
Abstract
Mössbauer, Raman, X-ray diffraction and X-ray photoelectron spectroscopies were used to examine the effects of temperature on the structure of two aluminoceladonite samples. The process of oxidation of Fe2+ to Fe3+ ions started at about 350 °C for the sample richer in Al and at 300 °C for the sample somewhat lower Al-content. Mössbauer results show that this process may be associated with dehydroxylation or even initiate it. The first stage of dehydroxylation takes place at a temperature > 350 °C when the adjacent OH groups are replaced with a single residual oxygen atom. Up to ~500 °C, Fe ions do not migrate from cis-octahedra to trans-octahedra sites, but the coordination number of polyhedra changes from six to five. This temperature can be treated as the second stage of dehydroxylation. The temperature dependence on the integral intensity ratio between bands centered at ~590 and 705 cm−1 (I590/I705) clearly reflects the temperature at which six-coordinated polyhedra are transformed into five-coordinated polyhedra. X-ray photoelectron spectra obtained in the region of the Si2p, Al2p, Fe2p, K2p and O1s core levels, highlighted a route to identify the position of Si, Al, K and Fe cations in a structure of layered silicates with temperature. All the measurements show that the sample with a higher aluminum content and a lower iron content in octahedral sites starts to undergo a structural reorganization at a relatively higher temperature than the less aluminum-rich sample does. This suggests that iron may perform an important role in the initiation of the dehydroxylation of aluminoceladonites.
Similar content being viewed by others

Introduction
The hydrous layer silicates, commonly known as clay minerals, are widely used in many industrial applications such as paper production, foundries and the chemical- and food industries (Hradila et al. 2003; Murad and Fabris 2010; Moretto et al. 2011). A 2:1 layer is the basic structural unit of various 2:1 phyllosilicates. It consists of an edge-sharing octahedral sheet sandwiched by two corner-sharing tetrahedral sheets. In general, the octahedral sheet of a 2:1 layer contains three symmetrically independent sites differing in the mutual arrangement of OH groups and oxygen atoms coordinating octahedral cations. The unit cell contains two symmetrically independent octahedral, i.e., one trans-ostahedra (M1) and one cis-octahedra (M2) differing in the positions of OH groups. The adjacent cis-octahedra share an edge formed by two OH groups whereas, in trans-octahedra, hydroxyls are located opposite to each other (Muller et al. 1999; Zviagina et al. 2007).
The thermal behavior and related process of dehydroxylation of dioctahedral silicates has been the subject of many investigations (Tsipursky et al. 1985; Muller et al. 2000a, 2000b, 2000c). Dehydroxylation slightly modifies the coordinates of atoms other than the residual oxygen which replaces two hydroxyl groups and is located in the plane of the octahedral cations midway between them. This replacement induces a change of the coordination of the octahedral sheet cations, becoming five instead of six. Also, it has been shown quantitatively that the dehydroxylation of Fe, Mg-rich dioctahedral micas, when heated, is accompanied by a migration of the octahedral sheet cations from cis to trans-sites (Drits et al. 1995; Muller et al. 1998, 2000c; Dainyak et al. 2006). Generally, on heating phyllosilicates, four different processes may occur separately or concurrently, namely, oxidation of cations, loss of water, migration of cations to different sites and disintegration of the structure. However, many questions still remain unanswered about the coordination of the octahedral cations after dehydroxylation, the details of the reaction mechanism and the fate of dehydroxylates formed in nature.
In this study, X-ray diffraction, X-ray photoelectron spectroscopy, Mössbauer and Raman analysis were used to examine the effects of heating on the structure of Fe-bearing dioctahedral mica. The study tested conclusions about the structure of dehydroxylates and octahedral-cation migration in phyllosilicates and attempts a global explanation of the role of the content of Fe in the octahedral sheet in the dehydroxylation process. Additionally, X-ray photoelectron spectroscopy was used as a new tool to visualize cation arrangement in a structure of layer silicates at high temperatures.
Experimental details
Materials
Several secondary minerals have been found in the altered volcanic rocks of intermediate to basaltic composition in Lubiechowa near Świerzawa (Sudety Mountains) from which two types of clay mineral were collected from amygdales, one emerald-green, the other of dark-green. Based on electron-microprobe analyses, calculated formulas for these are K0.629Ca0.041(Mg0.545Fe2+0.180Mn0.002)(Fe3+0.430Al0.950)(Si3.755Al0.245)O10(OH)2 for emerald-green “celadonite” and K0.766Ca0.031(Mg0.584Fe2+0.134Mn0.001)(Fe3+0.562Al0.655)(Si3.962Al0.038)O10(OH)2 for dark-green “celadonite”, where FeO and Fe2O3 contents were determined by the use of the Fe3+/Fe2+ values obtained from Mössbauer spectra. According to criteria for the classification of clay minerals (Rieder et al. 1998; Drits et al. 1997; Tischendorf et al. 2007; Zviagina et al. 2015), the samples cannot be considered celadonite, but aluminoceladonite. Here, they are named as aluminoceladonites AlCel1 and AlCel2, respectively. Several types of measurements were performed for the studied samples.
X-ray diffraction
In-situ, high-temperature X-ray diffraction (XRD) measurements were performed by CuKα radiation using a PANalytical, X’Pert Pro Multi-Purpose Diffractometer with a Philips PW3040/60 X-ray generator and an Anton Paar HTK-16 thermal chamber. A nickel filter was placed in the diffracted-beam path to reduce the Kβ radiation. XRD patterns were collected in the range 4o2θ to 90 o2θ with a step size of 0.01o2θ and nominal time per step of 300 s, with the aid of a scanning X’Celerator detector (with an active length of 2.12o2θ).
Thermal analysis
The thermogravimetric analyses (TGA) and differential scanning calorimetry (DSC) were performed using the thermal analyzer NETZSCH Jupiter STA 449 F3. The curves were measured in the temperature range 25-800 °C at a heating rate of 5 °C/min. The samples (about 25–30 mg aliquots of powder) were measured in Al2O3 crucibles in a protective argon atmosphere.
57Fe Mössbauer spectroscopy
57Fe Mössbauer transmission spectra were recorded at room temperature using a MS96 spectrometer and a linear arrangement of a 57Co:Rh (25 mCi) source, a multichannel analyzer, an absorber and a detector. The spectrometer was calibrated at room temperature with a 30 μm thick α-Fe foil. The samples were pulverized in an agate mortar and heated for 8 h in air under static conditions in an electric furnace to a variety of temperatures in the range 100-600 °C. After each heating treatment, the Mössbauer measurements were taken. Numerical analysis of the Mössbauer spectra was performed using the WMOSS program (Prisecaru 2009-2016).
Raman spectroscopy
Raman spectra of the samples were obtained using a WITec confocal Raman microscope CRM alpha 300R equipped with a solid-state laser (λ = 532 nm) and a charge-coupled device (CCD) camera. The excitation laser radiation was coupled to the microscope through a single-mode optical fibre with a 50 μm diameter. An air Olympus MPLAN (100×/0.90NA) lens was used. The Raman spectra were initially laser-excited for a few hours to eliminate afterglow-type luminescence, and decrease the so-called Raman background effect. The spectrometer monochromator was calibrated using the emission lines of a Ne lamp. The temperature measurements were performed in-situ using a Linkam heating/cooling stage. The crystals of aluminosilicate were heated at a rate of 5 °C/min with a temperature accuracy of 0.1 °C in air.
X-ray photoelectron spectroscopy
XPS spectra were obtained using a PHI5700/660 Physical Electronics Photoelectron Spectrometer with monochromatic Al Kα X-ray radiation (1486.6 eV). The energy of the electrons was measured with a hemispherical analyzer with resolution of about 0.3 eV. Photoelectron emission was measured from a surface area with a diameter of 800 μm and at a take-off angle of 45 °. Due to the occurrence of a charge effect, a neutralizer was used for nonconductive samples. To determine the binding energy (BE), the C1s component was set at 285 eV. The solid sample was mounted on a PTS CLEAV RES/C-K sample holder (Prevac Company), cleaved in UHV conditions in a vacuum of about 3.10−9 Tr and transferred to the XPS chamber. The series of measurements in situ were performed at temperatures of 100, 300 and 550 °C. The vacuum in the XPS chamber during the temperature measurements did not exceed 5.10−8 Torr. The increase and stabilization of each temperature was executed over about 30 min using the Sample Heating Power Supply HEAT2-PS device and controlled by the use of EUROTHERM (Prevac Company). The HEAT2-PS worked in a resistive mode with a ramp of heating current of 1A/min.
Results
X-ray diffraction
High-temperature XRD patterns (Fig. S1 in the Electronic Supplementary Material (ESM)) were collected every 50 °C ranging from room temperature to 700 °C and refined by the use of Rietveld method. Table 1 shows the unit-cell parameters of the investigated samples obtained for the temperature range < 700 °C. The migration of octahedral cations from former cis-octahedra strongly decreases the repulsion between those cations bonded to residual oxygen anions and can be reflected by the temperature-dependence of the a and b parameter values (Table 1; Fig. 1). The decrease of a for AlCel1 is at 600 °C. For AlCel2, no significant decrease of a can be observed in this temperature region. The minimum values of the b parameters are observed near 650 °C and indicate that migration of octahedral cations takes place in these wide temperature ranges. Also, with the pure dehydroxylation increases the interlayer thickness, an increase which is reflected in the increase of the c parameter. This rapid increase of this parameter comes after heating at 550 °C for AlCel1 and 500 °C for AlCel2, which is also confirmed by the variation of the unit-cell volume versus temperature (Fig. 1). The relative displacement of the adjacent layers along the a axis is characterized by dependence of the Ic.cosβ/aI value on temperature (Fig. 1). The deviation of the observed Ic.cosβ/aI from the ideal −0.333 value (Drits et al. 2006) depends significantly on the difference in the sizes of the vacant and occupied octahedral site of the 2:1 layer. It is expected that increasing octahedral Al will be reflected in deviation decreases of the Ic.cosβ/aI values. The changes of this value from about 0.363 for the sample of AlCel1 heated at 550 °C to 0.303 for that heated at 700 °C, and from 0.367 for the sample of AlCel2 heated at 500 °C to 0.313 for that heated at 700 °C also indicate the transformation of cis into trans sites in these temperature ranges. More precisely, the coordination number of polyhedrons change from six into five. Detailed analysis of the unit-cell parameters (Table 1; Fig. 1) reveal additional effects which take place near temperatures of 100 °C and 250 °C for AlCel2 and, to a much weaker degree, at 350 °C for AlCel1. Changing values of the b and Ic.cosβ/aI parameters at 100 °C can be a result of the loss of bound molecular waters in the structure of the AlCel2. The decrease in unit-cell volume and unit-cell parameters at 350 °C for AlCel1, and at 250 °C for AlCel2, can be a result of deprotonation via Fe oxidation, which reduces the octahedral thickness.

The temperature-dependence of the a) b parameter, b) unit-cell volume, c) Ic.cosβ/aI values calculated from the XRD patterns for the investigated aluminoceladonites. The lines indicate the trends of the data and act as a guide to the eye
Thermal analysis
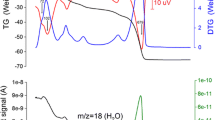
The dehydroxylation temperature can be determined from the DSC-TGA curves, as depicted in Fig. 2. These curves show some thermal movements that occur in the aluminoceladonite samples over a temperature range of ~30 °C to 800 °C. TGA/DSC experiments for the investigated aluminoceladonites show that a small loss of ~0.84 wt% occurs in the temperature range 50-100 °C for AlCel1 (Fig. 2a) and ~ 0.65 wt% between 50 and 150 °C for AlCel2 (Fig. 2a). The weight decrease, and the broad endothermic hump between the above temperature ranges, presumably result from the loss of loosely-bound molecular waters. For AlCel1, in the temperature ranges 100-550 °C and 550-780 °C, two areas of weight loss (3.15 wt% and 2.55 wt%) are observed and indicate the dehydroxylations. The endothermic events that occurred in the ranges 425-567 °C and 567-675 °C support this process. In the first region, the main sharp peak is seen at a temperature of 515 °C, preceded by a small peak at 478 °C. This region is deemed the first stage of dehydroxylation. It occurs in a wide temperature range, which is confirmed by a breadth of the endothermic hump which is about 142 °C wide. In the second temperature range, another endothermic peak related to the second stage of dehydroxylation is visible at 625 °C. For AlCel2, the dehydroxylations indicated by the mass change of <5.1% were detected from 200 to 660 °C. On the DSC curve for AlCel2, endothermic peaks though visible are not as sharp as for AlCel1 and appear at different temperatures than for AlCel1. The first peak is visible at 408 °C and a further two at 484 °C and 510 °C. For both samples, an endothermic peak was detected at 736 °C. This is probably the temperature at which the decomposition of the aluminoceladonites starts (Savko et al. 2015).

TGA/DTA curve of the investigated a) AlCel1 and b) AlCel2 samples
57Fe Mössbauer spectroscopy
Mössbauer spectra of the initial samples were fitted as a superposition of six doublets, the hyperfine parameters (Table S1 in the ESM) of which indicate iron located only in the octahedral sheet. The initial sample of AlCel1 contains about 27% Fe2+ ions. The Fe3+ ions prefer the cis-M2 site where 70% of them are located and the rest are present in trans-M1. In AlCel2, Fe2+ ions constitute 19% of total iron and are approximately equally divided between two octahedral sites with different local geometric environments around the Fe2+ ions. The hyperfine parameters of the ferric doublets indicate that the majority of the Fe3+ is located in the cis-M2 site in AlCel2. The remainder of these ions are in trans-M1 sites. It should be noted that, the ferrous doublets do not correspond to a specific crystallographic site (M1 or M2), but arise from local geometric and/or electronic distortion environments around the Fe atoms within the octahedral sheet of the micas (Rancourt 1994).
Selected Mössbauer spectra obtained after heating in air at definite temperatures are presented in Figs. 3 and 4. Figure 5 presents the changes in the hyperfine parameters for each component in the AlCel2 sample after heating at high temperatures. The fact that full line width at half maximum for all components connected with Fe3+ was the same during the fitting procedure for all Mössbauer spectra allows tracking of the changes in hyperfine parameters each component in. The full line width of doublets connected with Fe2+ changed, a change connected with visible changes in a shape of the Mössbauer spectrum and with processes taking place in the aluminoceladonite structure. Due to the dehydration stage, the escape of defects and changes that take place in neighboring octahedral sites, small changes in a shape of the Mössbauer spectra and hyperfine parameters of all components after heating at 100 °C is seen. After heating at 200 °C, an increase of abundance of Fe2+ that is generated by the presence of structural water is visible. The oxidation of Fe2+ to Fe3+ started at ~350 °C for AlCel1 and at 300 °C for AlCel2. This process took place over a wide temperature range and finished ~150 °C higher for each aluminoceladonite (Fig. 5). It may be assumed that at a temperature of ~450 °C for AlCel1 and 350 °C for AlCel2, the first stage of dehydroxylation takes place. Up to a temperature of ~550 °C for AlCel1 and 500 °C for AlCel2, the contribution of components connected with Fe3+ ions in M1 sites increases rapidly (Fig. 5c). To be precise, the two new doublets represent Fe3+ ions in M1 sites as a result of the decline of those connected with M2. The observed effects on the Mössbauer spectra at these temperatures can be treated as the second stage of dehydroxylation. It is also reflected by the changes in the contribution of Fe in M1 and M2 sites versus heating temperature (Fig. 6a and b). The average value of the isomer shifts (IS) in these new doublets is about 0.35 mm s−1 and the values of quadrupole splitting (QS) are about 1.65 mm s−1 and 2.15 mm s−1. The QS parameter does not depend on the total charge at each site but rather on the anisotropy in the charge distribution at the nucleus; the latter parameter increases with increasing octahedral distortion. For this reason, such large QS values indicate redistribution of the octahedral cations over all of the octahedral positions, with the result that the octahedral environments are very distorted. At 550 °C for AlCel1 and 500 °C for AlCel2 on the Mössbauer spectra (Figs. 2 and 3) and in Table S1 (in the ESM), doublets appear with values of isomer shift of about 0.19 mm s−1 and 0.25 mm s−1 that are characteristic for Fe3+ in tetrahedral coordination.

Room temperature Mössbauer spectra of AlCel1 obtained for the initial sample a) and after heating in air at temperature b) 400 °C and c) 600 °C. Quadrupole doublets, their assignment (M1, M2 – octahedral sites, T - dimeric pyramid) and contributions are indicated on the spectra

Room temperature Mössbauer spectra of AlCel2 obtained for a) the initial sample and after heating in air at temperature b) 200 °C, c) 300 °C, d) 400 °C, e) 500 °C and f) 600 °C. Quadrupole doublets, their assignment (M1, M2 – octahedral sites, T - dimeric pyramid) and contributions are indicated on the spectra

Plots of the a) isomer shift (IS), b) quadrupole splitting (QS) and c) fraction of Fe species visible on Mössbauer spectrum of AlCel2, as function of heating temperature. Errors bars are smaller than the symbols. The lines indicate the trends of the data and act as a guide to the eye

Raman spectra of the initial sample of aluminoceladonites as well as collected at different temperatures of sample obtained for a) AlCel1 (left column) and for b) AlCel2 (right column); the zone of hydroxyl stretching are added as insert. Bands that are highlighted dark grey correspond to the overlapping signals of tetrahedral and octahedral units, and bands that are highlighted light grey represent vibrations of aluminum-iron octahedrons and tetrahedrons
Raman spectroscopy
Raman spectra were collected every 50 °C ranging from room temperature to 600 °C. Post-processing analysis including baseline and luminescence correction as well as cosmic-ray removal was done using WitecProjectFour, and peak-fitting analysis using the GRAMS software package. Peak fitting was done by means of the Voigt function with a minimum number of components. The spectra of both investigated samples are presented in Fig. 6. In the hydroxyl-stretching range from 3400 to 3700 cm−1, five bands at 3627, 3608, 3582, 3564 and 3541 cm−1 for AlCel1 are seen, and five at 3620, 3609, 3588, 3567 and 3542 cm−1 for AlCel2 (Fig. 6; Table 2). The two bands with the highest Raman shifts at 3627 and 3620 cm−1 are interpreted as AlOHAl stretching vibrations. In the light of previous analysis for clay minerals (Zviagina et al. 2004, 2015, 2020), other bands represent AlMgOH, 2MgOH, MgFe3+OH and/or AlFe2+OH with a possible contribution from AlFe3+OH, and 2Fe3+OH. The temperature-dependent experiment revealed only slight alteration in the band arrangement in the hydroxyl region (3500–3600 cm−1). The main changes were found to be connected to modification of the band intensity, whereas the band position linked to the vibration of hydroxyl groups at tetrahedral and octahedral units, especially at MgFe2+OH or AlFe2+OH, AlFe3+OH, 2Fe3+OH and MgFe3+OH, remained unchanged. Interestingly, temperatures >500 °C (AlCel1) and > 400 °C (AlCel2) practically deactivate vibration originating from the OH modes due to the first stage of dehydroxylation. This effect can also be explained as a result of molecular mobility which, in the case of aluminoceladonite, may lead to the loss of the hydrogen-bonding scheme. This observation is strictly correlated with the analysis of bands ascribed to the stretching vibration within (AlnSi4-nO10)3− units, i.e., in the 1150–950 cm−1 region (Povarennykh 1978). Here, the intensity of the main bands centered at 1130 and 1083 cm−1 (AlCel1) and at 1144 and 1113 cm−1 (AlCel2) decrease up to 500 °C and to 400 °C, respectively. With both samples, those bands vanish completely above these temperatures.
The temperature-dependent experiment also revealed a decline in hydrogen bonding strength that may be linked to the molecular re-arrangement within the tetrahedral sheets, including mutual rotation of silica tetrahedra. The reason for this structural reconfiguration is not completely clear, especially in the context of band analysis of this 1150–950 cm−1 region. Therefore, to shed light on this problem, the analyses of the bands located below 950 cm−1 were taken into consideration. Here, huge differences were found in the position of bands and their intensities in analysis of the 750–500 cm−1 range - differences linked to the molecular vibration within tetrahedral and octahedral sheets (Fig. 6). For the record, the bands centred in between 710 and 690 cm−1 correspond to the overlapping signal of the stretching vibration of aluminum in tetrahedral and octahedral coordination as well as nonbridging and bridging oxygen in relation to the Ag symmetry mode (McKeown et al. 1999). The appearance of the band between 690 and 600 cm−1 in turn may originate from the translational modes of i) nonbridging oxygen in the yz plane, ii) bridging oxygen in the yz plane as for the Ag symmetry mode and iii) non-bridging oxygen in the xz plane as for the Bg symmetry mode (McKeown et al.1999; Wang et al. 2015). Similarly, well-defined bands located at 600–500 cm−1 derived from the overlapping signal of ions occupying the tetrahedral as well as octahedral coordination. Tetrahedrally-coordinated ions are involved in the bending motion related to the xy plane (Ag and Bg symmetry mode), while octahedrally-coordinated ions bending motion in the translational modes in the xz plane (McKeown et al. 1999). Based on temperature-dependent data, bands located at 549 cm−1 (AlCel1) or 553 cm−1 (AlCel2) in the initial samples were shifted, respectively, towards lower frequencies and are found at 523 cm−1 and 544 cm−1 in the samples annealed at 400 °C and 350 °C, respectively. At the same time, an increase of full width at half maximum (FWHM) suggests strong molecular reorganization due to the heating. It is notable that those alterations correlate quite well with modifications within the hydroxyl group region and within tetrahedral sheets. This may suggest that the decrease in the hydrogen bonding scheme induced simultaneous rotation of tetrahedral- and octahedral units. Furthermore, the octahedral positions are not fully charge-balanced due to isomorphic substitutions occurring between Al3+, Fe3+, Mg2+and Fe2+ that are atypical for celadonite micas. As a result, the temperature may additionally intensify variable structural effects by introducing structural point defects into the crystal structure, as is especially well seen in the case of AlCel1 where many new low intensity bands appear on the Raman spectrum (Fig. 6).
X-ray photoelectron spectroscopy
One of the most powerful direct methods for investigating electronic structure, chemical bonds, and the nearest surroundings of atoms in substances is X-ray photoelectron spectroscopy. As XPS signals arise from within about 1–10 nm of the outermost surface layers of investigated samples (Elmi et al. 2016), this method is generally treated as a surface sensitivity method. Discrepancies, e.g., apparent shifts of the photoelectron peaks, can be explained as due to selective attenuation of some signals (Bhattacharyya 1993). Shifts due to surface charging do not depend on the thickness of the sample, but on whether or not there is an adsorbed layer on the mica surface that reduces the charging and, hence, the shift is also reduced. Moreover, many solid systems can exhibit different compositions at their surfaces due to oxidation, layering, or other surface forces. However, the surface analysis of clay minerals is indeed representative of the overall bulk composition. This is to be expected since clay mineral layers should be of similar composition whether at the surface or in the center of an aggregate. Therefore, the interpretation of the XPS spectra of the investigated aluminoceladonite can provide information on the molecular environment, i.e., oxidation state, multiplet structure, chemical bonding, etc. with an error of <10%. This can enable visualization of cation arrangement in a layer-silicate structure at high temperatures. Reference survey spectra obtained in a wide range of energies, and high-resolution core lines of C1s, O1s, Si2p, Al2p, Fe2p and K2p, were recorded at room temperature. Then, the series of measurements in situ were performed at temperatures of 100, 300 and 550 °C. Quantification of XPS spectra utilizing peak area and peak height sensitivity factor was possible thanks to the Multipak Physical Electronics application. The XPS core level spectra were fitted with Gaussian-Lorentzian functions. XPS spectra obtained in the region of the Si2p, Al2p, Fe2p, K2p and O1s core levels for the aluminoceladonite AlCel2 are shown on Fig. 8.
The Si2p spectrum should show a doublet branching ratio of 2:1 for Si 2p3/2 to Si 2p1/2 with a spin-orbit splitting of about 1 eV difference. Values of Si2p binding energies show Si4+ in tetrahedral coordination (Bhattacharyya 1993; Biino and Gröning 1998; Elmi et al. 2014, 2016). These spectra in Fig. 8 present two Si2p (3/2, 1/2) doublets. The silicon binding energy shows that different populations of silicon correlate with substitutions in the tetrahedral sheet. A lower Si2p binding energy may result from a negative charge increase on the silicate framework as a result of Si4+ replacement by Al3+. Our results indicate that the tetrahedral sheet contains only Si in tetrahedral coordination with some Al3+ substitution. On the basis of the area ratio of Si2p components, the ratio of Si4+ in tetrahedral coordination to Al3+ can be calculated; for AlCel2 this ratio is 72/28. Because, the Al2p spectra have evident asymmetry in peak shape, they were decomposed into two Al2p doublets (Fig. 8). The obtained Al binding energy value is very close to both fourfold and sixfold coordination. However, an octahedral sheet must compensate for the charge of two relatively negative and adjacent tetrahedral sheets. The positive nature of the octahedral sheet coupled with the relatively negative silicate sheet results in more ionic Al–O bonds. The enhanced ionicity of the Al–O bond may be reflected in the relatively higher Al2p binding energy value. Also, the presence of OH and/or F enhances the positive character of the Al resulting in a moderate increase in the Al2p binding energy (Barr et al. (1997)). That is why the Al2p3/2 line located at a binding energy of 74.3 eV relates to Al present in octahedral coordination, while the other line with a BE of 72.8 eV arises from Al in tetrahedral coordination. The ratio of Al ions in tetrahedral to those in octahedral coordination is 27/73 and is very close to those obtained for Si2p, supporting the correctness of our interpretation. Heating at 100 °C causes the decrease of the intensity lines located at BE of 102.8 eV for the Si2p spectrum and 74.3 eV for the Al2p spectrum. The calculated ratio of Si4+ in tetrahedral coordination to Al3+ which substituted Si4+ is 85/15 and the ratio of Al ions in tetrahedral coordination to these in octahedral coordination is 14/86. Heating at 300 °C does not lead to significant changes in the Si2p and Al2p spectra but, after heating at a temperature of 550 °C, these spectra become almost symmetrical. The ratios of the intensity lines mentioned above for Si are 91/9 at 300 °C and 97/3 at 550 °C. For the Al2p spectra, the obtained ratios amount to 12/88 at 300 °C and 97/3 at 550 °C.
Potassium K2p has clearly spaced spin-orbit components (Fig. 8). Because, the K2p spectra have asymmetric peak shapes, they were decomposed into two K2p doublets. The K2p3/2 binding energies at 294.4 eV and 292.8 eV are connected with the K+ ions occupying interlayer sites between two 2:1 layers. The area ratio for the K2p doublets is 80/20 for AlCel2, which results from the ratio of K+ and Ca2+, respectively. Obtained the calcium content is too high and it may suggest that not only Ca2+ is substituting the K+ in interlayer sites. This explanation is supported by K2p spectra obtained at 100 °C and 300 °C. As is seen on Fig. 8, the intensity line of K2p3/2 with a binding energy of 294.4 eV decreases and the area ratio for the doublets of K2p is 88/12 at 100 °C and 95/5 at 300 °C. This is also supported by XPS spectra of O1s regions obtained at the studied temperatures (Fig. 8). The O1s core-level line of the initial sample of dark-green celadonite comprises three separate peaks located at binding energies of 530.2, 531.7 and 533 eV, which can be related to the existence of different forms of oxygen bondings. The most intense line at a binding energy of 531.7 eV is typical of lattice oxygen O2− (Kloprogge and Wood, 2018) and the peak positioned at the highest binding energy of 533 eV corresponds to hydroxyl oxygen OH− (Schingaro et al. 2013). The lowest intensity line at 530.2 eV is due to the presence of O-Me bonds or H2O or the sum of both. The intensity of this line decreases after heating at 100 °C, which confirms the presence of a small amount of structural water in interlayer sites in the initial sample. Heating at 100 °C also causes an increase of the intensity line corresponding to hydroxyl oxygen at a binding energy of 533 eV; this can be an effect of reaction of the water molecule H2O with oxygen O2− creating OH− and OH groups. At 300 °C, the ratio of the peaks located at binding energies of 531.7 and 533 eV is almost the same but, at 550 °C, the intensity of the hydroxyl oxygen line increases again.
The Fe2p spectra are characterised by doublet structures relating to the spin-orbit splitting between 2p1/2 and 2p3/2 states. In addition to the multiplet structures, shake-up satellite peaks arising from intrinsic energy losses when the photoelectron leaves the hosting atom, are present. Deconvolution of the Fe2p peak (Fig. 8) shows three main components. The first, a weaker component related to the Fe2+ valence state, is a doublet with binding energies Fe2p3/2 = 709.5 eV and Fe2p1/2 = 723 eV. The second, the highest component, is characteristic of the Fe3+ state observed as a multiplet with binding energies of the main component Fe2p3/2 = 712 eV and Fe2p1/2 = 725.5 eV. The third component at a binding energy = 714.4 eV is associated with the Fe3+ state. The Fe2+/Fe3+ ratio was determined to be 18/82. The area ratio of the peaks at binding energies = 712 eV and 714.2 eV is 62/20 and reflects the M2/M1 ratio of Fe3+ ions in octahedral positions. The ~2 eV distance between these components can reflect dehydroxylated sites. Small changes of the Fe2p spectra are observed at 300 °C where the obtained Fe2+/Fe3+ ratio is 22/78 and the ratio M2/M1 of Fe3+ ions is 56/22, based on the area ratio of the Fe2p lines. The main changes of the Fe2p spectrum are observe at 550 °C where the binding energies of the peaks and their intensities change. All of the doublets observed are connected with Fe3+ ions. The two peaks located at 710.4 and 712.5 eV can be associated with Fe in M1 sites and that with a binding energy of 714.2 eV with a small contribution of iron in dimeric pyramid sites. The area ratio of peaks at bonding energies = 710.4 eV and 712.5 to that at 714.2 eV is 83/17, which is similar to the ratio 87/13 obtained from the Mössbauer studies at this temperature.
Discussion
TGA-DSC analysis reveal a small weight loss in the temperature range 50-150 °C that can result of the loss of bound molecular waters in the structure of the investigated aluminoceladonites. The endothermic events, areas of weight loss and the temperature-dependence of the unit-cell parameters indicate two-stage process of the dehydroxylation. The first stage of dehydroxylation takes place at a temperature of about 450 °C for AlCel1 and at a 350 °C for AlCel2, when the adjacent OH groups are replaced with a single residual oxygen atom. In turn, temperature about 550 °C for AlCel1 and 500 °C for AlCel2 can be treated as the temperature where the second stage of dehydroxylation started. Finally, sample decomposition process starts at 736 °C.
The crystal-chemical models obtained due to 57Fe Mössbauer spectroscopy (Tsipursky and Drits (1984); Dainyak et al. 2004, 2006, 2009, 2013; Zviagina et al. 2015) have shown that the values of the fitted quadrupole doublets Fe3+ and Fe2+ (visible on the Mössbauer spectra) may correspond to specific local cationic arrangements around these ions. Such cation distribution reconstructions can indicate random or ordered distribution of octahedral cations and can also display Fe clustering or domain structure. Due to above, the inner doublets with quadrupole splitting of 0.17 mm s−1 and 0.19 mm s−1 is assigned to Fe3+ with the three nearest Fe3+ ions. The quadrupole splitting of 0.49 mm s−1 and 0.54 mm s−1 reflects Fe3+ in the arrangement of MgFe2+Fe3+ and 2AlFe3+, respectively. The value of QS, which is 0.81 mm s−1, is close to the theoretical value responsible for Fe3+ in the 3Al arrangement. Though AlCel1 contains more Al ions than AlCel2, no doublets reflecting Fe3+ ions with 3Al arrangement are observed on the Mössbauer spectra, suggesting that Al ions are steadily ordered in octahedral sites. The large values of quadrupole splitting, i.e., 1.01, 1.29 and 1.44 mm s−1, associated with the trans-OH (M1) sites, are the consequence of Fe3+ in arrangements AlMgFe3+, 2Fe3+Mg and 2AlMg, respectively. For AlCel1, a higher proportion of components related to the Al environment than in AlCel2 is observed due to the higher Al content. A similar interpretation of the quadrupole splitting can be made for the local cation arrangements around Fe2+ ions. The smallest value of QS (1.66 mm s−1) is connected with the arrangement of 2MgFe2+ ions and the highest value (2.77 mm s−1) with 2Fe3+Fe2+. Intermediate QS reflect an almost similar arrangement, i.e., 2.23 mm s−1 is the result of 2Fe2+Al and 2.66 mm s−1 is the result of AlMgFe2+ local cation distributions. The above interpretation of Mössbauer spectra suggests that Fe3+ ions are not randomly located in the structure of the studied samples but in some position within the preferred neighbourhood.
According to so-performed analysis, a determined temperatures of the dehydroxylation by analysis of Mössbauer spectra turned out to be in good agreement with the XRD and DSC-TGA results. However, Mössbauer spectroscopy results indicate additional factors responsible for initiation of the process of dehydroxylation in the investigated aluminoceladonite. Namely, at temperature ~ 350 °C for AlCel1 and at 300 °C for AlCel2 the oxidation of Fe2+ to Fe3 started. In the oxidation process, the charge balance can be provided by the loss of protons; the occurrence of proton jumps between neighboring OH− groups and O2− ions may be postulated. A proton jump or proton exchange causes a break-down of the ordering of the OH− groups and the reduction of bond strength between Fe and hydroxyl. In facilitating loss of one of these groups, oxidation of Fe2+ may thus be associated with dehydroxylation or even initiate the process. The first stage of dehydroxylation takes place as the adjacent OH groups are replaced by a single residual oxygen atom. In this stage of dehydroxylation Fe ions do not migrate from M2 to M1 sites, but coordination numbers undergo change. This replacement of OH by O induces a change in the coordination of the octahedral sheet cations, becoming five instead of six. Additionally, above temperature 550 °C for AlCel1 and 500 °C for AlCel2, where second stage of dehydroxylation take place, on Mössbauer spectra appear doublet characteristic for Fe3+ in tetrahedral coordination. This can be the effect of the remodelling of some M1 coordination polyhedrons. The changing of coordination number from five to four is associated with detachment of an OH group or of O with the result that a dimeric pyramid can be formed.
An interesting supply of the previously mentioned techniques provides temperature-dependent Raman and XPS data. First of all, Raman spectra reveal huge differences in the position of bands and their intensities in the 750–500 cm−1 range were found (Fig. 6). More precisely, a gradual decrease of the intensity of the band centered at 706 cm−1 (AlCel1) and 708 cm−1 (AlCel2) was correlated with an increase of the band at 567 cm−1 (AlCel1) and 575 cm−1 (AlCel2). Additionally, the peak-fitting analysis was used to estimate the integrated intensity of two bands at ~705 cm−1 and at ~590 cm−1 and calculation of the integrated intensity ratio I590/I705 (Fig. 7c). It is worth noting that this approach was purely hypothetical and not known from the literature. An interesting difference emerged, namely, the sample with higher aluminum started to undergo a structural reorganization at a higher temperature (400 °C) than the less aluminum-rich sample did (~300 °C). It may be speculated that the difference may reflect some change in the structural stability of the celadonite micas. What is more, a plot of temperature-dependent integral intensity revealed a jump of the I590/I705 values at temperatures of ~500 °C may indicate a phase transition of celadonite into ferri-phlogopite or ferri-biotite. However, the appearance of potentially new phases was countered by the XRD and Mössbauer results. The correlation of the Raman data with the Mössbauer spectroscopy outcomes does raise the possibility of analyzing the contribution of Fe in octahedral sites in relation to temperature. The Mössbauer spectroscopy data suggest that the I590/I705 pattern may reflect the increase in occupancy of the M1 site with increasing temperature (Fig. 7). The observed maximum of the intensity ratio of I590/I705 at 550 °C for AlCel1 and 500 °C for AlCel2 indicates that the polyhedra have been transformed from six-coordinated into five-coordinated polyhedra. Afterwards, a slow decrease in the curves with temperature (Fig. 7c) reflects the migration of cations from cis-sites to trans-sites. Those data are in good agreement with results obtained from the XRD analysis and reflect the conclusions taken from the Mössbauer data.

Plots of temperature dependence of average fraction of Fe species in M1 and M2 sites obtained from Mössbauer measurements for a) AlCel1 and b) AlCel2 and c) integral intensity ratio between bands centred at ~590 and ~ 705 cm−1 versus temperature obtained from Raman results. Note: values of fraction of Fe species in octahedral sites were obtained after cooling the sample from high to room temperature and the lines indicate only the trends of the data

XPS spectra showing (in rows) the a) Si2p, b) Al2p, c) K2p, d) O1s and f) Fe2p regions with relative peak deconvolutions obtained for AlCel2 initial sample as well as collected at temperature 300 °C and 550 °C. The binding energy value (BE) of each component is presents on the spectrum
Secondly, X-ray photoelectron spectroscopy were applied to look more precisely on the chemical environment and follow the impact of temperature on the modification of chemical states. According to XPS data, the two Si2p (3/2, 1/2) doublets indicate that the tetrahedral sheet contains only Si in tetrahedral coordination with some Al3+ substitution. On the basis of the area ratio of Si2p components, the ratio of Si4+ in tetrahedral coordination to Al3+ is 72/28. On the other hand, the Al2p spectrum indicate that Al ions are present in octahedral and in tetrahedral coordination in structure of AlCel2. The ratio of Al ions in tetrahedral to those in octahedral coordination is 27/73 and is very close to those obtained for Si2p. Heating the sample at 100 °C causes the change of the intensity lines for the Si2p and for the Al2p. This observation confirms the XRD results which indicated the loss of bound molecular waters in the structure of the AlCel2 close to this temperature. After heating at a temperature of 550 °C these spectra become almost symmetrical. The ratio of the intensity lines mentioned above for Si is 97/3 and for Al is 97/3, at 550 °C. These results suggest that at higher temperatures, the Al cations are mostly located in octahedral sites what confirm conclusions from XRD, Raman and Mössbauer analysis.
The K2p spectrum of initial sample AlCel2 were decomposed into two doublets. The origin of the extra K2p doublet is not well understood. On the basis of the chemical analysis, it might be concluded that it is the result of calcium substitution modifying the chemical environment in the interlayer configuration of the micas. The area ratio for the mentioned above components is 80/20, which results from the ratio of K+ and Ca2+, respectively. However, the calcium content is too high compared to the results of the chemical analysis. It may suggest that not only Ca2+ is substituting the K+ in interlayer sites but that in these sites also, a small amount of structural water is present. This explanation is supported by K2p spectra obtained at 100 °C and 300 °C. The intensity of extra doublet decreases during the heating and the area ratio for the doublets of K2p is 88/12 at 100 °C and 95/5 at 300 °C. This is the result of dehydration of interlayer water from inner surfaces with the increase in temperature. This is also supported by XPS spectra of O1s regions obtained at the studied temperatures.
The Fe2+/Fe3+ ratio, based on the area ratio of Fe2p, was determined to be 18/82, in very good agreement with the chemical analysis and with the Mössbauer results. Also, the area ratio of the peaks connected with Fe3+ is 62/20 and reflects the M2/M1 ratio of Fe3+ ions in octahedral positions. The main changes of the Fe2p spectrum are observe at 550 °C where all of the doublets observed are connected with Fe3+ ions. The two peaks can be associated with Fe in M1 sites and one, with a small contribution, of iron in dimeric pyramid sites. Relationship of these Fe2p peaks with specific Fe sites in structure of aluminoceladonite or more generally in structure of layer silicates was showed first time. The area ratio two first peaks to the last one is 83/17, which is similar to the ratio 87/13 obtained from the Mössbauer studies at this temperature.
Conclusions
We show that all of the measured data show that the aluminoceladonite with a higher aluminum content started to structurally reorganize at a higher temperature range than the less aluminum-rich sample. The oxidation of Fe2+ to Fe3+ ions started at ~350 °C for the Al-richer sample and at 300 °C for the other. This process may be associated with, or even initiate, dehydroxylation. Up to a temperature of ~500 °C, cation migration in the octahedral sheet is not seen. Based on Mössbauer results, visible changes in the coordination numbers of polyhedrons occurred; octahedrally-coordinated Fe3+ occupying M2 sites becomes 5-coordinated. This temperature can be viewed as the second stage of dehydroxylation. Based on Raman data, the proposed temperature-dependent integral intensity ratio between bands centred at ~590 and 750 cm−1 (I590/I705) clearly indicates the temperature at which six-coordinated polyhedra are transformed to five-coordinated. Above ~500 °C, cation migration from former cis-sites to former trans-sites take place. Changing of coordination number from five into four is associated with the detachment of OH or O and, as a result, dimetric pyramids can be formed.
References
Barr T, Seal S, Wozniak K, Klinowaki J (1997) ESCA studies of the coordination state of aluminium in oxide environments. J Chem Soc Faraday Trans 93:181–186
Biino G, Gröning P (1998) Cleavage mechanism and surface chemical characterization of phengitic muscovite and muscovite as constrained by X-ray photoelectron spectroscopy. Phys Chem Min 25:168–181
Bhattacharyya K (1993) XPS study of mica surfaces. J Electron Spectros Relat Phenomena 63:289–306
Dainyak L, Drits V, Lindgreen H (2004) Computer simulation of octahedral cation distribution and interpretation of the Mössbauer Fe2+ components in dioctahedral trans-vacant micas. Eur J Mineral 16:451–468
Dainyak L, Zviagina B, Rusakov V, Drits V (2006) Interpretation of the nontronite-dehydroxylate Mössbauer spectrum using EFG calculations. Eur J Mineral 18:753–764
Dainyak L, Rusakov V, Sukhorukov I, Zviagina B, Drits V(2009) An improved model for the interpretation of Mössbauer spectra of dioctahedral 2:1 trans-vacant Fe-rich micas: refinement of parameters. Eur J. Mineral 21(5):995–1008.
Dainyak L, Rusakov V, Sukhorukov I, Drits V (2013) Octahedral cation distribution in glauconites from southern Urals by combination of crystal-chemical model and quasi-continuous model-independent quadrupole splitting distributions (QSD) fitted to their Mössbauer spectra. Eur J Mineral 25(3):405–414
Drits V, Besson G, Muller F (1995) An improved model for structural transformations of heated-treated aluminous dioctahedral 2:1 layer silicates. Clays Clay Mineral 43(6):718–731
Drits V, Dainyak G, Muller F, Besson G, Manceaut A (1997) Isomorphous cation distribution in celadonites, glauconites and Fe-illites determined by infrared, Mössbauer and EXAFs spectroscopies. Clay Miner 32:153–179
Drits V, McCarty D, Zviagina B (2006) Crystal-chemical factors responsible for the distribution of octahedral cations over trans- and cis-sites in dioctahedral 2:1 layer silicates. Clays Clay Mineral 54(2):131–152
Elmi C, Brigatti M, Guggenheim S, Pasquali S, Montecchi M, Nannarone S (2014) Crystal chemistry and surface configurations of two polylithionite-1M crystals. Am Mineral 99:2049–2059
Elmi C, Guggenheim S, Gier R (2016) Surface crystal chemistry of phyllosilicates using X-ray photoelectron spectroscopy: a review. Clays Clay Mineral 64:537–551
Hradila D, Grygara T, Hradilova J, Bezdicka P (2003) Clay and iron oxide pigments in the history of painting. Appl Clay Sci 22:223–236
Kloprogge J.T, Wood B (2018) X-ray photoelectron spectroscopy and Raman microscopy of a ferroan platinum crystal from the Kondyor Massif, Russian Far East. Spectrosc Lett 52(1) 21:43–48. https://doi.org/10.1080/00387010.2018.1546745
McKeown D, Bell M, Etz E (1999) Vibrational analysis of the dioctahedral mica: 2M1 muscovite. Am mineral 84:1041–1048. 0003-004X/99/0708–1041$05.00
Moretto L, Orsega E, Mazzocchin G (2011) Spectroscopic methods for the analysis of celadonite and glauconite in Roman green wall paintings. J Cult Herit 12:384–391
Muller F, Plançon A, Drits V, Besson G (1998) Modelisation of X-ray powder diffraction patterns for the study of heat-treated Fe-rich dioctahedral 2:1 layer silicates. J Phys IV France 8:91–98
Muller F, Plançon A, Besson G, Drits V (1999) Nature, proportion and distribution of stacking faults in celadonite minerals. Mater Struct 6(2):129–134
Muller F, Drits V, Plançon A, Besson G (2000a) Dehydroxylation of Fe3+, mg-rich dioctahedral micas: (I) structural transformation. Clay Miner 35(3):491–504
Muller F, Drits V, Tsipursky S, Plançon A (2000b) Dehydroxylation of Fe3+, mg-rich dioctahedral micas: (II) cation migration. Clay Miner 35(3):505–514
Muller F, Drits V, Plançon A, Robert J-P (2000c) Structural transformation of 2:1 dioctahedral layer silicates during dehydroxylation-rehydroxylation reactions. Clay Clay Miner 48:572–585
Murad E, Fabris J (2010) Kaolin mining and beneficiation: the role of iron. J Phys: Conference Series 217:012066. https://doi.org/10.1088/1742-6596/217/1/012066
Ospitali F, Bersani D, Di Lonardo G, Lottici P (2008) ‘Green earths’: vibrational and elemental characterization of glauconites, celadonites and historical pigments. J Raman Spectrosc 39:1066–1073
Povarennykh A (1978) The use of infrared for the determination of minerals. Am Mineral 63:956–959
Prisecaru I (2009-2016) WMOSS4 Mössbauer spectral analysis software. Available at www.wmoss.org ()
Rancourt D (1994) Mössbauer spectroscopy of minerals II. Problem of resolving cis and trans octahedral Fe2+ sites. Phys Chem Miner 21:250–257
Rieder M, Cavazzini G, D’Yakonov Y, Frank-Kameneckii V, Gottardi G, Geggenheim S, Koval P, Müller G, Neiva A, Radoslovich E, Robert J, Sassi F, Takeda H, Weiss Z, Wones D (1998) Nomenclature of the micas. Can Mineral 36:905–912
Savko K, Piliugin S, Bazikov N (2015) Experimental data for high-temperature decomposition of natural celadonite from banded iron formation. Chin J Geochem 34(4):507–514
Schingaro E, Lacalamita M, Scordari F, Mesto E (2013) 3T-phlogopite from Kasenyi kamafugite (SW Uganda): EPMA, XPS, FTIR, and SCXRD study. Am Mineral 98:709–717
Tischendorf G, Forster H, Gottesmann B, Rieder M (2007) True and brittle micas: composition and solid-solution series. Mineral Mag 71(3):285–320
Tsipursky S, Drits V (1984) The distribution of octahedral cations in the 2:1 layers of dioctahedral smectites studied by oblique-texture electron diffraction. Clay Miner 19:177–193
Tsipursky S, Kameneva M, Drits V (1985) Structural transformation of Fe3+ containing dioctahedral 2:1 phyllosilicates in the course of dehydroxylation. In J. Konta, Ed., proceedings of the 5th meeting of the European clay groups, Prague, 569-577
Wang A, Freeman J, Jolliff B (2015) Understanding the Raman spectral features of phyllosilicates. J Raman Spectrosc 46(10):829–845
Zviagina B, McCarty D, Środoń J, Drits V (2004) Interpretation of infrared spectra of dioctahedral smectites in the region of OH-stretching vibrations. Clays Clay Mineral 52(4):399–410. 0.1346/CCMN.2004.0520401
Zviagina B, Sakharov B, Drits V (2007) X-ray diffraction criteria for the identification of trans- and cis-vacant varieties of dioctahedral micas. Clays Clay Mineral 55(5):467–480
Zviagina B, Drits V, Środoń J, McCarty D, Dorzhieva O (2015) The illite-aluminoceladonite series: distinguishing features and identification criteria from X-ray diffraction and infrared spectroscopy data. Clays Clay Mineral 63(5):378–394
Zviagina B, Drits V, Dorzhieva O (2020) Distinguishing features and identification criteria for K-Dioctahedral 1M micas (Illite-Aluminoceladonite and Illite-Glauconite-Celadonite series) from middle-infrared spectroscopy data. Minerals 10:153
Acknowledgments
Constructive comments of two anonymous reviewers and journal editor Anton Beran are gratefully acknowledged.
This work was supported by the National Science Centre, Poland (grant DEC- 2018/02/X/ST10/00662).
Author information
Authors and Affiliations
Corresponding author
Additional information
Editorial handling: A. Beran
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 995 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kądziołka-Gaweł, M., Czaja, M., Dulski, M. et al. Impact of high temperatures on aluminoceladonite studied by Mössbauer, Raman, X-ray diffraction and X-ray photoelectron spectroscopy. Miner Petrol 115, 431–444 (2021). https://doi.org/10.1007/s00710-021-00753-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00710-021-00753-z