
Abstract
Three new iridium aluminum intermetallics CaAl4Ir2, SrAl4Ir2, and EuAl4Ir2 were synthesized from the elements using silica or tantalum ampoules. They crystallize in the tetragonal crystal system with space group P4/ncc and lattice parameters of a = 782.20(1) and c = 779.14(2) pm for CaAl4Ir2, a = 797.62(1) and c = 772.75(2) pm for SrAl4Ir2, and finally a = 791.78(5) and c = 773.31(5) pm for EuAl4Ir2. All compounds crystallize isostructurally and adopt a new structure type that can be derived from the KAu4In2 type structure. To compare the structures from a crystallographic point of view, a group–subgroup relation between KAu4In2 and EuAl4Ir2 as well as KAu4In2 and KAu4Sn2 have been established using the Bärnighausen formalism. Finally, quantum-chemical calculations have been conducted, showing that in all three title compounds, a polyanionic [Al4Ir2]δ– network exists with significant (polar) bonding interactions, while the respective Caδ+, Srδ+, and Euδ+ cations are located in octagonal channels.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rare earth (RE) and transition metals (T) form numerous ternary intermetallic compounds with aluminum [1]. Besides fundamental research with respect to phase analyses and determination of isothermal sections of the RE-T-Al phase diagrams, these RExTyAlz phases are of interest with respect to their broadly varying crystal chemistry and magnetic properties. Many of these phases find, e.g., application as precipitation hardening in modern light-weight aluminum-based alloys [2]. Usually, the rare earth elements are in a stable trivalent oxidation state. Europium, in contrast, prefers the stable divalent state due to a half-filled 4f shell with an electron configuration of [Xe] 4f7. It is, thus, isoelectronic with Gd3+ and exhibits a high magnetic moment of 7.94 µB per Eu/Gd atom, a good prerequisite for interesting magnetic properties. However, so far, no complete isothermal section of the Eu–T–Al systems was studied. The only work is the partial section from 0 to 33.3 at% Eu for the Eu–Ag–Al system [3]. In view of the magnetic properties, representative examples are the antiferromagnets EuTi2Al20 (TN = 3.6 K) and EuV2Al20 (TN = 5.5 K) [4] and the quaternary silicides EuRhAl4Si2 (TC = 11 K) and EuIrAl4Si2 (TC = 15 K) [5].
When it comes to the 5d metals, the information is scarce. Only the Eu–Au–Al system was studied in some more detail and the compounds were reported: equiatomic EuAuAl which orders antiferromagnetically at TC = 50 K [6], EuAu6.09Al5.91 which structurally derives from the NaZn13 type [7], EuAuAl3 [8], Eu2Au6Al3 [9], EuAu4.82Al2.18 [7], EuAu2Al2 [10, 11], and the TC = 16.5 K ferromagnet EuAu3Al2 [12].
In contrast, hardly any compounds were reported for the ternary systems with T = Os, Ir, and Pt [1]; however, recent phase analytical studies revealed highly interesting results with respect to new compounds. MgCuAl2 type EuPtAl2 orders ferromagnetically at TC = 54 K [13] and an abrupt valence change at T ~ 45 K was observed in Eu2Pt6Al15 [14]. A really remarkable compound is Eu2Ir3Al9 [15], one of the rare examples of an intermetallic phase with purely trivalent europium.
The synthesis of such europium-based intermetallic phases deserves careful reaction conditions, since europium metal has a comparatively low boiling temperature of only 1870 K [16]. Some syntheses were conducted in quasi-open arc-melting furnaces (e.g., for EuAu2Al2 [11]), leading to europium evaporation, and thus changes of the initial sample composition. Better results can be obtained in sealed high-melting metal ampoules (niobium or tantalum, e. g., for Eu2Au6Al3 [9]) or by synthesis in a low-melting metal flux (e.g., for EuIrAl4Si2 [5]). In the case of low Eu-content compounds, the constituent elements can be wrapped in Al foil and carefully arc melted (Eu2Pt6Al15 [14]).
In the course of our systematic phase analytical work in the Eu–T–Al systems with the 5d metals, EuAl4Ir2 could be obtained. Synthetic attempts to obtain isostructural alkaline earth representatives yielded also CaAl4Ir2 and SrAl4Ir2. Herein, we report on their crystal chemistry and the structural relationship with the aristotype KAu4In2 [17].
Results and discussion
Phase analysis
Synthetic attempts to yield phase pure samples of CaAl4Ir2 (82(1) wt.-%) resulted in the formation of the title phases along with binary IrAl (11(1) wt.-%) and Ca2Ir3Al9 (7(1) wt.-%), in the case SrAl4Ir2 (80(1) wt.-%), IrAl (20(1) wt.-%) and traces of a yet unidentified phase were observed. Weight fractions were obtained by Rietveld refinement. For EuAl4Ir2, only traces of the title compound could be observed, the main product was Eu2Ir3Al9 along with binary IrAl. When comparing the product composition with the weighed rations, it becomes evident that Ca/Sr is lost. This can often be found in the lid of the refractory metal ampoules; however, even an excess of alkaline-earth metal does not enable the synthesis of phase pure samples.
Crystal chemistry
EuAl4Ir2, CaAl4Ir2, and SrAl4Ir2 crystallize with a new structure type, in the tetragonal crystal system with space group P4/ncc, Pearson symbol tP28 and Wyckoff sequence gfc. A view of the EuAl4Ir2 structure along the c axis is presented in Fig. 1. Each iridium atom is coordinated by eight aluminum atoms in the shape of a bicapped trigonal prism with Ir–Al distances ranging from 253 to 275 pm, the shortest distances found in the structure. These distances are a bit longer than the sum of the covalent radii [16] of 251 pm for Ir + Al. In addition, homoatomic Al–Al interactions can be observed (vide infra). Therefore, both the Ir–Al and Al–Al contacts can be considered bonding interactions with the iridium and aluminum atoms building an [Al4Ir2]δ– network, which leaves large channels (eight-membered rings) that are filled by the europium atoms. The Eu–Ir and Eu–Al distances are 316 pm and range from 321 to 354 pm, which are slightly to well above the sum of the covalent radii (Eu + Ir = 311 pm / Eu + Al = 310 pm) suggesting rather weak bonding interactions.

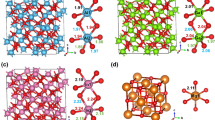
View of the KAu4In2 [17] and EuAl4Ir2 structures along the c axes. Potassium (europium), gold (iridium), and indium (aluminum) atoms are drawn as medium gray, blue, and magenta circles, respectively. Displacement ellipsoids are drawn at a 99% level. The [Au4In2]δ– and [Al4Ir2]δ– networks are emphasized
The structural motif readily reminds of the KAu4In2 structure [17] and its isotypic compounds RbAu4In2 [17], CaNi4Sn2 [18], SrNi4Sn2 [18], SrCu4Sn2 [19], and EuCu4Sn2 [20]. The KAu4In2 type structure [17], however, crystallizes with the body centered space group I4/mcm. Consequently, EuAl4Ir2 could be a superstructure variant of KAu4In2. The corresponding group–subgroup scheme, proving their direct relationship, is shown in Fig. 2 using the Bärnighausen formalism [21,22,23,24,25]. Space group P4/ncc is a klassengleiche subgroup of index 2 of I4/mcm.

Group–subgroup scheme in the Bärnighausen formalism [21,22,23,24,25] for the structures of KAu4In2 [17] and EuAl4Ir2. The index for the klassengleiche (k) symmetry reduction, the origin shift and the evolution of the atomic parameters are given. Note that the EuAl4Ir2 structure is described in the setting O2
The main shift in the atomic parameters in the lower symmetric EuAl4Ir2 structure concerns the europium atoms. The inverse coloring of the transition metal and p element positions in the pair KAu4In2 / EuAl4Ir2 leads to a significantly different bonding situation, resulting in a shift of the europium atoms off the subcell mirror plane. For the aluminum position a decoupling of the x and y parameters is observed. These displacements allow for (i) the adjustment of the different size of the transition metal and the p element and (ii) an optimized bonding of europium with the network.
Figure 3 presents cutouts of the channels (eight-membered rings) of the KAu4In2 and EuAl4Ir2 structures. Although the transition metals are the most electronegative elements in both compounds (Au: 2.54 and Ir: 2.20 on the Pauling scale [16] vs. In: 1.78 and Al 1.61), we observe the inverse coloring of the polyanion. Another striking point is the charge difference between K+ and Eu2+. In KAu4In2, the potassium atoms are located on mirror planes perpendicular to the c axis and are coordinated by eight gold atoms (338 pm K–Au). The coordination shell is enhanced by eight indium atoms at the much longer K–In distances of 392 pm. The situation is different in EuAl4Ir2. As a consequence of the exchanges of Au → Al and In → Ir the europium atoms shift in c direction and are coordinated by four iridium (316 pm) and twelve aluminum (321–354 pm) neighbors (Fig. 4, left). The different cation coordination can be considered as a kind of puckering effect for bond maximization.

Cutout of the channels (eight-membered rings) in the structures of KAu4In2 [17] and EuAl4Ir2. Potassium (europium), gold (iridium), and indium (aluminum) atoms are drawn as medium gray, blue, and magenta circles, respectively. Displacement ellipsoids are drawn at an 80% level for KAu4In2 and at 99% for EuAl4Ir2

Coordination polyhedra surrounding the Eu (left), Ir (middle), and Al (right) atoms in the structure of EuAl4Ir2. Europium, iridium, and aluminum atoms are drawn as medium gray, blue, and magenta circles, respectively. The site symmetries are given
CaAl4Ir2 and SrAl4Ir2 exhibit smaller / larger unit cells compared to EuAl4Ir2 (see Table 1), in line with the differing ionic radii (ionic radius Ca2+: 112, Sr2+: 126 pm, Eu2+: 125 pm, all CN = 8 [26]). Therefore, displacements of the Ca / Sr atoms are observed; however, only small variations in the Ir and Al positions take place (Table 2). Since Ca2+ is slightly smaller compared to Eu2+ shorter Ca–Ir (307 pm) and Ca–Al (319–345 pm) distances are found nicely underlining the geometric influence of the cation in the cavities. In contrast, the Sr–Ir (317 pm) and Sr–Al (323–355 pm) distances are a bit elongated (Table 3), in line with the slightly larger ionic radius.
This geometric consideration is nicely underpinned by another closely related crystal structure. The stannide KAu4Sn2 reported by Sinnen and Schuster [27] crystallizes in the non-centrosymmetric space group I\(\bar{4}\)c2. The latter is a translationengleiche subgroup of I4/mcm. The underlying group-subgroup scheme is shown in Fig. 5. KAu4Sn2 keeps a body-centered lattice; however, similar to the EuAl4Ir2 structure, a decoupling of the x and y parameters on the 16i site takes place. This way, the gold atoms can react on the slightly smaller size (covalent radius 139 pm [16]) of the tin atoms as compared to indium (covalent radius 142 pm [16]). The distortions in KAu4Sn2 are much more subtle compared to EuAl4Ir2.

Finally, we draw back to the [Al4Ir2]δ– network in the EuAl4Ir2 structure. Besides substantial Ir–Al bonding, we observe a range of Al–Al homoatomic interactions with Al–Al distances of 255–273 pm, all shorter than in fcc aluminum (12 × 286 pm [28]) but slightly longer than the sum of the covalent radii (250 pm) [16]. These interactions substantially stabilize the EuAl4Ir2 structure. We address these features in more detail in the following chapter on the basis of electronic structure calculations.
The valence electron count (VEC) in contrast is not simple to discuss. It increases from SrNi4Sn2 (VEC = 50), via KAu4In2 (VEC = 51) and KAu4Sn2 (VEC = 53) to SrCu4Sn2 (VEC = 54). With the inverse coloring for EuAl4Ir2 the VEC has the much lower value of 32, underpinning the different bonding situation.
Electronic structure calculations
The experimental crystallographic data obtained in this work was used for electronic structure calculations. The performed calculations of the electronic structure confirm the existence of an [Al4Ir2] network in all three phases. Higher electron localization (red regions) is observed around the Al and Ir atoms, which are the most electronegative component (Fig. 6) compared to Ca, Sr, or Eu which donate their valence electrons. However, it should be noted, that the values of the maxima of the electronic localization functions (ELF) are somewhat different, in particular ELF = 0.863 (for CaAl4Ir2), 0.740 (for SrAl4Ir2), and 0.655 (for EuAl4Ir2), respectively. The alkaline earth metals (Ca and Sr) donate their valence electrons more easily than the rare earth metal europium. The ELF iso-surfaces presented in Fig. 6 suggest that polyatomic network is partially negatively charged according to [Al4Ir2]δ– that is compensated by positively charged alkaline earth or rare earth metals.

Electron localization function (ELF) mapping around the particular atoms in CaAl4Ir2, SrAl4Ir2, and EuAl4Ir2 (top) and iso-surfaces of ELF (bottom). The isosurfaces of ELF around the atoms for all structures are drawn at the same levels: isosurface value is 75%, opacity value 70%
These results are in line with the electronegativities (χ(Ca) = 1.00; χ(Sr) = 0.95; χ(Eu) = 1.2; χ(Al) = 1.61; χ(Ir) = 2.20 [16]) and are confirmed by Bader charges calculated for Ca2Ir3Al9 [29].
The integrated crystal orbital Hamilton population (–iCOHP) values confirm that the strongest bonding is between the aluminum and iridium atoms. For the Ir–Al bond the –iCOHP values are 2.317 eV (for CaAl4Ir2), 2.141 eV (for SrAl4Ir2), and 2.093 eV (for EuAl4Ir2), respectively. For the homoatomic Al–Al bonds, the –iCOHP values are much smaller, almost by a factor of two.
The total and partial densities of states (DOS) for all structures are shown in Fig. 7. For EuAl4Ir2, the 4f orbitals were taken into calculations which give the locations of the occupied and unoccupied 4f states to be −6 and 3.8 eV with respect to EF, respectively. In Fig. 7, the Eu partial DOS consists of a corresponding large peak at 3.8 eV and a small but broadened peak −6 eV (extend from −5 to −7 eV). Visible density of electronic states at the Fermi level for these compounds indicates metallic behavior. A common feature of these kind of structures is a very intense peak due to the overlap of the d-orbital of Ir and the p-orbitals of Al in the valence band between −2 to −4 eV (for CaAl4Ir2 and SrAl4Ir2). For EuAl4Ir2, the significant shift of Ir-5d states up to a range from −5 to −7 eV compared to the range from −2 to −4 eV in the Ca- and Sr-compounds is obviously caused by the presence of additional interactions of the electrons of the occupied 5d orbital of iridium with the electrons of the occupied 4f orbital of europium, the states of which are actually in the interval from −5 to −7 eV.

Density of states in CaAl4Ir2, SrAl4Ir2, and EuAl4Ir2
Conclusion
CaAl4Ir2, SrAl4Ir2, and EuAl4Ir2 could be synthesized from the elements and were characterized via single crystal and powder X-ray diffraction experiments. They all crystallize with space group P4/ncc and adopt a new structure type which can be derived from the KAu4In2 type structure (I4/mcm). The title compounds, however, exhibit an inverse coloring of the [Al4Ir2]δ– network with significant polar bonding interactions. Within the polyanion, octagonal channels are observed that host the Caδ+, Srδ+, and Euδ+ cations. The structural relation was established using a group-subgroup scheme. Analysis of the electronic localization function shows, that the proposed network in all three title compounds exhibits a significant negative charge, with larger absolute charges for the alkaline earth compounds, in line with their lower electronegativities.
Experimental
Synthesis
Starting materials for the synthesis of the samples were europium ingots (American Elements, 99.99%), calcium and strontium pieces (Alfa Aesar, 99.9%), iridium powder (Agosi, 99.9%), and aluminum turnings (Koch Chemicals, 99.9%). For the synthesis of EuAl4Ir2, the elements were initially weighed in the atomic ratio 1:2:1 and sealed in an evacuated silica ampoule. The latter was placed in a muffle furnace, heated to 1373 K, kept at that temperature for 6 h, followed by rapid cooling to 873 K and a further annealing step at that temperature for 240 h.
For CaAl4Ir2 and SrAl4Ir2, the constituent metals were also used in their elemental form and weighed in the ideal stoichiometric ratios. Pieces of the elements were loaded into tantalum tubes which were subsequently arc-welded [30] under an argon pressure of about 800 mbar. The argon gas was purified over titanium sponge (873 K), molecular sieves and silica gel. The respective alkaline earth metals and europium pieces were cleaned mechanically under dried (Na) cyclohexane and were kept in Schlenk tubes under argon prior to use. For sealing the ampoules, the elements were added to the tantalum tube in an argon counter-flow. The gas-tight sealed containers were then placed into the water-cooled reaction chamber of an induction furnace (Hüttinger Elektronik, type 1.5/300 or TRUMPF Hüttinger TruHeat HF 5010) and rapidly heated twice to about 1573 K for 20 min interrupted by switching off the power supply for one minute [31]. The temperature was controlled using a Sensor Therm Methis MS09 pyrometer with a stated accuracy of ± 50 K. The tantalum tubes were quenched; no reaction of the samples with the container material was observed.
EDX data
The single crystals of CaAl4Ir2 and EuAl4Ir2, investigated on the diffractometer, were semiquantitatively analyzed by EDX in a Zeiss EVO® MA10 scanning electron microscope (variable pressure mode (60 Pa) and W cathode) using CaF2, EuF3, Ir, and Al2O3 as standards (ZAF correction algorithm (Z = atomic number, A = absorption, F = fluorescence) in combination with fixed X-ray intensity values for the energies of the respective standards, the so-called standard-free analyses). The average of three point measurements on the irregular (conchoidal fracture) crystal surfaces of 16 ± 2 at.-% Ca: 29 ± 2 at.-% Ir: 55 ± 2 at.-% Al and 16 ± 2 at.-% Eu: 30 ± 2 at.-% Ir: 54 ± 2 at.-% Al confirm the ideal compositions (14.3 at.-% M: 28.6 at.-% Ir: 57.1 at.-% Al) refined from the single crystal X-ray data. No impurity elements heavier than sodium (detection limit of the instrument) were detected.
X-ray diffraction on powders and single crystals
To check the phase-purity, the polycrystalline samples of all samples were analyzed by powder X-ray diffraction: Guinier technique, image plate system Fujifilm, BAS-1800, Cu-Kα1 radiation, and α-quartz (a = 491.30 pm, c = 540.46 pm) as an internal standard.
Powder X-ray diffraction (PXRD) patterns of the pulverized samples used for Rietveld refinements were recorded at room temperature on a D8-A25-Advance diffractometer (Bruker, Karlsruhe, Germany) in Bragg–Brentano θ–θ-geometry (goniometer radius 280 mm) with Cu Kα radiation (λ = 154.0596 pm). A 12 µm Ni foil working as Kβ filter and a variable divergence slit were mounted at the primary beam side. A LYNXEYE detector with 192 channels was used at the secondary beam side. Experiments were carried out in a 2θ range of 6–130° with a step size of 0.013° and a total scan time of 1 h. The recorded data was evaluated using the Bruker TOPAS 5.0 software [32], with the observed reflections being treated via single-line fits.
Irregularly shaped crystal splinters were selected from the crushed annealed samples and glued to glass fibers with beeswax. For a first-quality check, Laue patterns were collected on a Buerger precession camera (white Mo radiation, Fuji-film imaging plate). Complete data sets of suitable crystals were recorded at room temperature using a Stoe IPDS-II image plate system (graphite monochromatized Mo radiation; λ = 71.073 pm) in oscillation mode. A numerical absorption correction was applied to the data sets. Details on the crystallographic data are summarized in Table 1.
Structure refinements
The data sets showed a primitive tetragonal lattice with high Laue symmetry. Careful examination of the systematic extinctions revealed compatibility with space group P4/ncc or its subgroups. The centrosymmetric group P4/ncc was found to be correct. The starting atomic parameters were deduced with the charge flipping algorithm (program Superflip [33]) and the structures of CaAl4Ir2, SrAl4Ir2, and EuAl4Ir2 were refined by least squares on F2 using the program Jana 2006 [34] with anisotropic displacement parameters for all atoms. For the final refinement cycles the atomic coordinates were transformed to the setting listed in the Bärnighausen tree. As a check for the correct compositions, the occupancy parameters of all sites were refined in separate series of least-squares cycles. All sites were fully occupied within two standard deviations. The final difference Fourier syntheses was contour-less. The refined atomic coordinates, displacement parameters and interatomic distances are listed in Tables 2 and 3.
CCDCs 2182276 (EuAl4Ir2), 2182277 (SrAl4Ir2), and 2182278 (CaAl4Ir2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Theoretical studies
All calculations were conducted by means of the tight-binding linear muffin-tin orbital method in the atomic spheres approximation (TB–LMTO–ASA) [35,36,37]. The exchange and correlation effects were accounted for in terms of the local density approximation (LDA) [38]. The density of states (DOS) and the integrated crystal orbital Hamilton population (iCOHP) [39] values were used for chemical bonding analyses. The electron localization function (ELF) map was calculated as described in Ref. [40]. The visualization of electronic structure calculation data (ELF, DOS) was generated using the wxDragon program [41].
References
Villars P, Cenzual K (2020) Pearson's Crystal Data: Crystal Structure Database for Inorganic Compounds (release 2020/21). ASM International®, Materials Park, Ohio (USA)
Kammer C (2001) Aluminium-Taschenbuch 1: Grundlagen und Werkstoffe, 16th edn. Beuth Verlag, Berlin
Verbovytskyy Y, Pereira Gonçalves A (2013) Intermetallics 43:103
Ramesh Kumar K, Nair HS, Christian R, Thamizhavel A, Strydom AM (2016) J Phys: Condens Matter 28:436002
Maurya A, Thamizhavel A, Provino A, Pani M, Manfrinetti P, Paudyal D, Dhar SK (2014) Inorg Chem 53:1443
Hulliger F (1993) J Alloys Compd 200:75
Smetana V, Steinberg S, Mudryk Y, Pecharsky V, Miller GJ, Mudring A-V (2015) Inorg Chem 54:10296
Hulliger F (1995) J Alloys Compd 218:255
Gerke B, Pöttgen R (2014) Z Naturforsch 69b:121
Hulliger F, Nissen H-U, Wessicken R (1994) J Alloys Compd 206:263
Xue B, Hulliger F, Schwer H (1995) J Alloys Compd 221:L6
Schmiegel J-P, Block T, Gerke B, Fickenscher T, Touzani RS, Fokwa BPT, Janka O (2016) Inorg Chem 55:9057
Stegemann F, Block T, Klenner S, Zhang Y, Fokwa BPT, Timmer A, Mönig H, Doerenkamp C, Eckert H, Janka O (2019) Chem Eur J 25:10735
Radzieowski M, Stegemann F, Block T, Stahl J, Johrendt D, Janka O (2018) J Am Chem Soc 140:8950
Stegemann F, Block T, Klenner S, Janka O (2019) Chem Eur J 25:3505
Emsley J (1999) The Elements. Oxford University Press, Oxford
Li B, Corbett JD (2006) J Am Chem Soc 128:12392
Hlukhyy V, Raif F, Claus P, Fässler TF (2008) Chem Eur J 14:3737
Pani M, Fornasini ML, Manfrinetti P, Merlo F (2011) Intermetallics 19:957
Mazzone D, Paulose PL, Dhar SK, Fornasini ML, Manfrinetti P (2008) J Alloys Compd 453:24
Bärnighausen H (1980) Commun Math Chem 9:139
Müller U (2004) Z Anorg Allg Chem 630:1519
Müller U (2010) International Tables for Crystallography, Vol. A1, Symmetry relations between space groups. John Wiley and Sons, Chichester, United Kingdom
Müller U (2012) Symmetriebeziehungen zwischen verwandten Kristallstrukturen. Vieweg + Teubner Verlag, Wiesbaden, Germany
Block T, Seidel S, Pöttgen R (2022) Z Kristallogr 237:215
Shannon RD, Prewitt CT (1969) Acta Crystallogr B 25:925
Sinnen H-D, Schuster U-U (1978) Z Naturforsch 33b:1077
Donohue J (1974) The Structures of the Elements. Wiley, New York
Stegemann F, Zhang Y, Fokwa BPT, Janka O (2020) Dalton Trans 49:6398
Pöttgen R, Gulden T, Simon A (1999) GIT Labor-Fachz 43:133
Pöttgen R, Lang A, Hoffmann R-D, Künnen B, Kotzyba G, Müllmann R, Mosel BD, Rosenhahn C (1999) Z Kristallogr 214:143
Bruker AXS Inc (2014) TOPAS 5.0, Karlsruhe, Germany
Palatinus L, Chapuis G (2007) J Appl Crystallogr 40:786
Petříček V, Dušek M, Palatinus L (2014) Z Kristallogr 229:345
Andersen OK (1975) Phys Rev B 12:3060
Andersen OK, Jepsen O (1984) Phys Rev Lett 53:2571
Andersen OK, Pawlowska Z, Jepsen O (1986) Phys Rev B 34:5253
von Barth U, Hedin L (1972) J Phys C 5:1629
Dronskowski R, Blöchl PE (1993) J Phys Chem 97:8617
Becke AD, Edgecombe KE (1990) J Chem Phys 92:5397
Eck B (2103) wxDragon, RWTH Aachen, Aachen, Germany 2013
Acknowledgements
This work was supported by the DAAD (Deutscher Akademischer Austauschdienst), grant no. 91619802. Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and German Science Foundation (project number INST 256/349-1). Funding has also been provided by the Deutsche Forschungsgemeinschaft DFG (JA 1891-10-1).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zaremba, N., Pavlyuk, V., Stegemann, F. et al. MAl4Ir2 (M = Ca, Sr, Eu): superstructures of the KAu4In2 type. Monatsh Chem 154, 43–52 (2023). https://doi.org/10.1007/s00706-022-03005-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-022-03005-8