
Abstract
Sixteen specimens of historical remains of inorganic pharmaceuticals dating back to the eighteenth century were analyzed by a combination of spectroscopy (ICP–MS, atomic absorption/emission, UV–Vis, infrared and Raman), capillary zone electrophoresis, and chemical methods (titration and gravimetric analysis). The results obtained confirmed the identity of 14 of the specimens analyzed. With the exception of one, impurities were found in all specimens, often at relatively high concentrations. Based on these impurities, it was possible to confirm the origin of six substances as naturally occurring minerals. The other specimens were probably prepared by period apothecaries or artisans through chemical reactions. In two specimens, a recipe of the time, based on the ignition of metal with sulfur, could be confirmed. For anatron, a substance that originated as a by-product of glass melting, it was possible to determine its composition as a mixture of alkaline sulfates, carbonates, and chlorides (the first such analysis described in the literature). On the other hand, for two specimens, it was found that the substance was mistaken for a completely different compound than would be expected from the Latin inscription on the apothecary jar.
Graphical abstract

Similar content being viewed by others


Introduction
The identity of a substance and its purity are a key factor in chemistry. After all, a chemical substance is defined as a form of matter having constant and definite composition best characterized by the entities (molecules, formula units, atoms) it is composed of [1]. Uncertainty in the identity of a substance or its confusion with another can lead not only to the failure of an expected chemical reaction but also to an accident or disaster.
Both identity and purity of a substance play a very crucial role in pharmacy, where confusion of a drug or the mere impurity contained in it (even in very small quantities) can have fatal consequences, in extreme cases even death. While today, the identity and purity of substances used in pharmacy are strictly prescribed by the pharmacopoeia and carefully controlled, this was certainly not the case in the past. Therefore, as part of our project of analyzing the historical remains of pharmaceuticals, we have studied the identity and purity of sixteen inorganic pharmaceuticals from the eighteenth century. The samples analyzed were obtained from the exceptionally preserved Baroque pharmacy of the Capuchin monastery in Prague at Hradčany (now in the collections of the National Museum in Prague) [2]. Thanks to a remarkable coincidence, the remains of pharmaceuticals from this pharmacy have been untouched by human hands since the end of the eighteenth century. In our previous works, we have studied preparations containing herbal substances from this pharmacy; for example, chicory extract [3], senna extract [4], preparations containing opium [5, 6] or ipecacuanha [7] and baroque ointments [8].
The analysis of historical remains of inorganic pharmaceuticals has been discussed several times in the literature; we give an overview of such analyzes in our review [9]. Vázquez de Ágredos Pascual et al. [10, 11] recently analyzed several dozen inorganic compounds from the nineteenth century pharmacy of Santa Maria della Scala in Rome. In the past, a number of inorganic drugs were also used by alchemists [12]. In fact, iatrochemistry, which in turn applied alchemical ideas and working practices to the production of medicines, emerged from this milieu. An eminent example of the analysis of iatrochemical medicine is Principe’s work [13], in which, based on a replication of an alchemical procedure of the time, he demonstrated that the impurities contained in the mineral raw materials used in the production of a medicine can significantly affect the outcome of the preparation of such a medicine.
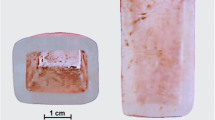
An overview of the historical remains of inorganic pharmaceuticals analyzed in this work is presented in Fig. 1 and Table 1.

Baroque apothecary jars from which samples were taken for analysis (inventory number in the collection of the National Museum, transcription of the Latin inscription on the jar) a H2-4714 Vitriolum album, b H2-4763 Alumen crudum, c H2-4765 Chalybs limatura, d H2-4773 Plumbum ustum, e H2-4794 Nihilum album, f H2-4802 Anatron, g H2-4817 Chalybs Martis, h H2-4819 Aes viride, i H2-4836 Alumen ustum, j H2-4851 Chalybs cum sulphure praeparatio, k H2-4857 Lapis calaminaris, l H2-4902 Vitriolum Martis calcinatum, m H2-9507 Viride cupri, n H2-9543 Sacchcarum Saturni, o H2-9582 Nitrum depuratum, p H2-9643 Borax veneta (Color figure online)
Results
Elemental composition in samples analyzed
To determine the elemental composition of the historical remains of inorganic pharmaceuticals analyzed, their solutions in 2% nitric acid were used. Samples that did not completely dissolve in this medium were dissolved in aqua regia (that is, samples H2-4765, H2-4773, H2-4817, H2-4819, H2-4851, H2-4857, and H-9507). The elemental composition was determined by inductively coupled plasma mass spectrometry (ICP-MS) as the main method. The content of elements present at higher concentrations (more than 1%) was simultaneously determined and confirmed by atomic absorption spectroscopy (AAS), atomic emission spectroscopy (AES), ultraviolet–visible (UV/Vis) spectroscopy, or titration. The quantification of the elemental content of the samples analyzed is presented in Table 1, which also gives the theoretical composition based on the Latin inscription on the apothecary jar from which the sample originated. According to the detected concentrations, the elements were divided into (i) macroelements, which were considered to be elements present in concentrations greater than 1%, these elements forming a major component of the sample analyzed or its significant impurity, and (ii) microelements, that is, elements present in concentrations less than 1%, which can, therefore, be considered as impurities.
Quantification of anions in samples analyzed
For the historical remains of inorganic pharmaceuticals analyzed, which were expected to have significantly represented anions, quantification of these anions in aqueous sample solutions was performed by capillary zone electrophoresis. The exception was sample H2-4773, which was dissolved in aqua regia, for which it could be expected that the sulfide anion present in the sample would be oxidized to sulfate anion by aqua regia. The sample H2-9643 was subjected to simultaneous determination of the borate anion by acidimetric titration. The results obtained are presented in Table 2.
Infrared and Raman spectroscopy of samples analyzed
The nature of the selected specimens of the historical remains of inorganic pharmaceuticals analyzed was investigated by FT-IR and Raman spectroscopy. The recorded IR spectra of samples H2-4773 and H2-4817 indicate the presence of at least trace amounts of sulfates. This conclusion is based on the presence of bands of ν3 SO42− modes (wide band at 1080 cm−1 for H2-4817 and asymmetric doublet at 1163 and 1044 cm−1 for H2-4773), ν1 SO42− mode (weak band at 965 cm−1 for H2-4773) and ν4 SO42− modes (asymmetric doublet at 624 and 591 cm−1 for H2-4773).
The Raman spectra of the sample H2-4794, depicted in Fig. 2, clearly show that the main component of the sample is potassium sulfate (characteristic bands at 1145, 1108, 1093, 963, 626, 619 and 455 cm−1). The Raman spectrum of sample H2-4851 (Fig. 3) manifests the presence of elemental sulfur S8 by the characteristic bands at 472, 247, 218 and 153 cm−1 (cf. [14]).

Raman spectrum of sample H2-4794 Nihilum album (red line) compared to the reference spectrum of potassium sulfate (blue line) (Color figure online)

Raman spectrum of sample H2-4851 Chalybs cum sulphure praeparatio (red line) compared to the reference spectrum of cyclooctasulfur molecule (blue line) (Color figure online)
The vibrational spectra of the sample H2-4857 exhibit manifestations of carbonate anions with lowered symmetry; i.e., splitted bands of ν3 CO32− modes at 1479 and 1418 cm−1 (IR spectrum), ν1 CO32− mode at 1086 cm−1 (Raman spectrum), ν2 CO32− mode at 875 cm−1 (IR spectrum) and ν4 CO32− mode at 712 cm−1 (both spectra). However, the IR spectrum also shows features characteristic to hydroxides (stretching O–H vibrations); i.e., wide structured bands in the region 3500 − 2700 cm−1 and a sharp peak at 3648 cm−1.
The recorded IR spectra of the sample H2-9507 (Fig. 4) show that this is an acetate-type verdigris [15, 16]. The comparison with the reference spectrum [17] of synthetic verdigris (composition Cu(CH3COO)2⋅2Cu(OH)2) clearly indicates the close relationship of the spectra. The spectrum of sample H2-9507 contains a broad band of stretching O–H vibrations (maximum at ~ 3200 cm−1) and a pair of strong bands at 1567 and 1397 cm−1 belonging to antisymmetric and symmetric COO− vibrations, respectively. The increased relative intensity of the band at 1397 cm−1 and the presence of a sharp band at 1494 cm−1 also indicate the presence of carbonate anions (splitted bands of ν3 CO32− modes) in the sample.

Infrared spectrum of sample H2-9507 Viride cupri
Solubility and waters of hydration of the samples analyzed
Two specimens of historical remains of inorganic pharmaceuticals analyzed, which according to the theoretical or established composition should be well soluble in water, were found to contain a water-insoluble fraction. This fraction was quantified gravimetrically; for sample H2-4794 the water-insoluble fraction was determined to be 2.66%, for sample H2-4802 the water-insoluble fraction was determined to be 2.23%.
For three specimens analyzed, the content of hydrate water was determined by gravimetric method, and the results of the determinations are summarized in Table 3.
Discussion
The historical remains of inorganic pharmaceuticals analyzed in this work can generally be broadly divided according to their composition determined into (i) raw materials of natural origin, that is, minerals obtained by collecting or mining, and (ii) substances prepared by chemical reactions. Classification into the first group is usually sanctioned by the higher impurity content of the analyzed pharmaceutical. The apothecaries of the time did not, of course, analyze such substances of natural origin, but accepted their identity as given solely on the basis of the origin of the substance (declared by the supplier) or on the basis of characteristic features such as consistency, color, or solubility. This could easily lead to confusion. This is the reason why the two samples analyzed from the study set, H2-4794 and H2-4857, were mistaken by a period apothecary for a substance completely different than the Latin inscription on the apothecary jar suggests. Next, the theoretical and determined composition of the analyzed historical remains of inorganic pharmaceuticals is discussed, set in a brief pharmaceutical-historical context. The analyzed samples are grouped alphabetically according to the content of the main element.
Aluminium
Sample H2-4763 and similar sample H2-4836 were confirmed as aluminium potassium sulfate (trivially called “alum”), used in the eighteenth century to treat scurvy, as a scar remover and as a component of topical preparations [18, 19]. A higher aluminium content was found in sample H2-4836, which is consistent with the Latin inscription on the apothecary jar Alumen ustum, suggesting that it is ignited alum (ignition reduces the hydrate water content) [19]. The impurities present indicate that the source of both specimens analyzed was a mineral, probably alunite [20], which was, therefore, used as either raw (sample H2-4763) or ignited (sample H2-4836).
Boron
Of the samples in the studied set, the specimen with boron content was represented in only one case. For this sample H2-9643, its identity as an alkaline tetraborate (trivially called “borax”) was confirmed by analysis, consistent with the Latin inscription on the apothecary jar. The sample is a mixture of hydrated sodium tetraborate and potassium tetraborate, with admixtures of both sodium/potassium carbonate and sodium/potassium sulfate. Borax was usually obtained as a mineral from various natural deposits and then purified (especially in Venice, hence the name of the substance Borax veneta) by crystallization [20, 21]. It was used as an abortive, diuretic, and in preparations for the treatment of arthritis and female disease [18, 19].
Copper
The Latin inscription Aes viride on the apothecary jar of sample H2-4819 suggests that it is the copper salts of acetic acid, also known by the trivial name “verdigris”. It has been used for centuries as a pigment in painting [15, 16], and has had an equally important place in pharmacy, where it was applied as a disinfectant in topical preparations since the time of ancient Egypt [19, 21]. It was prepared by dissolving copper compounds (oxide, carbonate) in acetic acid by various methods. The analysis of the specimen confirmed the high copper content of the sample, but due to the small amount of preserved sample, further characterization by spectroscopy was not possible. On the contrary, for the second copper-bearing historical remains analyzed, sample H2-9507, it was possible not only to determine the high copper content but also to confirm by IR spectroscopy that this specimen is indeed acetate-type verdigris.
Iron
There were four samples with iron as the main component. In sample H2-4765, a high iron content corresponding to a mixture of its oxides was found. The substance was prepared by sawing metallic iron, hence the literal translation of the Latin inscription Chalybs limatura on the apothecary jar “sawed iron”. It was used in preparations for liver disease and anemia [19, 21]. The action of air oxygen naturally oxidized such iron filings over the centuries. Schröder found similar content in another historical relic of Chalybs limatura dated to the seventeenth century [22]. Practically the same conclusions were found for sample H2-4817 Chalybs Martis (literally “the iron of the god Mars”). Again, this is a mixture of iron oxides and iron oxide-hydroxides, moreover in a mixture with sulfate, as shown by the IR spectra. In the third sample analyzed, H2-4851 Chalybs cum sulphure praeparatio, ferrous sulfide was confirmed. It was produced by calcination of iron with sulfur and was used as an astringent, roborant, and antacid [18, 19]. Compared to the theoretical composition, only 72% FeS was found in the sample, this is probably the result of an incomplete reaction during preparation; the rest of the sample consists of unreacted sulfur and impurities. This is confirmed by the presence of elementary sulfur in the sample as detected by Raman spectroscopy. Analysis of a fourth iron-containing sample, H2-4902, confirmed the presence of ferrous sulfate (trivially called green vitriol). The compound was used in the eighteenth century pharmacy to treat tuberculosis and to produce bleeding-stopping agents [19, 21]. According to the Latin inscription Vitriolum Martis calcinatum on the apothecary jar, it is supposed to be ferrous sulfate calcined. However, the iron content found corresponds roughly to that in ferrous sulfate heptahydrate. In addition, the sample contains a small proportion of sulfates and carbonates impurities, particularly copper and zinc. The source of the substance could have been either a mineral (especially melanterite or rozenite [20]) or the chemical reaction of iron or its compounds (oxides) with sulfuric acid.
Lead
The first sample analyzed, H2-4773, bears the Latin name Plumbum ustum, literally “burnt lead”. According to the historical literature [18, 19], this substance was prepared by placing lead sheets in a crucible together with sulfur and by strong ignition a brown powder was obtained. Lead sulfide was thus formed, and it was used in preparations for skin diseases, scar healing, and hemorrhoids. This corresponds to an observed lead content of 86%; the theoretical lead content in lead sulfide is 86.6%. After dissolution of the sample in aqua regia for analysis, the sulfur of the sulfide anion was oxidized to sulfates, of which 45.5% was found (i.e., 113% of the theory). The use of sulfur in the preparation of this specimen is also evidenced by the traces of sulfate demonstrated by IR spectroscopy. The metallic impurities found (mainly antimony and arsenic) come from the ore used in the production of lead. The second historical lead-bearing sample analyzed, H2-9543, was confirmed to be pure lead acetate. The substance, known since antiquity for its sweet taste (hence the Latin name Saccharum Saturni, literally “Sugar of Saturn”), was prepared by dissolving lead or its compounds in acetic acid [19, 22]. Schröder [22] analyzed a sample from the nineteenth century and obtained a similar result.
Potassium
There were two potassium salts in the study set. An interesting substance used in the eighteenth century pharmacy was anatron (also called Fel vitri, literally “bile of glass”), a by-product of glass melting [19]. It was used mainly for its diuretic effect [21]. The identity of sample H2-4802 was confirmed and it was found that the sample contains alkaline sulfates (56.9%), carbonates (3.6%) and chlorides (0.41%). The water-insoluble fraction of 2.23% corresponds to the sulfates and carbonates of barium and calcium present. To the best of our knowledge, this is the first analysis of anatron described in the literature. The second potassium salt in the study set was sample H2-9582. In accordance with the Latin inscription on the apothecary jar Nitrum depuratum, the analysis confirmed the specimen composition as potassium nitrate with minor admixtures of alkaline carbonates and sulfates. It was obtained by purifying of the mineral niter [20] (hence depuratus, literally “purified”) or artificially prepared from organic residues [19, 21]. In the pharmacy of that time, it was used in anti-inflammatory preparations and as a digestant.
Zinc
Analysis of sample H2-4714 Vitriolum album confirmed that it is indeed zinc sulfate (trivially called “white vitriol”). In the eighteenth century pharmacy, it was used mainly as an emetic, as part of eye drops or topical preparations [18, 19, 22]. However, the specimen analyzed is not pure zinc sulfate, but a mixture of 74% ZnSO4·7H2O, 16% FeSO4·7H2O, 7% MnSO4·2H2O and 3% other admixtures (calculated based on the results of analyzes). This composition corresponds to the mineral goslarite [20], which was a natural source of zinc sulfate in the eighteenth century. The second specimen that was believed to contain a zinc compound was sample H2-4794. The Latin inscription on the apothecary jar Nihilum album (literally “white nothing”, or “white snow”) has been used for centuries for zinc oxide [19, 21]. It was available in nature as the mineral zincite [19, 20], but more often it was prepared synthetically as a by-product of the metallurgical processing of zinc ores. Zinc oxide was mainly used in topical preparations. However, the results of analyzes showed that in this case it is a completely different substance, which does not contain zinc at all. It is a mixture containing 73% K2SO4, 11% CaCO3 and 16% other admixtures (calculated based on the results of analyzes). It is the presence of calcium carbonate (which is poorly soluble in water) that can create the illusion that it is zinc oxide, which is also insoluble in water (if by chance the apothecary of the time tested the solubility of the substance in water). This is proven by the determination of the water-insoluble fraction, which is 2.66%. The third zinc-containing specimen was sample H2-4857, which according to Latin inscription on apothecary jar Lapis calaminaris should be mineral calamine (synonymously smithsonite, chemically zinc carbonate [20]). The substance, as the aforementioned zinc compounds, was used mainly in topical preparations [19, 21]. However, the analysis of the specimen shown that—also in this case—it was mistaken for another substance. Based on the results of the analysis, the composition of the specimen can be characterized as 48.9% CaCO3, 42.7% MgCO3 and 7.2% ZnCO3, the remaining 1.2% being other admixtures. Therefore, it is definitely not calamine, which contains 80.3% zinc, but most probably mineral dolomite [20].
Conclusions
The combination of several spectral techniques, capillary zone electrophoresis, and classical chemical methods of analysis provided an interesting insight into the pharmaceutical and artisanal practice of the eighteenth century with possible overlap into alchemy. In the studied set of 16 historical remains of inorganic pharmaceuticals, the identity of the main constituent was confirmed in 14 cases. In two cases, however, it turned out that period apothecaries or their suppliers had mistakenly replaced the alleged substance with a chemically completely different one. In all, except for one, numerous impurities were found in the specimens studied. Based on these impurities, it was possible to estimate the mineral sources of the substance in six cases. Analysis of two specimens allowed us to confirm a recipe of the time, which consisted of synthesis from metal and sulfur by ignition. For the first time, anatron, a substance commonly used in the eighteenth century pharmacy, was analyzed.
Experimental
Analyzed samples, chemicals
The historical remains of inorganic pharmaceuticals analyzed were collected directly from the original Baroque apothecary jars. The inventory numbers of the individual jars in the collection of the Department of Older Czech History, National Museum (Prague, Czech Republic) are given in Table 1. The appearance of the jars is specified in Fig. 1. Eleven jars were hollowed out of wood in the shape of a cylinder 140 mm high and 90 mm in diameter. The five vessels were in the shape of a glass cup 120−150 mm high and 50−70 mm in diameter, which was closed with an original leather lid secured with a string. After carefully opening the jar, three samples of the contents were taken from the jar using a porcelain spoon (one from the center and two from opposite sides). The exception was sample H2-4819, where only a small amount of residue was recovered from the vessel. The samples were kept in glass containers in the dark and homogenized in a porcelain mortar prior to analysis.
All of the chemicals used were of p.a. grade or higher. Deionized water, prepared on a Milli-Q instrument (Millipore, USA), a with a specific conductivity < 0.05 μS cm−1, was used.
Procedures, instrumentation
The ICP-MS measurements were performed on an Inductively Coupled Plasma Mass Spectrometer ICP-MS 7900 (Agilent, USA) equipped with a micromist glass nebulizer working at 1550 W (with a flow 1.03, 0.90, and 15.0 dm3 min−1 of nebulizer, auxiliary and plasma gas, respectively). Results were evaluated from the “No gas” mode. For calibration and sample solutions preparation, 2% (v/v) nitric acid was used. In the case of samples dissolved in aqua regia, the resulting solution was diluted by water so that the resulting nitric acid concentration was below 2%. Working standards were prepared by an appropriate diluting of two multielement certified reference solutions (i) AN9094MFN CRM Mix (As, Be, Ca, Cd, Co, Cr, Cu, Fe, Li, Mg, Mn, Mo, Ni, P, Pb, Sb, Se, Sn, Sr, Ti, Tl, V, Zn) with concentration of 100 mg dm−3 of each element in 5% HNO3 and 0.2% HF (Analytika, Czech Republic), and (ii) AN9087MC (Au, Ir, Os, Pd, Pt, Rh, Ru) with concentration of 100 mg dm−3 of each element in 20% HCl (Analytika, Czech Republic). The concentrations of all elements in the calibration solutions were in a concentration range of 1.6−1000 μg dm−3.
The High Resolution Continuum Source Atomic Absorption Spectrometer ContrAA700 (Analytik Jena, Germany) was used for the determination of Al, As, Ca, Cd, Fe, K, Mg, Na, Pb, and Zn using an external calibration approach. All calibration solutions were prepared from certified reference materials (Analytika, Czech Republic) with a concentration of determined element (1000 ± 2) mg dm−3. The samples and standards were diluted with 1% HCl or HNO3. For all measurements, 0.1% CsCl ionization buffer was used. Calcium, potassium, and sodium were determined in the atomic emission mode, and all other elements were determined in the atomic absorption arrangement. Only three pixels (central pixel ± 1 side pixel from the 200 observed) were used for the signal evaluation. In case of self-absorption observed, the sample was diluted as needed to avoid this unfavorable phenomenon. The wavelengths and other experimental conditions for the determination of individual elements are summarized in Table 4.
UV/Vis spectrometry was carried out on a single-beam diode array spectrometer HP-8453 (Agilent, USA) in a quartz cuvette with an absorption layer thickness of 1 cm. The 1,10-phenanthroline method was used for the determination of iron [24], and the aqueous ammonia method was used for the determination of copper [25].
For the chelatometric determination of the lead content, exactly about 0.05 g of the sample was weighed, diluted in a titration flask with 100 cm3 of deionized water and 3 g of hexamethylenetetramine were added. The resulting solution was titrated with 0.05 mol dm−3 volumetric solution of EDTA. Xylenol orange was used to indicate the equivalence point.
Electrophoretic measurements were carried out on an Agilent 7100 capillary electrophoresis instrument (Agilent Technologies, Germany) equipped with a capacitively coupled contactless conductivity detector (Admet, Czech Republic). The measurement temperature was maintained at 25 °C. A fused-silica capillary (Polymicro Technologies, USA) of 20 μm i.d., 375 μm o.d. was cut to 50.0 cm total length, 35.0 cm effective length. Prior to the first use, the capillary was flushed 10 min with 1.0 mol dm−3 aqueous solution of sodium hydroxide and then 10 min with deionized water. Between individual runs, the capillary was flushed 5 min with the background electrolyte. The sample was introduced by a pressure of 5 kPa for 20 s. The determination of chloride, sulfate, nitrate, and phosphate was carried out in a background electrolyte consisting of 1.0 mol dm−3 aqueous solution of formic acid. During separation, the voltage was set at − 25 kV (current was 8 μA). Quantification was based on a 7-point calibration curve with standards ranging from 0.05 to 5.00 mmol dm−3 concentration. Peak areas were related to the peak area of benzenesulfonate, added as an internal standard at a concentration of 1 mmol dm−3. The determination of carbonate, acetate, and borate ions was carried out in a background electrolyte consisting of an aqueous solution of 50 mmol dm−3 sodium hydroxide, 22.5 mmol dm−3 disodium phosphate, and 0.2 mmol dm−3 cetyltrimethylamonium bromide. During separation, the voltage was set at − 15 kV (current was 9 μA) and a pressure of 100 mbar was applied to the inlet vial. Quantification was based on a 7-point calibration curve with standards ranging from 0.05 to 5.00 mmol dm−3 concentration. Peak areas were related to the peak area of 2-(N-morpholino)ethanesulfonic acid, added as an internal standard at a concentration of 5 mmol dm−3.
For the acidimetric determination of the borates, exactly about 0.1 g of the sample was weighed and titrated with a 0.1 mol dm−3 volumetric solution of hydrochloric acid. The equivalence point was indicated by methyl red.
Infrared spectra were recorded on a Thermo Fisher Scientific Nicolet iS50 FTIR spectrometer (4 cm−1 resolution, Happ–Genzel apodization) in the 400−4000 cm−1 region (KBr beamsplitter) using the ATR (diamond crystal) technique. Standard ATR correction (Thermo Nicolet Omnic software, version 9.2.89) was applied to the recorded spectra. Raman spectra were recorded on a Thermo Nicolet 6700 FTIR spectrometer equipped with the Nicolet Nexus FT Raman module (4 cm−1 resolution, Happ–Genzel apodization, CaF2 beamsplitter, 1064 nm Nd:YVO4 laser excitation, 400 mW power at the sample) in the 100−3700 cm−1 region.
The water-insoluble fraction was determined by weighing exactly about 0.5 g of the sample in a volumetric flask of 50.00 cm3. The flask was filled to the mark with deionized water and left to stand for 24 h with occasional shaking. The insoluble fraction was determined by filtration through a filter cup (previously dried to constant weight) and subsequent drying at 105 °C.
The hydrate water content was determined gravimetrically by weighing exactly about 0.25 g of the sample into an ignition crucible (previously ignited to constant weight) and then ignited at 350 °C to constant weight.
All measurements were performed at least three times and were statistically evaluated at a 95% confidence level.
Data availability
The experimental data supporting the findings of this study are available from the corresponding author, K.N., upon reasonable request.
References
IUPAC (1997) Compendium of chemical terminology. https://doi.org/10.1351/goldbook
Nesměrák K, Kunešová J (2015) Ces Slov Farm 64:79
Nesměrák K, Štícha M, Lener T, Červený V, Kunešová J (2022) Monatsh Chem 153:707
Nesměrák K, Kudláček K, Čambal P, Štícha M, Kozlík P, Červený V (2020) Monatsh Chem 151:1241
Nesměrák K, Štícha M, Belianský M, Červený V, Kozlík P, Kudláček K, Kunešová J (2021) Monatsh Chem 152:1089
Nesměrák K, Kudláček K, Štícha M, Kozlík P, Červený V, Kunešová J (2019) Monatsh Chem 150:1593
Nesměrák K, Kudláček K, Štícha M, Červený V, Kunešová J, Yildiz I (2018) Monatsh Chem 149:1535
Kudláček K, La Nasa J, Ribechini E, Colombini MP, Nesměrák K (2023) Microchem J 190:108680
Nesměrák K, Kudláček K, Babica J (2017) Monatsh Chem 148:1557
Cavallo G, Vázquez de Ágredos Pascual ML (2018) Powder Diffr 33:270
Vázquez de Ágredos Pascual ML, Cavallo G, Pagioti R, Iranzo LR, Martín MS, Walter P, Van-Elslande E, Izzo FC (2018) Eur J Sci Theol 14:3
Chalupa R, Nesměrák K (2021) Monatsh Chem 152:1019
Principe L (1987) Ambix 34:21
Ward AT (1968) J Phys Chem 72:4133
Buse J, Otero V, Melo MJ (2019) Heritage 2:1614
de la Roja JM, Baonza VG, San Andrés M (2007) Spectrochim Acta A 68:1120
https://spectra.chem.ut.ee/paint/pigments/verdigris/. Accessed 4 May 2023
Collegium Pharmaceuticum (1729) Dispensatorium pharmaceuticum Austriaco-Viennense. Kürner, Vienna
Pomet P (1748) A complete history of drugs. Bonwicke, London
https://handbookofmineralogy.org/. Accessed 4 May 2023
Triller DW (1764) Dispensatorium pharmaceuticum universale. Varrentrapp, Francofurtum
Schröder G (1957) Die pharmazeutisch-chemischen Produkte deutscher Apotheken im Zeitalter der Chemiatrie. Technische Hochschule Braunschweig, Bremen
Marschner K, Pétursdóttir ÁH, Bücker P, Raab A, Feldmann J, Mester Z, Matoušek T, Musil S (2019) Anal Chim Acta 1049:20
Thompson JM (1953) Anal Chem 25:1231
Mehlig JP (1941) Ind Eng Chem Anal Ed 13:533
Acknowledgements
The financial support by the project Cooperatio Chemistry of Charles University is gratefully acknowledged.
Funding
Open access publishing supported by the National Technical Library in Prague. This research was funded by Univerzita Karlova, the project Cooperatio Chemistry.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nesměrák, K., Janoušková, E., Červený, V. et al. Identity and purity of historical remains of inorganic pharmaceuticals from the eighteenth century. Monatsh Chem 154, 1003–1011 (2023). https://doi.org/10.1007/s00706-023-03092-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-023-03092-1