is a catabolic and evolutionary conversed process in which cellular components and damaged organelles are degraded or recycled through lysosomal activity (Mizushima 2007). Hence, this process provides cells the ability to adapt to stressful conditions and prevent cellular damage and cell death. (Patergnani et al. 2015; Verma et al. 2021).
In recent times, substantial advances regarding the therapeutic potential of autophagy in cancer have led to several clinical trials expected to block autophagy in cancer. However, the role of autophagy in cancer remains elusive and varies in different contexts and stages of the disease (Singh et al. 2018). Specifically, while autophagy acts as a cytoprotective mechanism to enhance tumor progression, autophagy can promote the sensitivity of tumors to therapeutic agents (Folkerts et al. 2019; Deng et al. 2020; Yu et al. 2021; Ramakrishnan et al. 2012; Kim et al. 2014). Stem from these, to develop an appropriate therapeutic approach based on autophagy modulation in cancer, a comprehensive understanding of the connections existing between autophagy and cancer together with a clear identification of the molecular mechanisms characterizing this axis is important. This review seeks to provide insights into the recent advances regarding the dual role of autophagy in cancer treatment.
Autophagy-related genes and secreted factors promote drug resistance via autophagy activation
Autophagy-related genes play key roles in therapeutic resistance and hence approaches to silence these genes have recently emerged. For example, suppressing autophagy through genetic knockdown of autophagy‑related 4A (ATG4A) in MCF7 significantly improved the response of breast cancer cells to tamoxifen (Li and Zan 2022). Zhang et al. have proven in breast cancer that adaptor SH3BGRL can potentiate autophagy-induced doxorubicin resistance by promoting PIK3C3 translation and ATG12 stability. Furthermore, the genetic knockdown of either PIK3C3 or ATG12 attenuated autophagy and re-sensitized breast cancer cells to doxorubicin (Zhang et al. 2022). A mechanistic study has revealed that ecotropic viral integration site 1 (EVI1) upregulates ATG7 expression to impede the sensitivity of leukemia cells to imatinib treatment (Niu et al. 2020). In addition, autophagy induction through ATG5 upregulation promotes resistance of gastric cancer cells to fluorouracil (5-FU) (Pei et al. 2018). In a recent study, Tang et al. found that increased expression of Sestrin2 induces autophagy, inhibits apoptosis of tumor cells, and promotes doxorubicin, cisplatin, and methotrexate resistance in osteosarcoma (Tang et al. 2021). Tumor-derived cytokines are important regulators of autophagy-induced drug resistance. For example, autophagy induced by IL6-mediated activation of JAK2 promoted the resistance of colorectal cancer cells to oxaliplatin (Hu et al. 2021). In other experimental settings, the coordinative effect of exosomes and autophagy in drug resistance has been described. According to Pan et al. exosomal circATG4B activated autophagy to induce oxaliplatin resistance in colorectal cancer (Pan et al. 2022) (see Table 1).
Circulating RNAs promote drug resistance through autophagy activation
Circulating RNAs play an essential role in metastasis, proliferation, apoptosis resistance and chemoresistance through autophagy induction (Yuan et al. 2022; Liu et al. 2019). In breast cancer, Hsa_circ_0092276 was found to promote proliferation, protection of tumors from apoptosis and doxorubicin resistance (Wang et al. 2021). It has also been established that cirRNAs can promote chemoresistance by sponging miRNAs to upregulate downstream genes (Chen et al. 2019). In colorectal cancer, activation of autophagy through the circHIPK3/miR-637/STAT3 pathway suppressed the sensitivity of cancer cells to oxaliplatin (Zhang et al. 2019).
Long-noncoding RNAs promote drug resistance through autophagy activation
Emerging evidence have demonstrated that long-noncoding RNNs (LncRNAs) participate in autophagy-induced chemoresistance. In ovarian cancer, autophagy induction by lncRNA XIST promoted the resistance of cancer cells to carboplatin treatment (Xia et al. 2022). According to Hu et al. induction of autophagy via LINC00641/miR‑582‑5p axis promotes the resistance of gastric cells to oxaliplatin (Hu et al. 2020). In a similar study by Hou et al. LINC00963-induced autophagy promoted oxaliplatin resistance (Hou et al. 2021). Relatedly, activation of autophagy through H19/SAHH/DNMT3B promoted resistance of breast cancer cells to tamoxifen (Wang et al. 2019). Zhou et al. investigated how lncRNA PVT1 regulates autophagy-induced gemcitabine in pancreatic cancer cells and showed that PVT1 activated Wnt/β-catenin and enhanced autophagic activity leading to the resistance of cancer cells to gemcitabine (Zhou et al. 2020).
The tumor microenvironment promotes drug resistance through autophagy activation
Previous studies have demonstrated that immune cells infiltrating tumors can be influenced by interactions in the tumor microenvironment to support tumor progression (Kwantwi 2023b, 2023c; Sheng et al. 2023; Peng et al. 2021; Cai et al. 2020; Kwantwi et al. 2021a; Li et al. 2021). Notably, autophagy induction can shape immune cells to gain protumor functions leading to therapeutic resistance and tumor progression (Bustos et al. 2020; Ishimwe et al. 2020; Jiang et al. 2019; Janji et al. 2018). Emerging evidence indicates that autophagy can promote the tumor-promoting functions of TAMs which makes them attractive targets in TAMs-targeted therapies (Luo et al. 2020; Shan et al. 2017). In laryngeal cancer, autophagy inhibition using chloroquine (CQ) promoted the repolarization of M2 macrophage towards the M1 phenotype. Furthermore, these M1 TAMs showed higher phagocytotic activity towards Hep-2 laryngeal tumor cells and re-sensitized Hep-2 cells to cisplatin treatment, suggesting a role for autophagy activation in cisplatin resistance (Guo et al. 2019). Targeting autophagy through lysosomal inhibitors has shown to be a promising strategy for overcoming therapeutic resistance (Geisslinger et al. 2020; Vyas et al. 2022). In other experimental settings, the link between autophagy activation and impaired antigen presentation and their implication on therapeutic efficacy has been established. According to Li et al. autophagy activation is associated with the degradation of histocompatibility complex class I (MHC-I) and impaired antigen presentation which culminates in anti-PD-1 and anti-CTLA4 resistance in pancreatic ductal adenocarcinoma (Yamamoto et al. 2020). This provides the rationale that a combination of autophagy inhibitors may synergize with immune checkpoint inhibitors to improve treatment response (Li et al. 2017).
PD-L1 is known to suppress the antitumor immunity of the host, promoting tumor progression (Kwantwi et al. 2021b; Kwantwi 2023a). Specifically, tumor intrinsic PD-L1 can promote autophagy induction (Chen et al. 2022; Brogden et al. 2016; Clark et al. 2016) which negatively regulates treatment response. In bladder cancer, tumor cell-intrinsic PD-L1 activated mTORC1 and autophagy to promote cis‐platinum resistance (Zhang et al. 2021a).
Autophagy activation by stress promotes therapeutic resistance
Endoplasmic reticulum (ER) stress factors which include hypoxia, chemical factors, nutrient deficiency, and intracellular oxygen reactive species can trigger autophagy (Huang et al. 2018; Mrakovcic and Fröhlich 2019). Bouznad et al. recently investigated how IRE1A and XBP-1 regulate the malignant behavior and autophagy-mediated drug resistance in colorectal cancer and revealed that IRE1A-induced hypoxia upregulates XBP-1 and downregulates miR-34a. This promotes EMT and autophagy thereby inducing 5-FU resistance and tumor metastasis, particularly in p53 deficient tumors (Bouznad et al. 2023). Collectively, targeting p53 may be a novel strategy to overcome autophagy-mediated therapeutic resistance in cancer.
Nutrients including but not limited to glucose, amino acids, vitamins, inorganic salts, and lipids are essential for the growth of all cell types and for the maintenance of a steady state in response to adverse environmental conditions (Fan et al. 2022; Butler et al. 2021). Accordingly, since interfering with specific nutrients can be lethal to cells such as cancer, therapeutic strategies to target nutrient dependency in cancer have emerged. This is supported by a recent study conducted to evaluate the relationship between vitamin D and therapeutic resistance. Li et al. uncovered that activation of vitamin D receptor by calcitriol or its analog EB1089 sensitized MCF-7-derived antiestrogen-resistance LCC9 human breast cancer cells to tamoxifen treatment by blocking IRE1α–JNK-mediated autophagy (Li et al. 2021).
Autophagy activation by Insulin resistance promotes therapeutic resistance
Several lines of evidence strongly suggest that cancer-associated co-morbidities can contribute to the pathophysiology of cancer (Panigrahi and Ambs 2021; Anwar et al. 2021). According to Li et al. autophagy induced by insulin resistance in HCC cells can regulate ER stress to maintain homeostasis thereby promoting the resistance of HCC cells to cisplatin. Furthermore, inhibition of autophagy impaired IR-mediated cisplatin drug in HCC cells (Li et al. 2018a).
Autophagy activation by microbiota promotes drug resistance
The microbiota is known to play an important role in tumor development and progression (Cheng et al. 2021). Notably, evidence suggest that gut microbiota can modulate the immune response and affect therapeutic outcomes (Sivan et al. 2015; Viaud et al. 2013). Particularly, fusobacterium nucleatum has been implicated in autophagy-mediated therapeutic resistance. According to Yu and colleagues, downregulation of miR-18a and miR-4802 mediated by fusobacterium nucleatum impede the sensitivity of colorectal cancer to oxaliplatin- and 5-FU through TLR4 and MYD-mediated autophagy (Yu et al. 2017).
Autophagy activation promotes therapeutic efficacy
Besides autophagy-mediated therapeutic resistance already discussed, a huge body of evidence have also established that autophagy activation can improve therapeutic outcomes. Placenta specific 8 (PLAC8) which was first identified through the genomic-wide gene expression analysis of mid-gestation and placentas of embryos is restricted to the spongiotrophoblast layer of the mouse placenta. According to Chen et al. while PLCA8-induced autophagy inhibition promoted breast cancer resistance to adriamycin, activation of autophagy using rapamycin significantly improved the response of breast cancer to adriamycin treatment (Chen et al. 2021).In gastric cancer, autophagy suppression by Rab5a induces resistance of cancer cells to cisplatin treatment but knockdown of Rab5a activates autophagy and enhances the sensitivity of cancer cells to cisplatin treatment (Xu et al. 2018). According to Li et al. autophagy activation by miR-519a improves the sensitivity of glioblastoma to temozolomide treatment (Li et al. 2018b). Furthermore, ADRB2 inhibition in HCC led to enhanced efficacy of HCC cells to sorafenib (Wu et al. 2016). Relatedly, miR-21 inhibition activated autophagy and enhanced the sensitivity of HCC cells to sorafenib treatment (He et al. 2015). High expression of PARD3 in laryngeal squamous cell carcinoma (LSCC) inhibited autophagy and promoted LSCC sensitivity to chemoresistance (Gao et al. 2020). In breast cancer, autophagy activation through fucoidan/mTOR/p70S6K/TFEB pathway enhanced cancer cells to doxorubicin (Zhang et al. 2021b). Zhou et al. showed that chrysin inhibits carbonyl reductase 1 (CBR1) and triggers autophagy-dependent ferroptosis, enhancing the sensitivity of pancreatic cancer cells to gemcitabine (Zhou et al. 2021). In addition to the above, miR‑199a‑5p targeted p62 and inhibited ATG5-mediated autophagy through PI3K/Akt/mTOR pathway activation. This decreased the sensitivity of lung cancer cells to multiple chemotherapeutic drugs, including PTX, taxotere, topotecan, SN38, oxaliplatin, and vinorelbine. However, autophagy activation re-sensitized lung cancer cells to these chemotherapies (Zeng et al. 2021).
Accumulating evidence have shown that autophagy activation can improve treatment response to immune checkpoint inhibitors. It has been found that autophagy activation by V9302, a small-molecule inhibitor of glutamine metabolism, decreases B7H3 expression to boost antitumor immunity of CD8 + T cells. More importantly, anti-PD-1 together with V9302 treatment provided a synergistic effect in patients insensitive to anti-PD-1 therapy (Li et al. 2022a). Autophagy activation can promote the sensitivity of cancer cells to temozolomide by enhancing the polarization of M2 macrophages to M1 phenotype (Li et al. 2022b). In addition, over-expression of MAGE-A downregulated autophagy which resulted in melanoma resistance to CTLA-4 blockade (Shukla et al. 2018), re-enforcing the concept of autophagy activation as a valuable approach to increase the efficiency of immunotherapies.
Conclusion
Autophagy has become a hotspot in cancer research given its dual role in therapeutic responses. The evidence provided in his review suggests that inhibiting or activating autophagy for therapeutic purposes needs careful consideration. Either case can positively or negatively impact treatment efficacy. Concerning this difficulty, it remains unclear when and how autophagy must be activated or blocked to increase treatment efficiency in patients. However, although autophagy inhibitors would benefit patients with upregulated autophagy, while autophagy activation would be efficient for patients with downregulated autophagy, this approach requires further investigations and specific evaluation.
Data availability
Not applicable.
References
Anwar SL, Cahyono R, Prabowo D, Avanti WS, Choridah L, Dwianingsih EK et al (2021) Metabolic comorbidities and the association with risks of recurrent metastatic disease in breast cancer survivors. BMC Cancer 21(1):590
Bouznad N, Rokavec M, Öner MG, Hermeking H (2023) miR-34a and IRE1A/XBP-1(S) form a double-negative feedback loop to regulate hypoxia-induced EMT, metastasis, chemo-resistance and autophagy. Cancers 15(4):1143
Brogden KA, Vali S, Abbasi T (2016) PD-L1 is a diverse molecule regulating both tumor-intrinsic signaling and adaptive immunosuppression. Transl Cancer Res 5:S1396–S1399
Bustos SO, Antunes F, Rangel MC, Chammas R (2020) Emerging autophagy functions shape the tumor microenvironment and play a role in cancer progression—implications for cancer therapy. Front Oncol 10:606436
Butler M, van der Meer LT, van Leeuwen FN (2021) Amino acid depletion therapies: starving cancer cells to death. Trends Endocrinol Metab 32(6):367–381
Cai Z, Zhang M, Boafo Kwantwi L, Bi X, Zhang C, Cheng Z et al (2020) Breast cancer cells promote self-migration by secreting interleukin 8 to induce NET formation. Gene 754:144902
Chen H, Liu S, Li M, Huang P, Li X (2019) circ_0003418 inhibits tumorigenesis and cisplatin chemoresistance through Wnt/β-catenin pathway in hepatocellular carcinoma. Onco Targets Ther 12:9539–9549
Chen Y, Jia Y, Mao M, Gu Y, Xu C, Yang J et al (2021) PLAC8 promotes adriamycin resistance via blocking autophagy in breast cancer. J Cell Mol Med 25(14):6948–6962
Chen Z, Liu S, Xie P, Zhang B, Yu M, Yan J et al (2022) Tumor-derived PD1 and PD-L1 could promote hepatocellular carcinoma growth through autophagy induction in vitro. Biochem Biophys Res Commun 605:82–89
Cheng P, Shen P, Shan Y, Yang Y, Deng R, Chen W, et al (2021) Gut microbiota-mediated modulation of cancer progression and therapy efficacy. Front Cell Dev Biol 9
Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B et al (2016) Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Can Res 76(23):6964–6974
Deng Z, Lim J, Wang Q, Purtell K, Wu S, Palomo GM et al (2020) ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 16(5):917–931
Fan K, Liu Z, Gao M, Tu K, Xu Q, Zhang Y (2022) Targeting nutrient dependency in cancer treatment. Front Oncol 12:820173
Folkerts H, Hilgendorf S, Vellenga E, Bremer E, Wiersma VR (2019) The multifaceted role of autophagy in cancer and the microenvironment. Med Res Rev 39(2):517–560
Gao W, Guo H, Niu M, Zheng X, Zhang Y, Xue X et al (2020) circPARD3 drives malignant progression and chemoresistance of laryngeal squamous cell carcinoma by inhibiting autophagy through the PRKCI-Akt-mTOR pathway. Mol Cancer 19(1):166
Geisslinger F, Müller M, Vollmar AM, Bartel K (2020) Targeting lysosomes in cancer as promising strategy to overcome chemoresistance—a mini review. Front Oncol 10
Guo Y, Feng Y, Cui X, Wang Q, Pan X (2019) Autophagy inhibition induces the repolarisation of tumour-associated macrophages and enhances chemosensitivity of laryngeal cancer cells to cisplatin in mice. Cancer Immunol Immunother CII 68(12):1909–1920
He C, Dong X, Zhai B, Jiang X, Dong D, Li B et al (2015) MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget 6(30):28867–28881
Hou M, Li C, Dong S (2021) LINC00963/miR-4458 regulates the effect of oxaliplatin in gastric cancer by mediating autophagic flux through targeting of ATG16L1. Sci Rep 11(1):20951
Hu Y, Su Y, Lei X, Zhao H, Wang L, Xu T et al (2020) LINC00641/miR-582-5p mediate oxaliplatin resistance by activating autophagy in gastric adenocarcinoma. Sci Rep 10(1):14981
Hu F, Song D, Yan Y, Huang C, Shen C, Lan J et al (2021) IL-6 regulates autophagy and chemotherapy resistance by promoting BECN1 phosphorylation. Nat Commun 12(1):3651
Huang K, Chen Y, Zhang R, Wu Y, Ma Y, Fang X et al (2018) Honokiol induces apoptosis and autophagy via the ROS/ERK1/2 signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis 9(2):157
Ishimwe N, Zhang W, Qian J, Zhang Y, Wen L (2020) Autophagy regulation as a promising approach for improving cancer immunotherapy. Cancer Lett 475:34–42
Janji B, Berchem G, Chouaib S (2018) Targeting autophagy in the tumor microenvironment: new challenges and opportunities for regulating tumor immunity. Front Immunol 9:887
Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ et al (2019) The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer 18(1):17
Kim S, Ramakrishnan R, Lavilla-Alonso S, Chinnaiyan P, Rao N, Fowler E et al (2014) Radiation-induced autophagy potentiates immunotherapy of cancer via up-regulation of mannose 6-phosphate receptor on tumor cells in mice. Cancer Immunol Immunother CII 63(10):1009–1021
Kwantwi LB (2023a) Interplay between tumor-derived factors and tumor-associated neutrophils: opportunities for therapeutic interventions in cancer. Clin Transl Oncol 25(7):1963–1976
Kwantwi LB (2023b) Genetic alterations shape innate immune cells to foster immunosuppression and cancer immunotherapy resistance. Clin Exp Med 23(8):4289–4296
Kwantwi LB (2023c) The dual and multifaceted role of relaxin-2 in cancer. Clin Transl Oncol 25(10):2763–2771
Kwantwi LB, Wang S, Sheng Y, Wu Q (2021a) Multifaceted roles of CCL20 (C-C motif chemokine ligand 20): mechanisms and communication networks in breast cancer progression. Bioengineered 12(1):6923–6934
Kwantwi LB, Wang S, Zhang W, Peng W, Cai Z, Sheng Y et al (2021b) Tumor-associated neutrophils activated by tumor-derived CCL20 (C-C motif chemokine ligand 20) promote T cell immunosuppression via programmed death-ligand 1 (PD-L1) in breast cancer. Bioengineered 12(1):6996–7006
Li H, Li X, Liu S, Guo L, Zhang B, Zhang J et al (2017) Programmed cell death-1 (PD-1) checkpoint blockade in combination with a mammalian target of rapamycin inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology 66(6):1920–1933
Li L, Liu X, Zhou L, Wang W, Liu Z, Cheng Y et al (2018a) Autophagy plays a critical role in insulin resistance- mediated chemoresistance in hepatocellular carcinoma cells by regulating the ER stress. J Cancer 9(23):4314–4324
Li H, Chen L, Li JJ, Zhou Q, Huang A, Liu WW et al (2018b) miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J Hematol Oncol 11(1):70
Li M, Fang L, Kwantwi LB, He G, Luo W, Yang L et al (2021) N-Myc promotes angiogenesis and therapeutic resistance of prostate cancer by TEM8. Med Oncol 38(10):127
Li Q, Zhong X, Yao W, Yu J, Wang C, Li Z et al (2022a) Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity. J Biol Chem 298(4):101753
Li Z, Fu WJ, Chen XQ, Wang S, Deng RS, Tang XP et al (2022b) Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J Exp Clin Cancer Res CR 41(1):74
Li Q, Zan L (2022) Knockdown of ATG4A inhibits breast cancer progression and promotes tamoxifen chemosensitivity by suppressing autophagy. Mol Med Rep 25(3)
Li Y, Cook KL, Yu W, Jin L, Bouker KB, Clarke R, et al (2021) Inhibition of antiestrogen-promoted pro-survival autophagy and tamoxifen resistance in breast cancer through vitamin D receptor. Nutrients 13(5)
Liu Z, Zhou Y, Liang G, Ling Y, Tan W, Tan L et al (2019) Circular RNA hsa_circ_001783 regulates breast cancer progression via sponging miR-200c-3p. Cell Death Dis 10(2):55
Luo J, He Y, Meng F, Yan N, Zhang Y, Song W (2020) The role of autophagy in M2 polarization of macrophages induced by micro/nano topography. Int J Nanomed 15:7763–7774
Mizushima N (2007) Autophagy: process and function. Genes Dev 21(22):2861–2873
Mrakovcic M, Fröhlich LF (2019) Molecular determinants of cancer therapy resistance to HDAC inhibitor-induced autophagy. Cancers (Basel) 12(1)
Niu Y, Yang X, Chen Y, Jin X, Li L, Guo Y et al (2020) EVI1 induces autophagy to promote drug resistance via regulation of ATG7 expression in leukemia cells. Carcinogenesis 41(7):961–971
Pan Z, Zheng J, Zhang J, Lin J, Lai J, Lyu Z et al (2022) A novel protein encoded by exosomal CircATG4B induces oxaliplatin resistance in colorectal cancer by promoting autophagy. Adv Sci 9(35):2204513
Panigrahi G, Ambs S (2021) How comorbidities shape cancer biology and survival. Trends Cancer 7(6):488–495
Patergnani S, Missiroli S, Marchi S, Giorgi C (2015) Mitochondria-associated endoplasmic reticulum membranes microenvironment: targeting autophagic and apoptotic pathways in cancer therapy. Front Oncol 5:173
Pei G, Luo M, Ni X, Wu J, Wang S, Ma Y et al (2018) Autophagy facilitates metadherin-induced chemotherapy resistance through the AMPK/ATG5 pathway in gastric cancer. Cell Physiol Biochem 46(2):847–859
Peng W, Sheng Y, Xiao H, Ye Y, Kwantwi LB, Cheng L et al (2021) Lung adenocarcinoma cells promote self-migration and self-invasion by activating neutrophils to upregulate Notch3 expression of cancer cells. Front Mol Biosci 8:762729
Ramakrishnan R, Huang C, Cho HI, Lloyd M, Johnson J, Ren X et al (2012) Autophagy induced by conventional chemotherapy mediates tumor cell sensitivity to immunotherapy. Can Res 72(21):5483–5493
Shan M, Qin J, Jin F, Han X, Guan H, Li X et al (2017) Autophagy suppresses isoprenaline-induced M2 macrophage polarization via the ROS/ERK and mTOR signaling pathway. Free Radical Biol Med 110:432–443
Sheng Y, Peng W, Huang Y, Cheng L, Meng Y, Kwantwi LB et al (2023) Tumor-activated neutrophils promote metastasis in breast cancer via the G-CSF-RLN2-MMP-9 axis. J Leukoc Biol 113(4):383–399
Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C et al (2018) Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 173(3):624–33.e8
Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS et al (2018) Dual role of autophagy in hallmarks of cancer. Oncogene 37(9):1142–1158
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM et al (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (new York, NY) 350(6264):1084–1089
Tang Z, Wei X, Li T, Wang W, Wu H, Dong H et al (2021) Sestrin2-mediated autophagy contributes to drug resistance via endoplasmic reticulum stress in human osteosarcoma. Front Cell Dev Biol 9:722960
Verma AK, Bharti PS, Rafat S, Bhatt D, Goyal Y, Pandey KK et al (2021) Autophagy paradox of cancer: role, regulation, and duality. Oxid Med Cell Longev 2021:8832541
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D et al (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science (new York, NY) 342(6161):971–976
Vyas A, Gomez-Casal R, Cruz-Rangel S, Villanueva H, Sikora AG, Rajagopalan P et al (2022) Lysosomal inhibition sensitizes TMEM16A-expressing cancer cells to chemotherapy. Proc Natl Acad Sci 119(12):e2100670119
Wang J, Xie S, Yang J, Xiong H, Jia Y, Zhou Y et al (2019) The long noncoding RNA H19 promotes tamoxifen resistance in breast cancer via autophagy. J Hematol Oncol 12(1):81
Wang Q, Liang D, Shen P, Yu Y, Yan Y, You W (2021) Hsa_circ_0092276 promotes doxorubicin resistance in breast cancer cells by regulating autophagy via miR-348/ATG7 axis. Transl Oncol 14(8):101045
Wu FQ, Fang T, Yu LX, Lv GS, Lv HW, Liang D et al (2016) ADRB2 signaling promotes HCC progression and sorafenib resistance by inhibiting autophagic degradation of HIF1α. J Hepatol 65(2):314–324
Xia X, Li Z, Li Y, Ye F, Zhou X (2022) LncRNA XIST promotes carboplatin resistance of ovarian cancer through activating autophagy via targeting miR-506-3p/FOXP1 axis. Journal of Gynecologic Oncology 33(6):e81
Xu W, Shi Q, Qian X, Zhou B, Xu J, Zhu L et al (2018) Rab5a suppresses autophagy to promote drug resistance in cancer cells. Am J Transl Res 10(4):1229–1236
Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S et al (2020) Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581(7806):100–105
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J et al (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170(3):548–63.e16
Yu L, Shi Q, Jin Y, Liu Z, Li J, Sun W (2021) Blockage of AMPK-ULK1 pathway mediated autophagy promotes cell apoptosis to increase doxorubicin sensitivity in breast cancer (BC) cells: an in vitro study. BMC Cancer 21(1):195
Yuan Y, Zhang X, Fan X, Peng Y, Jin Z (2022) The emerging roles of circular RNA-mediated autophagy in tumorigenesis and cancer progression. Cell Death Discovery 8(1):385
Zeng T, Xu M, Zhang W, Gu X, Zhao F, Liu X, et al (2021) Autophagy inhibition and microRNA‑199a‑5p upregulation in paclitaxel‑resistant A549/T lung cancer cells. Oncol Rep 46(1)
Zhang Y, Li C, Liu X, Wang Y, Zhao R, Yang Y et al (2019) circHIPK3 promotes oxaliplatin-resistance in colorectal cancer through autophagy by sponging miR-637. EBioMedicine 48:277–288
Zhang D, Reyes RM, Osta E, Kari S, Gupta HB, Padron AS et al (2021a) Bladder cancer cell-intrinsic PD-L1 signals promote mTOR and autophagy activation that can be inhibited to improve cytotoxic chemotherapy. Cancer Med 10(6):2137–2152
Zhang N, Xue M, Wang Q, Liang H, Yang J, Pei Z et al (2021b) Inhibition of fucoidan on breast cancer cells and potential enhancement of their sensitivity to chemotherapy by regulating autophagy. Phytother Res 35(12):6904–6917
Zhang S, Liu X, Abdulmomen Ali Mohammed S, Li H, Cai W, Guan W et al (2022) Adaptor SH3BGRL drives autophagy-mediated chemoresistance through promoting PIK3C3 translation and ATG12 stability in breast cancers. Autophagy 18(8):1822–1840
Zhou C, Yi C, Yi Y, Qin W, Yan Y, Dong X et al (2020) LncRNA PVT1 promotes gemcitabine resistance of pancreatic cancer via activating Wnt/β-catenin and autophagy pathway through modulating the miR-619-5p/Pygo2 and miR-619-5p/ATG14 axes. Mol Cancer 19(1):118
Zhou L, Yang C, Zhong W, Wang Q, Zhang D, Zhang J et al (2021) Chrysin induces autophagy-dependent ferroptosis to increase chemosensitivity to gemcitabine by targeting CBR1 in pancreatic cancer cells. Biochem Pharmacol 193:114813
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium. No financial support was received for this study.
Author information
Authors and Affiliations
Contributions
LBK conceived the idea, wrote and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflict of interest
There is no conflict of interest to declare.
Ethical approval and consent to participate
Not applicable.
Consent for publication
All authors declare that they agree to submit the article for publication.
Additional information
Handling editor: E. Closs.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kwantwi, L.B. The dual role of autophagy in the regulation of cancer treatment. Amino Acids 56, 7 (2024). https://doi.org/10.1007/s00726-023-03364-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00726-023-03364-4