
Abstract
A large number of signaling abnormalities have been implicated in the emergence and expression of l-DOPA-induced dyskinesia (LID). The primary cause for many of these changes is the development of sensitization at dopamine receptors located on striatal projection neurons (SPN). This initial priming, which is particularly evident at the level of dopamine D1 receptors (D1R), can be viewed as a homeostatic response to dopamine depletion and is further exacerbated by chronic administration of l-DOPA, through a variety of mechanisms affecting various components of the G-protein-coupled receptor machinery. Sensitization of dopamine receptors in combination with pulsatile administration of l-DOPA leads to intermittent and coordinated hyperactivation of signal transduction cascades, ultimately resulting in long-term modifications of gene expression and protein synthesis. A detailed mapping of these pathological changes and of their involvement in LID has been produced during the last decade. According to this emerging picture, activation of sensitized D1R results in the stimulation of cAMP-dependent protein kinase and of the dopamine- and cAMP-regulated phosphoprotein of 32 kDa. This, in turn, activates the extracellular signal-regulated kinases 1 and 2 (ERK), leading to chromatin remodeling and aberrant gene transcription. Dysregulated ERK results also in the stimulation of the mammalian target of rapamycin complex 1, which promotes protein synthesis. Enhanced levels of multiple effector targets, including several transcription factors have been implicated in LID and associated changes in synaptic plasticity and morphology. This article provides an overview of the intracellular modifications occurring in SPN and associated with LID.
Similar content being viewed by others


Introduction
The last two decades have witnessed a remarkable progress in the understanding of the mechanisms mediating the effects of dopamine replacement therapies in Parkinson’s disease (PD). This is particularly evident with regard to l-DOPA, which is still the most effective therapy to control the motor symptoms of PD. A number of molecular changes produced in response to administration of l-DOPA have been identified and many of them have been tested for their involvement in the development and expression of dyskinesia. Most of this information has been obtained from studies centered on the striatum, which is the main component of the basal ganglia and the principal target of anti-parkinsonian medications. This article provides an overview of the signaling abnormalities implicated in l-DOPA-induced dyskinesia (LID), with special emphasis on postsynaptic mechanisms occurring in striatal neurons.
Striatal projection neurons and control of motor function
In PD, the effects of l-DOPA are particularly evident at the level of the GABAergic projection neurons that connect the striatum to the efferent nuclei of the basal ganglia (internal segment of the globus pallidus and substantia nigra pars reticulata). Striatal projection neurons (SPN) express high levels of two major classes of dopamine receptors: the dopamine D1 receptors (D1R), whose activation increases the production of cAMP, and the dopamine D2 receptors (D2R), which inhibit cAMP synthesis. The opposite actions of these receptors on cAMP depend on their coupling to distinct G-proteins. Thus, D1R activate a Gαolf protein that stimulates the activity of adenylyl cyclase (AC), whereas D2R are coupled to Gαi/o-mediated inhibition of AC (Herve et al. 1993; Stoof and Kebabian 1981; Zhuang et al. 2000).
A fundamental feature of the basal ganglia circuitry is that striatal D1R and D2R are for the most part segregated into two separate populations of SPN, which innervate directly, or indirectly the output structures of the basal ganglia. This difference in connectivity confers to the SPN of the direct pathway (dSPN) the ability to promote motor activity, whereas the SPN of the indirect pathway (iSPN) suppresses motor activity. In addition, the selective expression of D1R in dSPN, and of D2R in iSPN, confers to dopamine the ability to enhance excitability of dSPN and to reduce excitability of iSPN, thereby promoting motor activity (Albin et al. 1989; DeLong 1990; Gerfen 1992).
Dopamine receptor sensitization: a fundamental step toward the development of dyskinesia
A major causal factor at the basis of LID is the development of sensitization at striatal dopamine receptors. This phenomenon, which can be viewed as a homeostatic response to the progressive loss of dopamine input to the basal ganglia, has been characterized in particular detail at the level of the D1R located on dSPN, but is not necessarily limited to this neuronal population.
Initial studies in toxin-based models of PD did not detect any increase in the number and affinity of striatal D1R (Aubert et al. 2005; Breese et al. 1987; Joyce 1991; Marshall et al. 1989; Savasta et al. 1988). This conclusion was supported by clinical studies in post-mortem samples from Parkinsonian patients (Hurley et al. 2001; Pimoule et al. 1985; Shinotoh et al. 1993). However, more recent investigations have identified a number of complex modifications affecting D1R and their associated signaling machinery, which prime this system to respond abnormally when exposed to dopaminergic drugs. This original priming is reinforced and maintained by additional molecular alterations produced by chronic administration of l-DOPA, which constitutes a secondary priming event (Fig. 1).

Schematic diagram illustrating the main mechanisms implicated in the sensitization of D1R associated with LID. In PD, the loss of dopamine input to the striatum increases the expression of the Gαolf protein and adenylyl cyclase type 5 (AC5). This change enhances the ability of D1R, which are expressed in the striatal projection neurons of the direct pathway (dSPN), to stimulate the synthesis of cAMP, a primary mediator of dopamine transmission. The increases in Gαolf protein and AC5 are paralleled by reduced expression of the G-protein-coupled receptor kinase 6 (GRK6), which decreases D1R phosphorylation, β-arrestin binding and receptor internalization. Dopamine depletion is also accompanied by increased expression of β-arrestin, which may counteract D1R sensitization. However, this effect is reversed by administration of l-DOPA. In contrast, overexpression of AC5 and, at least in part, Gαolf protein, as well as downregulation of GRK6, persists in dSPN even following administration of l-DOPA. Importantly, treatment with l-DOPA increases the levels of the postsynaptic density 95 (PSD-95) and dopamine D3 receptors (D3R). This further reinforces D1R-mediated transmission, leading to activation of the cAMP/PKA/DARPP-32, ERK and mTORC1 signaling cascades. The importance of these mechanisms is underscored by several studies, indicating that overexpression of GRK6 and β-arrestin, or downregulation of AC5 and PSD-95, decrease dyskinesia. In line with these findings, and further supporting the crucial role played by receptor sensitization in LID, this condition is also reduced by overexpression of the regulator of G-protein signaling 9-2 (RGS9-2), or by combining the administration of l-DOPA with D3R antagonists
Abnormal D1R trafficking and cell surface localization
Studies in non-human primates and rodents have shown that LID is associated with accumulation of D1R at the cell surface and that this effect is caused by disrupted receptor internalization, rather than overexpression (Aubert et al. 2005; Berthet et al. 2009; Guigoni et al. 2007). Notably, l-DOPA-mediated activation of D1R has been shown to impair the ubiquitin–proteasome system, thereby contributing to maintain D1R at the plasma membrane (Berthet et al. 2012). This phenomenon has been associated with the disassembling and reduced catalytic activity of the 26S proteasome (Barroso-Chinea et al. 2015).
The abnormal D1R trafficking implicated in LID has also been attributed to increased striatal levels of the postsynaptic density 95 (PSD-95), a member of the membrane-associated guanylate kinase proteins (Nash et al. 2005; Porras et al. 2012). PSD-95 acts as a postsynaptic scaffold, interacting with both D1R and N-methyl-D-aspartate (NMDA) glutamate receptors (Zhang et al. 2009; Fiorentini et al. 2003) (see article by Mellone and Gardoni in this issue for a detailed description of the involvement of glutamate receptors in dyskinesia). In experimental models of PD, lentiviral-mediated downregulation of PSD-95 enhances the lateral diffusion and internalization of D1R, leading to reduced receptor content at the cell surface. Notably, the same intervention diminishes the severity of LID (Porras et al. 2012).
The sensitized response of D1R to l-DOPA is associated with increased expression of dopamine D3 receptors in dSPN (Bezard et al. 2003; Bordet et al. 1997; Guillin et al. 2001). This event has significant functional repercussions, since the formation of heteromers with dopamine D3 receptors strengthens D1R transmission (Fiorentini et al. 2008; Marcellino et al. 2008). Thus, the up-regulation of dopamine D3 receptors may represent a parallel pathological mechanism, which further exacerbates D1R sensitization, contributing to the development of LID. In line with this possibility, several antagonists at dopamine D3 receptors have been shown to reduce the dyskinetic response to l-DOPA (Bezard et al. 2003; Kumar et al. 2009; Visanji et al. 2009; but see also Mela et al. 2010).
Controlling D1R sensitized transmission and LID via G-protein coupled receptor kinases
The impact on LID of abnormal recruitment of D1R at the cell surface is highlighted by a series of studies based on the overexpression of the G-protein-coupled receptor kinase (GRK) 6. GRK6 is highly expressed in the striatum and involved in the desensitization of D1R (Ahmed et al. 2008; Li et al. 2015). Activated D1R are phosphorylated by GRK6 (Li et al. 2015), which leads to binding of β-arrestin and internalization (Ferguson et al. 1996). In the rat 6-hydroxydopamine (6-OHDA) model of PD, GRK6 expression is reduced (an effect which persists even after administration of l-DOPA) (Ahmed et al. 2008), suggesting a possible involvement in D1R sensitization. In support of this possibility, overexpression of GRK6 by lentiviral delivery promotes D1R internalization, thereby normalizing cell surface expression and counteracting sensitized transmission (Ahmed et al. 2010, 2015a). Importantly, enhanced expression of GRK6 has been shown to reduce LID in rodent and non-human primate models of PD (Ahmed et al. 2010).
An analogous approach to control D1R sensitization and LID is based on targeting β-arrestin. The potential efficacy of this intervention was initially suggested by the observation that dopamine depletion increases the levels of β-arrestin2 in the brain of 1-methyl-4-phenyl 1,2,3,6-tetrahydropyridine (MPTP)-treated monkeys and that this effect is reversed by l-DOPA (Bezard et al. 2005; Price et al. 2010). Subsequent studies performed in several models of PD and LID have shown that overexpression of β-arrestin2 reduces some of the biochemical markers associated with LID and attenuates this condition. In contrast, deletion of β-arrestin2 enhances the dyskinetic effect of l-DOPA (Urs et al. 2015).
Similarly to GRK6, also GRK3 is highly expressed in the striatum and down-regulated in response to dopamine depletion (Ahmed et al. 2008). Interestingly, GRK3 has been found to reduce LID independently of its ability to phosphorylate dopamine receptors and initiate their internalization (Ahmed et al. 2015b). The effect of GRK3 is instead mediated via its regulator of G-protein signaling (RGS) homology domain, which binds to and sequesters active Gαq/11 protein (Carman et al. 1999). This G-protein couples receptors to stimulation of protein kinase C and calcium/calmodulin protein kinase II (Hepler and Gilman 1992), which are, therefore, inhibited by overexpression of GRK3. The downstream mechanism at the basis of the ability of GRK3 overexpression to reduce dyskinesia is not yet clear, but it has been proposed that reduced expression of the transcription factor ∆FosB, which is implicated in LID (see below), may be involved (Ahmed et al. 2015b).
Gαolf protein dysregulation in LID
D1R-mediated signal transduction is linked to the activation of a Gαolf protein positively coupled to AC (Herve et al. 2001; Zhuang et al. 2000). Several studies examined the involvement of this G-protein in the sensitization of D1R associated with PD. Work with experimental models and post-mortem samples from PD patients showed that the loss of striatal dopamine leads to up-regulation of Gαolf protein (Corvol et al. 2004; Herve et al. 1993; Penit-Soria et al. 1997; Marcotte et al. 1994; Alcacer et al. 2012; Rangel-Barajas et al. 2011; Ruiz-DeDiego et al. 2015b; Morigaki et al. 2017). It was also shown that this effect subsides in response to administration of l-DOPA (Corvol et al. 2004; Rangel-Barajas et al. 2011). However, in a recent study Morigaki et al. (2017) show that, in 6-OHDA lesion mice, the levels of Gαolf protein coupled to D1R, although reduced by l-DOPA, remain elevated in comparison with naïve mice. The same authors also show that a different regulation occurs in D2R-expressing iSPN, where the Gαolf protein is coupled to adenosine A2A receptors. In this case, administration of l-DOPA to PD mice down-regulates the Gαolf protein below the levels observed in naïve mice. This, in turn, promotes D2R transmission, which is antagonized by tonic activation of A2A receptors (Schiffmann et al. 2007). Thus, the overall effect of these distinct, cell-specific regulations of the Gαolf protein is to potentiate the effects of l-DOPA and dopamine receptor agonists on both D1R- and D2R-mediated transmission.
In spite of these findings, it has been reported that partial genetic inactivation of the Gnal gene, which fully counteracts the up-regulation of Gαolf protein associated with dopamine depletion, does not modify LID (Alcacer et al. 2012). Further studies will be necessary to characterize the potential impact of the Gαolf protein on dyskinesia. For instance, based on its different regulation in D1R- and D2R-expressing SPN (Morigaki et al. 2017), it would be interesting to examine the effects of selective downregulation of Gnal in dSPN and iSPN.
Attenuating sensitization and dyskinesia via RGS proteins
RGS proteins typically act by promoting the GTPase activity of Gα-proteins, thereby hastening their inactivation (Ross and Wilkie 2000). The RGS9-2 protein is particularly abundant in dopamine-innervated regions, such as the striatum (Rahman et al. 1999), where it reduces dopamine receptor-mediated motor responses (Rahman et al. 2003). In the MPTP non-human primate model of PD, viral-vector-mediated overexpression of RGS9-2 diminishes LID, an effect which has been proposed to depend on reduced sensitization and normalization of transmission at both D1R and D2R (Gold et al. 2007).
The RGS4 is expressed in both dSPN and iSPN (Taymans et al. 2004). Chronic administration of l-DOPA increases RGS4 mRNA in the sensorimotor striatum, and this effect positively correlates with the severity of dyskinetic behavior (Ko et al. 2014). Furthermore, intracerebral infusion of antisense oligonucleotides against RGS4, in parallel with repeated administration of l-DOPA, reduces the development of dyskinesia. Therefore, the way by which RGS4 modulates LID differs from that of RGS9-2 and remains to be fully clarified. In this regard, a modification of the interaction between RGS4 and the metabotropic glutamate type 5 receptor (mGlu5R) has been proposed as a potential mechanism (Ko et al. 2014).
Role of adenylyl cyclase type 5 in LID
In the striatum, the Gαolf protein couples D1R to stimulation of AC type 5 (AC5), which is highly expressed in SPN (Glatt and Snyder 1993; Mons and Cooper 1994). Work performed in the 6-OHDA rat model of PD shows that dopamine depletion leads to overexpression of AC5, which persists even after repeated administration of l-DOPA and correlates with the severity of dyskinesia (Rangel-Barajas et al. 2011). Up-regulation of AC5 is causally linked to LID. Thus, knockout, or lentiviral-mediated downregulation of AC5 in the dorsal striatum were found to attenuate LID in a PD mouse model (Park et al. 2014).
From sensitized D1R to abnormal cAMP signaling
As outlined above, activation of striatal D1R leads to Gαolf-mediated stimulation of AC5 and increased synthesis of cAMP. The sensitization caused by dopamine depletion strongly enhances the ability of l-DOPA to promote this response, thereby hyper-activating cAMP-dependent protein kinase (PKA). This effect is implicated in LID, which is counteracted by administration of the PKA inhibitor, Rp-cAMPS (Lebel et al. 2010). In SPN, cAMP signaling depends on the dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32), which amplifies responses produced by stimulation of PKA through inhibition of protein phosphatase-1 (PP-1) (Fienberg et al. 1998; Greengard 2001). Abnormal activation of DARPP-32, which is mediated by PKA-dependent phosphorylation at Thr34, has been described in rodents and non-human primate models of LID (Picconi et al. 2003; Santini et al. 2007, 2010). In line with these observations, DARPP-32-deficient mice, or mice expressing a mutated form of DARPP-32 lacking Thr34, are less prone to develop LID (Santini et al. 2007, 2012). Attenuated LID has also been described in mice lacking DARPP-32 specifically in the D1R-enriched dSPN (Bateup et al. 2010). In contrast, genetic inactivation of DARPP-32 in iSPN does not modify the dyskinetic response to l-DOPA (Bateup et al. 2010).
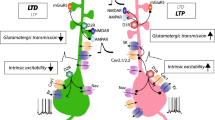
The dysregulation of the PKA/DARPP-32 signaling cascade associated with LID is linked to impaired synaptic plasticity. In particular, abnormal PKA-mediated phosphorylation of DARPP-32, leading to suppression of PP-1 activity, has been implicated in the loss of depotentiation (i.e. in the inability to revert long-term potentiation; LTP) at glutamatergic corticostriatal terminals (Picconi et al. 2003). The absence of bidirectional control of synaptic plasticity is a general feature of dyskinesia and has been shown to occur at both dSPN and iSPN (Calabresi et al. 2016; Picconi et al. 2003; Thiele et al. 2014) (see article by Picconi et al. in this issue for a detailed description of the alterations of synaptic plasticity in LID).
Extracellular signal-regulated kinases 1/2: central players in LID
The hyperactivation of cAMP/PKA/DARPP-32 produced by l-DOPA in the dopamine-depleted striatum results in the dysregulation of multiple signaling pathways ultimately implicated in dyskinesia. Among these downstream cascades, the extracellular signal-regulated kinases 1/2 (ERK) pathway, plays a particularly relevant role and has been the subject of intense investigation (Fig. 2). Gerfen et al. (2002) showed that the loss of dopamine input to the striatum results in a large increase in ERK phosphorylation, in response to administration of a D1R agonist. A few years later Pavon et al. (2006) reported that a similar effect occurred also in response to administration of l-DOPA and in association with LID. The increase in ERK phosphorylation induced by l-DOPA was then localized to dSPN and shown to occur in response to activation of D1R (Darmopil et al. 2009; Santini et al. 2009a; Westin et al. 2007).

Diagram summarizing the central role played by abnormal ERK signaling in LID. Hyperactivation of ERK in response to stimulation of sensitized dopamine D1 receptors is emerging as a central point of convergence for several signaling pathways implicated in dyskinesia, including the cAMP/PKA/DARPP-32, the Ras-GRF1/Ras/MEK, the phospholipase C (PLC)/protein kinase C (PKC) and the Shp2/Src cascades. Pharmacological or genetic inhibition of various constituents of these intracellular pathways has been shown to reduce LID. Activation of ERK generates additional pathological responses, such as abnormal Rhes/mTORC1 signaling, and overexpression of ∆FosB (mediated by MSK1) and Narp. Several interventions counteracting these changes have been shown to reduce dyskinesia
In neuronal cells, ERK activity is modulated by exchange factors, such as the Ras-guanyl nucleotide releasing factor 1 (Ras-GRF1) and the calcium- and diacylglycerol-regulated guanine nucleotide exchange factor II (CalDAG-GEFII), which is particularly abundant in SPN. Calcium-dependent activation of Ras-GRF1 and CalDAG-GEFII induces the exchange of GDP for GTP on the small G-protein Ras (Farnsworth et al. 1995; Toki et al. 2001), which in turn stimulates the protein kinase Raf. Raf phosphorylates the mitogen-activated protein kinase/ERK kinase (MEK), ultimately leading to phosphorylation/activation of ERK.
The first demonstration of the involvement of D1R-dependent activation of ERK in LID was obtained in a mouse model of PD using SL327, a systemically deliverable MEK inhibitor (Santini et al. 2007). Subsequent studies further corroborated the importance of ERK in dyskinesia. For instance, reduced ERK activation and concomitant attenuation of LID were achieved with lovastatin (Schuster et al. 2008), an inhibitor of Ras (Li et al. 2005b). Genetic inactivation or lentiviral-mediated downregulation of Ras-GRF1 resulted in the concomitant reduction of l-DOPA-induced phosphorylation of ERK and dyskinesia (Fasano et al. 2010; Bido et al. 2015). Furthermore, increased expression of CalDAG-GEFII, potentially linked to abnormal activation of ERK, was found in the striata of dyskinetic mice (Crittenden et al. 2009).
The increase in ERK phosphorylation produced by D1R requires intact PKA activity and phosphorylation of DARPP-32 at Thr34 (Lebel et al. 2010; Santini et al. 2007, 2012); but see also (Gerfen et al. 2008). Based on these observations, it was suggested that the inhibition of PP-1 exerted by DARPP-32 would decrease the activity of the striatal-enriched protein tyrosine phosphatase (STEP), thereby counteracting the dephosphorylation of ERK (Paul et al. 2003; Valjent et al. 2005). Additional work showed that the increase in ERK phosphorylation depends also on the interaction of D1R with the Src homology 2 domain-containing phosphatase 2 (Shp2) (Fiorentini et al. 2011, 2013). In particular, it was proposed that D1R-mediated activation of PKA leads to stimulation of Shp2. This, in turn, would disinhibit Src kinases, which positively regulate the Ras/Raf/ERK signaling cascade (Fiorentini et al. 2011; Yang et al. 2006). In accordance with this possibility, persistent Shp2/ERK activation was found in the striata of hemi-parkinsonian rats displaying dyskinetic behavior in response to l-DOPA or a D1R agonist (Fiorentini et al. 2013). Furthermore, lentiviral-mediated downregulation of Shp2 decreased ERK phosphorylation and attenuated LID (Fiorentini et al. 2016).
The increase in ERK activity mediated by l-DOPA or a D1R agonist is reduced by pharmacological inhibition of the mGlu5R, an intervention which also attenuates dyskinesia (Rylander et al. 2009). The mGlu5R-dependent stimulation of ERK is likely to occur via Gαq protein-mediated activation of phospholipase C and calcium-dependent signaling (Fieblinger et al. 2014; Rylander et al. 2009), thereby suggesting a further potential mechanism underlying the pathological activation of ERK in LID.
Studies performed in various models of PD indicate that the abnormal regulation of ERK observed in SPN subsides during the course of chronic l-DOPA administration, suggesting its primary involvement in the development, rather than in the maintenance of dyskinesia (Ding et al. 2011; Santini et al. 2010). Interestingly, this normalization is paralleled by increased ERK phosphorylation in striatal cholinergic interneurons (Chl), which may, therefore, represent an additional neural substrate specifically implicated in the long-term manifestation of dyskinesia (Ding et al. 2011). In line with this possibility, diphtheria toxin-mediated ablation of Chl attenuates LID, without affecting the anti-akinetic properties of l-DOPA (Won et al. 2014). The increase in ERK activity observed in Chl is indicative of increased excitability, which may be causally related to LID. This is supported by the observation that, in a mouse model of PD, short-pulse optogenetic stimulation of Chl exacerbates the dyskinetic response to l-DOPA (Bordia et al. 2016). Conversely, in the same PD model, LID is reduced by interventions that counteract Chl hyper-excitability by antagonizing excitatory histamine H2 receptors, whose activity is pathologically enhanced in the Chl of dyskinetic mice (Lim et al. 2015).
Activation of ERK results in the stimulation of complex intracellular cascades, involving distinct signaling pathways linked to the regulation of mRNA translation and gene transcription. A large body of evidence indicates that these downstream events represent an essential causal factor in the emergence of LID and several components of these intracellular pathways have been identified as potential targets for anti-dyskinetic interventions (Fig. 2).
Involvement of the mammalian target of rapamycin complex 1 in LID
One interesting downstream effector target of ERK is the mammalian target of rapamycin complex 1 (mTORC1) signaling cascade, which is involved in the regulation of protein synthesis. mTORC1 is activated by the small G proteins, Ras homolog enriched in brain (Rheb) and Ras homolog enriched in striatum (Rhes) (Long et al. 2005; Subramaniam et al. 2011), which are inhibited by the GTPase-activating heterodimer composed of tuberous sclerosis (TSC) 1 and 2 (Laplante and Sabatini 2012). ERK inhibits the TSC1/TSC2 complex, thereby promoting mTORC1 activity (Ma et al. 2005; Roux et al. 2004; Tsokas et al. 2007). This results in the control of downstream effectors, including the p70 ribosomal S6 kinases (Ruvinsky and Meyuhas 2006; Thomas et al. 1979) and the initiation factor 4E-binding protein 1 (Gingras et al. 2001), leading to enhanced mRNA translation (Laplante and Sabatini 2012). Augmented signaling along the mTORC1 cascade has been described in rodent models of dyskinesia (Decressac and Bjorklund 2013; Santini et al. 2009b; Subramaniam et al. 2011). Importantly, administration of mTORC1 inhibitors, such as rapamycin, or its analog temsirolimus (CCI-779), was shown to reduce the development and expression of LID in rodent models of PD (Decressac and Bjorklund 2013; Santini et al. 2009b; Urs et al. 2015). The importance of abnormal mTORC1 activation in dyskinesia is further substantiated by the observation that LID is reduced in Rhes-deficient mice (Subramaniam et al. 2011).
Abnormal gene transcription in LID
Upon activation, a large proportion of ERK translocates to the nucleus (Chen et al. 1992), where it controls a number of effectors implicated in various aspects of transcriptional regulation (Treisman 1996; Whitmarsh 2007). One major group of ERK downstream targets is represented by early response transcription factors, among which FosB has attracted considerable interest with regard to its involvement in LID. Chronic administration of l-DOPA induces a progressive accumulation of stable isoforms of FosB, collectively denominated ∆FosB, in the striata of animal models of LID and dyskinetic patients (Andersson et al. 1999; Berton et al. 2009; Darmopil et al. 2009; Fasano et al. 2010; Feyder et al. 2016; Lindgren et al. 2011; Pavon et al. 2006; Tekumalla et al. 2001). The increase in ∆FosB associated with dyskinesia is limited to the D1R-expressing dSPN and is mediated by activation of cAMP/PKA and ERK (Andersson et al. 1999; Darmopil et al. 2009; Feyder et al. 2016; Fasano et al. 2010; Lebel et al. 2010; Schuster et al. 2008; Chen et al. 2017).
∆FosB accumulation plays a causal role in LID. Initial experiments showing that antisense oligonucleotides against fosB reduced dyskinesia were followed by several studies in which LID was exacerbated or reduced by modulating FosB-dependent transcriptional activity. ∆FosB forms heterodimers with members of the jun family of transcription factors (typically JunD), which promote the expression of late response genes by binding to their activator protein-1 (AP-1) consensus sites (Hope et al. 1994). Striatal overexpression of a truncated form of JunD (∆JunD), lacking the transactivation domain and acting as a ∆FosB dominant-negative, reduced LID in the MPTP non-human primate model of PD (Berton et al. 2009). In contrast, overexpression of ∆FosB in the dopamine-depleted striatum of rats with a unilateral 6-OHDA lesion was sufficient to elicit abnormal involuntary movements indicative of dyskinetic behavior (Cao et al. 2010). In another study, selective silencing of ∆FosB expressing SPN, using the Daun02 inactivation method (Ghosh et al. 2000), was found to decrease the severity of LID (Engeln et al. 2016).
Work in the 6-OHDA mouse model of PD showed that overexpression of ∆FosB induced specifically in dSPN exacerbates LID. Conversely, dyskinesia is reduced by overexpression of ∆cJun, a truncated form of cJun lacking transcriptional activity (Feyder et al. 2016). The ability of ∆cJun to reduce LID suggests a possible involvement of dysregulated cJun in dyskinesia. This idea is supported by recent work showing that administration of l-DOPA to mice with a 6-OHDA lesion increases cJun phosphorylation (Spigolon et al. 2018). This effect occurs selectively in dSPN and is linked to increased strength at corticostriatal synapses, which has been associated with LID (Calabresi et al. 2016; Spigolon et al. 2018).
∆FosB expression and LID are modulated by genetic manipulation of the downstream regulatory element antagonistic modulator (DREAM). DREAM is activated by calcium and cAMP, and functions as a transcriptional repressor by binding to a regulatory element located downstream of the transcription initiation site of several genes, including members of the fos family. In a mouse model of PD, DREAM deficiency exacerbates the accumulation of ∆FosB and the severity of LID, whereas overexpression of a dominant-active DREAM mutant reduces both ∆FosB expression and dyskinesia (Ruiz-DeDiego et al. 2015a).
LID is also associated with abnormal expression of the transcription factor Zif268 (also called NGFI-A, Krox24, or Egr1) (Bastide et al. 2014; Carta et al. 2005; Fisone 2010; Carta et al. 2010). This effect may depend on concomitant activation of ERK, which mediates the increase in zif268 expression produced by cocaine in the striatum (Valjent et al. 2006). In the rat model of PD, repeated administration of l-DOPA produces a persistent elevation of zif268 mRNA expression in dSPN (Carta et al. 2005). Increased Zif268 may in turn lead to augmented expression of the activity-regulated cytoskeleton-associated protein (Arc, or Arg3.1) (Li et al. 2005a), which is also up-regulated in the dSPN of dyskinetic rats (Sgambato-Faure et al. 2005).
Aberrant regulation of ∆FosB, Zif268 and Arc is likely to contribute to the changes in synaptic plasticity described in experimental models of LID. In fact, these transcription factors regulate LTP and synaptic morphology (Guzowski et al. 2000; Jones et al. 2001; Nestler et al. 2001; Peebles et al. 2010; Plath et al. 2006), which undergo profound changes in dyskinesia. For instance, they could be implicated in the impairment of synaptic downscaling proposed to mediate dyskinetic behavior (Calabresi et al. 2016).
Dopamine depletion is associated with a dramatic loss of synaptic spines in dSPN and iSPN (Day et al. 2006; Suarez et al. 2014, 2016). Administration of l-DOPA re-establishes normal spine density in D2R expressing iSPN (Suarez et al. 2014, 2016). However, these newly formed synapses display thinner neck diameter and smaller PSD. Consequently, their numeric recovery does not rescue overall synaptic strength (Suarez et al. 2016). In contrast, l-DOPA normalizes synaptic strength in D1R-expressing dSPN, in spite of persistent reduction in spine density. This paradoxical effect has been ascribed to increased spine volume and PSD length (Suarez et al. 2016), possibly caused by the parallel increase in ∆FosB, Zif268 and Arc in dSPN.
A number of studies have provided additional information concerning the involvement of abnormal gene expression in LID. Analysis based on DNA microarray technology revealed major differences in striatal gene expression between dyskinetic and non-dyskinetic rats, both receiving l-DOPA. In particular, dyskinesia was associated with up-regulation of genes involved in calcium homeostasis, synaptic activity and remodeling, and downregulation of genes related to energy metabolism, or encoding ribosomal proteins (Konradi et al. 2004).
Using a similar approach, gene expression changes associated with de novo or long-term administration of l-DOPA were compared in dopamine-depleted rats. The overall transcriptomic response was higher following chronic l-DOPA treatment. Moreover, distinct clusters of genes were specifically deregulated in the two conditions, with only a small set of genes (including arc) affected by both treatments (El Atifi-Borel et al. 2009). Interestingly, genes involved in signal transduction and synaptic transmission were up-regulated by acute treatment and potentially involved in the initial response necessary for the development of LID. Other genes related to structural and cellular alterations were instead up-regulated by chronic treatment and possibly necessary for the persistent modifications involved in the maintenance of dyskinesia (El Atifi-Borel et al. 2009).
Recent work focused on the identification of early changes in gene expression implicated in the development of dyskinesia. A time course of the effect of l-DOPA in the 6-OHDA mouse model of PD identified different sets of genes deregulated at 1 h (immediate early genes, mitogen-activated protein kinase signaling-related and synapse-associated genes), 3 h (long-term depression-related genes) and 6 h (genes regulating protein phosphatase activity) (Charbonnier-Beaupel et al. 2015). Among these changes, those requiring ERK activation were selected as more likely molecular markers involved in the early development of LID. The study then focused on the immediate early gene, nptx2, whose overexpression was found to be ERK-dependent and associated with a high dyskinetic response (Charbonnier-Beaupel et al. 2015). Nptx2 codes for the neuronal activity-regulated pentraxin (Narp), which is involved in synaptic plasticity (Xu et al. 2003) and may represent a potential target for anti-dyskinetic therapy. Accordingly, Narp knockout mice showed a reduction of dyskinesia, while virus-mediated overexpression of Nptx2 worsened LID (Charbonnier-Beaupel et al. 2015).
A cell-type specific analysis of gene expression changes in dSPNs and iSPNs following chronic l-DOPA was provided by Heiman et al. (2014) using the translating ribosome affinity purification (TRAP) method. Dopamine depletion alone induced gene alterations in both classes of SPNs. However, chronic l-DOPA treatment strongly affected the expression profile in dSPNs, but induced only small changes in iSPNs. Besides increased levels of mitogen-activated protein kinase-related genes, dSPN showed up-regulation of several markers involved in homeostatic processes (e.g. Rgs6, Sstr2, Kcnn3), which were most likely activated to counteract abnormal D1R transmission. A noteworthy finding observed exclusively in dSPNs was the correlation between expression of genes such as Nurr1, Itch, Fra-1 and Dusp and the severity of the dyskinetic response to l-DOPA (Heiman et al. 2014).
Chromatin remodeling in LID
The abnormal D1R signaling produced by the combination of dopamine depletion and chronic administration of l-DOPA results in a number of chromatin modifications associated with dyskinesia (Fig. 3). Activation of mitogen-activated protein kinases, including ERK, results in the phosphorylation of histone H3 at Ser10 and Ser28, which mediates the rapid induction of c-jun and c-fos (Clayton and Mahadevan 2003; Heiman et al. 2014). In LID, the hyperactivation of cAMP and ERK signaling is accompanied by a similar response, which requires D1R and occurs selectively in dSPN (Feyder et al. 2016; Sodersten et al. 2014; Santini et al. 2007, 2012; Darmopil et al. 2009; Ruiz-DeDiego et al. 2015a).

Summary of the main chromatin modifications associated with LID. During dyskinesia, ERK-mediated activation of mitogen- and stress-activated kinase 1 (MSK1) results in the phosphorylation of histone H3 at Ser10 and 28. Both modifications promote gene transcription. Enrichment of histone H3 phosphorylated at Ser10 has been reported to occur at the fosB promoter and may, therefore, participate in the accumulation of ∆FosB associated with LID. ∆FosB forms heterodimers with members of the jun family of transcription factors which bind to the activator protein-1 (AP-1) sites of late response genes to regulate their transcription. Phosphorylation of histone H3 at Ser28 counteracts the action of Polycomb group (PcG) proteins, which suppress gene transcription via methylation at Lys27. Enrichment of Ser28 phosphorylation, which leads to displacement of PcG, has been found at the promoters of several genes up-regulated in the striata of dyskinetic mice. LID is also accompanied by increased expression of DNA demethylases, such as Tet3 and Gadd45b. This regulation is paralleled by reduced methylation of CpG dense regions of DNA, which is indicative of increased transcriptional activity
Studies in the mouse showed that the increase in ∆FosB implicated in LID depends on the mitogen- and stress-activated kinase 1 (MSK1), a major downstream target of ERK. Chromatin immunoprecipitation (ChIP) analysis showed that ERK-mediated activation of MSK1 leads to the accumulation of histone H3 phosphorylated on Ser10, at the promoter of fosB (Feyder et al. 2016). These findings provide a possible mechanism explaining the ability of sensitized D1R to promote ∆FosB expression. In line with the involvement of MSK1 in the regulation of ∆FosB, MSK1 knockout mice display attenuated dyskinesia in response to l-DOPA (Feyder et al. 2016) but see also (Alcacer et al. 2014).
The abnormal activation of PKA/DARPP-32 and ERK/MSK1 caused by l-DOPA in experimental parkinsonism is also leading to hyper-phosphorylation of histone H3 at Ser28 (Sodersten et al. 2014). This modification counteracts the repressive action exerted on transcription by Polycomb group (PcG) proteins (Gehani et al. 2010). Polycomb repressive complex 2 (PRC2) catalyzes di- and trimethylation of Lys27 on histone H3 at the promoters of specific genes, thereby establishing repression. In dyskinesia, DARPP-32- and MSK1-mediated phosphorylation at Ser28 leads to dissociation of PcG and de-repression of gene transcription (Sodersten et al. 2014). Thus, analysis of data from RNA- and ChIP-sequencing shows that induction of Ser28 phosphorylation on histone H3 correlates with genome-wide increase of mRNA transcripts from loci marked by Lys27 trimethylation (Sodersten et al. 2014).
Among the up-regulated genes positive for histone H3 Ser28 phosphorylation and Lys27 trimethylation were transcription factors involved in dopamine-mediated responses and potentially implicated in LID, such as atf3 and npas4 (Sodersten et al. 2014). Increased atf3 expression in the dorsal striatum is also triggered by chronic administration of amphetamine, a drug which augments dopamine release and produces a strong motor stimulant effect (Green et al. 2008). Npas4 is also interesting in the context of LID, since it promotes the expression of other immediate early genes associated with dyskinesia, such as arc, zif268 and c-fos (Ramamoorthi et al. 2011).
A particularly stable form of chromatin remodeling is produced by the methylation of DNA within short interspersed sequences enriched in cytosine and guanine, called CpG islands. Methylation of cytosines within CpG islands, which are thought to represent transcription initiation sites, is typically associated with silencing of gene expression (Deaton and Bird 2011). Regulation of DNA methylation has been intensively investigated with regard to its involvement in memory, addictive behavior and synaptic plasticity (Halder et al. 2016; LaPlant et al. 2010; Levenson et al. 2006; Miller et al. 2008) and recent studies provide information concerning its impact on LID. Chronic administration of l-DOPA in combination with dopamine depletion results in up-regulation of two DNA demethylases, Tet3 and Gadd45b, in the striata of dyskinetic rodents (Figge et al. 2016; Park et al. 2016). In line with this finding, genome-wide analysis of methylation identified a subset of CpG-enriched sequences, in which changes in methylation occurred in response to both dopamine depletion and administration of l-DOPA. This subset was enriched in transcripts aberrantly transcribed in the D1R expressing dSPN (Heiman et al. 2014), indicating a correlation between decreased DNA methylation and activation of genes associated with LID, including fosB and arc (Figge et al. 2016). Remarkably, the CpG-enriched sequences undergoing changes in methylation were mainly located in regulatory regions at long distance from the promoters (Gray et al. 2015) and implicated in activity-dependent regulation of immediate early genes, including fos (Malik et al. 2014).
The aforementioned findings suggest that interventions which control abnormal gene activation by promoting DNA methylation may represent potential strategies for the treatment of LID (Figge et al. 2016). Thus, administration of methionine, which enhances DNA methylation, reduces LID in rats, whereas RG-108, a DNA methyltransferase inhibitor, exacerbates this condition (Figge et al. 2016). However, genetic inactivation of Gadd45b, an intervention that should reduce DNA demethylation, has been found to enhance LID (Park et al. 2016). In contrast, increased expression of Gadd45b, which should decrease DNA methylation, thereby aggravating abnormal gene expression, reduces dyskinesia (Park et al. 2016). Therefore, the dynamics of the regulation of DNA methylation during LID, as well as the identification of specific targets for the development of effective therapeutic strategies, remain to be fully defined.
Conclusions
The use of animal models has been essential to discover numerous signaling abnormalities potentially implicated in LID, thereby providing useful information for the generation of new strategies to efficiently control the motor symptoms of PD. Drugs targeting various components of the Ras-ERK and mTORC1 signaling cascades are currently used, or under clinical investigation as anti-tumoral agents. Inhibitors of Raf, such as sorafenib, vemurafenib and dabrafenib, and MEK 1/2, such as trametinib, have been approved for the treatment of various types of cancers. Additional compounds with a similar pharmacological profile have entered clinical trials (Mandal et al. 2016). Different classes of mTOR inhibitors, including analogs of rapamycin (e.g. everolimus) and ATP-competitive inhibitors (AZD2014), have also been approved, or are in clinical trials as anticancer therapies (Xie et al. 2016).
The relative clinical safety of these compounds represents an encouraging feature with regard to their possible use for the treatment of neurodegenerative and neuropsychiatric diseases. However, the use of these drugs is still complicated by the risk of negative side effects, since they must be administered chronically and at relatively high doses. This problem could be circumvented combining low doses of drugs acting on distinct targets. The resulting cumulative effect exerted by partial inhibition of multiple signaling pathways may be sufficient to counteract LID, without abolishing intracellular processes necessary for normal cell function. In support of this approach, various combinations of Raf, MEK1/2 and mTORC1 inhibitors are commonly used in cancer treatment (Mandal et al. 2016).
An additional way to minimize the risk of complications is to identify effector targets located downstream of critical elements implicated in LID. This approach is exemplified by the search for signaling pathways (e.g. mTORC1), or genes (e.g. ∆FosB and Narp) controlled by ERK, whose dysregulation is ultimately implicated in dyskinesia. By the same reasoning, it will be important to distinguish specific constituents of the chromatin remodeling machinery, particularly enzymes catalyzing covalent modifications of both histones and DNA, which may represent viable druggable targets.
Progress in the understanding of the mechanisms of LID has also led to the identification of several molecular events, such as activation of key signaling components and overexpression of specific genes, which can be utilized to evaluate the potential efficacy of various types of interventions. For instance, continuous, as opposed to pulsatile, administration of l-DOPA, which significantly reduces the risk of developing LID, is not associated with increased phosphorylation of DARPP-32 and ERK, or with accumulation of ∆FosB in striatal neurons (Lebel et al. 2010). Similarly, administration of antagonists at mGlu5R, which attenuates dyskinesia in experimental PD, is associated with reduced striatal levels of phosphorylated ERK and MSK1 (Rylander et al. 2009). Altogether, these biochemical markers represent important tools for the rapid assessment of putative anti-dyskinetic therapies.
References
Ahmed MR, Bychkov E, Gurevich VV, Benovic JL, Gurevich EV (2008) Altered expression and subcellular distribution of GRK subtypes in the dopamine-depleted rat basal ganglia is not normalized by l-DOPA treatment. J Neurochem 104:1622–1636
Ahmed MR, Berthet A, Bychkov E, Porras G, Li Q, Bioulac BH, Carl YT, Bloch B, Kook S, Aubert I, Dovero S, Doudnikoff E, Gurevich VV, Gurevich EV, Bezard E (2010) Lentiviral overexpression of GRK6 alleviates l-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci Transl Med 2:28ra28. https://doi.org/10.1126/scitranslmed.3000664
Ahmed MR, Bychkov E, Kook S, Zurkovsky L, Dalby KN, Gurevich EV (2015a) Overexpression of GRK6 rescues l-DOPA-induced signaling abnormalities in the dopamine-depleted striatum of hemiparkinsonian rats. Exp Neurol 266:42–54. https://doi.org/10.1016/j.expneurol.2015.02.008
Ahmed MR, Bychkov E, Li L, Gurevich VV, Gurevich EV (2015b) GRK3 suppresses l-DOPA-induced dyskinesia in the rat model of Parkinson’s disease via its RGS homology domain. Sci Rep 5:10920. https://doi.org/10.1038/srep10920
Albin RL, Young AB, Penney JB (1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12:366–375
Alcacer C, Santini E, Valjent E, Gaven F, Girault JA, Herve D (2012) Galphaolf mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in l-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci 32:5900–5910. https://doi.org/10.1523/jneurosci.0837-12.2012
Alcacer C, Charbonnier-Beaupel F, Corvol JC, Girault JA, Herve D (2014) Mitogen- and stress-activated protein kinase 1 is required for specific signaling responses in dopamine-denervated mouse striatum, but is not necessary for l-DOPA-induced dyskinesia. Neurosci Lett 583:76–80. https://doi.org/10.1016/j.neulet.2014.09.018
Andersson M, Hilbertson A, Cenci MA (1999) Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol Dis 6:461–474
Aubert I, Guigoni C, Hakansson K, Li Q, Dovero S, Barthe N, Bioulac BH, Gross CE, Fisone G, Bloch B, Bezard E (2005) Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol 57:17–26. https://doi.org/10.1002/ana.20296
Barroso-Chinea P, Thiolat ML, Bido S, Martinez A, Doudnikoff E, Baufreton J, Bourdenx M, Bloch B, Bezard E, Martin-Negrier ML (2015) D1 dopamine receptor stimulation impairs striatal proteasome activity in Parkinsonism through 26S proteasome disassembly. Neurobiol Dis 78:77–87. https://doi.org/10.1016/j.nbd.2015.02.024
Bastide MF, Dovero S, Charron G, Porras G, Gross CE, Fernagut PO, Bezard E (2014) Immediate-early gene expression in structures outside the basal ganglia is associated to l-DOPA-induced dyskinesia. Neurobiol Dis 62:179–192. https://doi.org/10.1016/j.nbd.2013.09.020
Bateup HS, Santini E, Shen W, Birnbaum S, Valjent E, Surmeier DJ, Fisone G, Nestler EJ, Greengard P (2010) Distinct subclasses of medium spiny neurons differentially regulate striatal motor behaviors. Proc Natl Acad Sci USA 107:14845–14850. https://doi.org/10.1073/pnas.1009874107
Berthet A, Porras G, Doudnikoff E, Stark H, Cador M, Bezard E, Bloch B (2009) Pharmacological analysis demonstrates dramatic alteration of D1 dopamine receptor neuronal distribution in the rat analog of l-DOPA-induced dyskinesia. J Neurosci 29:4829–4835. https://doi.org/10.1523/jneurosci.5884-08.2009
Berthet A, Bezard E, Porras G, Fasano S, Barroso-Chinea P, Dehay B, Martinez A, Thiolat ML, Nosten-Bertrand M, Giros B, Baufreton J, Li Q, Bloch B, Martin-Negrier ML (2012) l-DOPA impairs proteasome activity in parkinsonism through D1 dopamine receptor. J Neurosci 32:681–691. https://doi.org/10.1523/JNEUROSCI.1541-11.2012
Berton O, Guigoni C, Li Q, Bioulac BH, Aubert I, Gross CE, Dileone RJ, Nestler EJ, Bezard E (2009) Striatal overexpression of DeltaJunD resets l-DOPA-induced dyskinesia in a primate model of Parkinson disease. Biol Psychiatry 66:554–561. https://doi.org/10.1016/j.biopsych.2009.04.005
Bezard E, Ferry S, Mach U, Stark H, Leriche L, Boraud T, Gross C, Sokoloff P (2003) Attenuation of levodopa-induced dyskinesia by normalizing dopamine D3 receptor function. Nat Med 9:762–767
Bezard E, Gross CE, Qin L, Gurevich VV, Benovic JL, Gurevich EV (2005) l-DOPA reverses the MPTP-induced elevation of the arrestin2 and GRK6 expression and enhanced ERK activation in monkey brain. Neurobiol Dis 18:323–335. https://doi.org/10.1016/j.nbd.2004.10.005
Bido S, Solari N, Indrigo M, D’Antoni A, Brambilla R, Morari M, Fasano S (2015) Differential involvement of Ras-GRF1 and Ras-GRF2 in l-DOPA-induced dyskinesia. Ann Clin Transl Neurol 2:662–678. https://doi.org/10.1002/acn3.202
Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC (1997) Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci USA 94:3363–3367
Bordia T, Perez XA, Heiss J, Zhang D, Quik M (2016) Optogenetic activation of striatal cholinergic interneurons regulates l-dopa-induced dyskinesias. Neurobiol Dis 91:47–58. https://doi.org/10.1016/j.nbd.2016.02.019
Breese GR, Duncan GE, Napier TC, Bondy SC, Iorio LC, Mueller RA (1987) 6-hydroxydopamine treatments enhance behavioral responses to intracerebral microinjection of D1- and D2-dopamine agonists into nucleus accumbens and striatum without changing dopamine antagonist binding. J Pharmacol Exp Ther 240:167–176
Calabresi P, Pisani A, Rothwell J, Ghiglieri V, Obeso JA, Picconi B (2016) Hyperkinetic disorders and loss of synaptic downscaling. Nat Neurosci 19:868–875. https://doi.org/10.1038/nn.4306
Cao X, Yasuda T, Uthayathas S, Watts RL, Mouradian MM, Mochizuki H, Papa SM (2010) Striatal overexpression of DeltaFosB reproduces chronic levodopa-induced involuntary movements. J Neurosci 30:7335–7343. https://doi.org/10.1523/jneurosci.0252
Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T (1999) Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem 274:34483–34492
Carta AR, Tronci E, Pinna A, Morelli M (2005) Different responsiveness of striatonigral and striatopallidal neurons to l-DOPA after a subchronic intermittent l-DOPA treatment. Eur J Neurosci 21:1196–1204
Carta AR, Frau L, Pinna A, Morelli M (2010) Dyskinetic potential of dopamine agonists is associated with different striatonigral/striatopallidal zif-268 expression. Exp Neurol 224:395–402. https://doi.org/10.1016/j.expneurol.20
Charbonnier-Beaupel F, Malerbi M, Alcacer C, Tahiri K, Carpentier W, Wang C, During M, Xu D, Worley PF, Girault JA, Herve D, Corvol JC (2015) Gene expression analyses identify Narp contribution in the development of l-DOPA-induced dyskinesia. J Neurosci 35:96–111. https://doi.org/10.1523/JNEUROSCI.5231-13.2015
Chen RH, Sarnecki C, Blenis J (1992) Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol 12:915–927
Chen G, Nie S, Han C, Ma K, Xu Y, Zhang Z, Papa SM, Cao X (2017) Antidyskinetic effects of MEK inhibitor are associated with multiple neurochemical alterations in the striatum of hemiparkinsonian rats. Front Neurosci 11:112. https://doi.org/10.3389/fnins.2017.00112
Clayton AL, Mahadevan LC (2003) MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett 546:51–58
Corvol JC, Muriel MP, Valjent E, Feger J, Hanoun N, Girault JA, Hirsch EC, Herve D (2004) Persistent increase in olfactory type G-protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J Neurosci 24:7007–7014
Crittenden JR, Cantuti-Castelvetri I, Saka E, Keller-McGandy CE, Hernandez LF, Kett LR, Young AB, Standaert DG, Graybiel AM (2009) Dysregulation of CalDAG-GEFI and CalDAG-GEFII predicts the severity of motor side-effects induced by anti-parkinsonian therapy. Proc Natl Acad Sci US A 106:2892–2896. https://doi.org/10.1073/pnas.0812822106
Darmopil S, Martin AB, De Diego IR, Ares S, Moratalla R (2009) Genetic inactivation of dopamine D1 but not D2 receptors inhibits l-DOPA-induced dyskinesia and histone activation. Biol Psychiatry 66:603–613. https://doi.org/10.1016/j.biopsych.2009.04.025
Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ (2006) Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci 9:251–259
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25:1010–1022. https://doi.org/10.1101/gad.2037511
Decressac M, Bjorklund A (2013) mTOR inhibition alleviates l-DOPA-induced dyskinesia in parkinsonian rats. J Parkinsons Dis 3:13–17. https://doi.org/10.3233/JPD-120155
DeLong MR (1990) Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13:281–285
Ding Y, Won L, Britt JP, Lim SA, McGehee DS, Kang UJ (2011) Enhanced striatal cholinergic neuronal activity mediates l-DOPA-induced dyskinesia in parkinsonian mice. Proc Natl Acad Sci USA 108:840–845. https://doi.org/10.1073/pnas.1006511108
El Atifi-Borel M, Buggia-Prevot V, Platet N, Benabid AL, Berger F, Sgambato-Faure V (2009) De novo and long-term l-Dopa induce both common and distinct striatal gene profiles in the hemiparkinsonian rat. Neurobiol Dis 34:340–350. https://doi.org/10.1016/j.nbd.2009.02.002
Engeln M, Bastide MF, Toulme E, Dehay B, Bourdenx M, Doudnikoff E, Li Q, Gross CE, Boue-Grabot E, Pisani A, Bezard E, Fernagut PO (2016) Selective inactivation of striatal FosB/DeltaFosB-expressing neurons alleviates l-DOPA-induced dyskinesia. Biol Psychiatry 79:354–361. https://doi.org/10.1016/j.biopsych.2014.07.007
Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA (1995) Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376:524–527
Fasano S, Bezard E, D’Antoni A, Francardo V, Indrigo M, Qin L, Dovero S, Cerovic M, Cenci MA, Brambilla R (2010) Inhibition of Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) signaling in the striatum reverts motor symptoms associated with l-dopa-induced dyskinesia. Proc Natl Acad Sci USA 107:21824–21829. https://doi.org/10.1073/pnas.1012071107
Ferguson SS, Downey WE 3rd, Colapietro AM, Barak LS, Menard L, Caron MG (1996) Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271:363–366
Feyder M, Sodersten E, Santini E, Vialou V, LaPlant Q, Watts EL, Spigolon G, Hansen K, Caboche J, Nestler EJ, Fisone G (2016) A role for mitogen- and stress-activated kinase 1 in l-DOPA-induced dyskinesia and FosB expression. Biol Psychiatry 79:362–371. https://doi.org/10.1016/j.biopsych.2014.07.019
Fieblinger T, Sebastianutto I, Alcacer C, Bimpisidis Z, Maslava N, Sandberg S, Engblom D, Cenci MA (2014) Mechanisms of dopamine D1 receptor-mediated ERK1/2 activation in the parkinsonian striatum and their modulation by metabotropic glutamate receptor type 5. J Neurosci 34:4728–4740. https://doi.org/10.1523/JNEUROSCI.2702-13.2014
Fienberg AA, Hiroi N, Mermelstein PG, Song W, Snyder GL, Nishi A, Cheramy A, O’Callaghan JP, Miller DB, Cole DG, Corbett R, Haile CN, Cooper DC, Onn SP, Grace AA, Ouimet CC, White FJ, Hyman SE, Surmeier DJ, Girault J, Nestler EJ, Greengard P (1998) DARPP-32: regulator of the efficacy of dopaminergic neurotransmission. Science 281:838–842
Figge DA, Eskow Jaunarajs KL, Standaert DG (2016) Dynamic DNA methylation regulates levodopa-induced dyskinesia. J Neurosci 36:6514–6524. https://doi.org/10.1523/JNEUROSCI.0683-16.2016
Fiorentini C, Gardoni F, Spano P, Di Luca M, Missale C (2003) Regulation of dopamine D1 receptor trafficking and desensitization by oligomerization with glutamate N-methyl-d-aspartate receptors. J Biol Chem 278:20196–20202
Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C (2008) Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol 74:59–69
Fiorentini C, Mattanza C, Collo G, Savoia P, Spano P, Missale C (2011) The tyrosine phosphatase Shp-2 interacts with the dopamine D(1) receptor and triggers D(1)-mediated Erk signaling in striatal neurons. J Neurochem 117:253–263. https://doi.org/10.1111/j.1471-4159.2011.07196.x
Fiorentini C, Savoia P, Savoldi D, Barbon A, Missale C (2013) Persistent activation of the D1R/Shp-2/Erk1/2 pathway in l-DOPA-induced dyskinesia in the 6-hydroxy-dopamine rat model of Parkinson’s disease. Neurobiol Dis 54:339–348. https://doi.org/10.1016/j.nbd.2013.01.005
Fiorentini C, Savoia P, Savoldi D, Bono F, Busi C, Barbon A, Missale C (2016) Shp-2 knockdown prevents l-dopa-induced dyskinesia in a rat model of Parkinson’s disease. Mov Disord 31:512–520. https://doi.org/10.1002/mds.26581
Fisone G (2010) Monitoring dyskinesia with Zif. Exp Neurol 226:11–14. https://doi.org/10.1016/j.expneurol.20
Gehani SS, Agrawal-Singh S, Dietrich N, Christophersen NS, Helin K, Hansen K (2010) Polycomb group protein displacement and gene activation through MSK-dependent H3K27me3S28 phosphorylation. Mol Cell 39:886–900. https://doi.org/10.1016/j.molcel.20
Gerfen CR (1992) The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci 15:285–320. https://doi.org/10.1146/annurev.ne.15.030192.001441
Gerfen CR, Miyachi S, Paletzki R, Brown P (2002) D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci 22:5042–5054
Gerfen CR, Paletzki R, Worley P (2008) Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J Neurosci 28:7113–7120. https://doi.org/10.1523/jneurosci.3952-07.2008
Ghosh AK, Khan S, Marini F, Nelson JA, Farquhar D (2000) A daunorubicin beta-galactoside prodrug for use in conjunction with gene-directed enzyme prodrug therapy. Tetrahedron Lett 41:4871–4874. https://doi.org/10.1016/S0040-4039(00)00742-5
Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, Sonenberg N (2001) Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev 15:2852–2864. https://doi.org/10.1101/gad.912401
Glatt CE, Snyder SH (1993) Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature 361:536–538
Gold SJ, Hoang CV, Potts BW, Porras G, Pioli E, Kim KW, Nadjar A, Qin C, LaHoste GJ, Li Q, Bioulac BH, Waugh JL, Gurevich E, Neve RL, Bezard E (2007) RGS9-2 negatively modulates l-3,4-dihydroxyphenylalanine-induced dyskinesia in experimental Parkinson’s disease. J Neurosci 27:14338–14348. https://doi.org/10.1523/jneurosci.4223-07.2007
Gray JM, Kim TK, West AE, Nord AS, Markenscoff-Papadimitriou E, Lomvardas S (2015) Genomic views of transcriptional enhancers: essential determinants of cellular identity and activity-dependent responses in the CNS. J Neurosci 35:13819–13826. https://doi.org/10.1523/JNEUROSCI.2622-15.2015
Green TA, Alibhai IN, Unterberg S, Neve RL, Ghose S, Tamminga CA, Nestler EJ (2008) Induction of activating transcription factors (ATFs) ATF2, ATF3, and ATF4 in the nucleus accumbens and their regulation of emotional behavior. J Neurosci 28:2025–2032. https://doi.org/10.1523/JNEUROSCI.5273-07.2008
Greengard P (2001) The neurobiology of slow synaptic transmission. Science 294:1024–1030
Guigoni C, Doudnikoff E, Li Q, Bloch B, Bezard E (2007) Altered D(1) dopamine receptor trafficking in parkinsonian and dyskinetic non-human primates. Neurobiol Dis 26:452–463. https://doi.org/10.1016/j.nbd.2007.02.001
Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P (2001) BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature 411:86–89
Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA (2000) Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci 20:3993–4001
Halder R, Hennion M, Vidal RO, Shomroni O, Rahman RU, Rajput A, Centeno TP, van Bebber F, Capece V, Garcia Vizcaino JC, Schuetz AL, Burkhardt S, Benito E, Navarro Sala M, Javan SB, Haass C, Schmid B, Fischer A, Bonn S (2016) DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci 19:102–110. https://doi.org/10.1038/nn.4194
Heiman M, Heilbut A, Francardo V, Kulicke R, Fenster RJ, Kolaczyk ED, Mesirov JP, Surmeier DJ, Cenci MA, Greengard P (2014) Molecular adaptations of striatal spiny projection neurons during levodopa-induced dyskinesia. Proc Natl Acad Sci USA 111:4578–4583. https://doi.org/10.1073/pnas.1401819111
Hepler JR, Gilman AG (1992) G proteins. Trends Biochem Sci 17:383–387
Herve D, Levi-Strauss M, Marey-Semper I, Verney C, Tassin JP, Glowinski J, Girault JA (1993) G(olf) and Gs in rat basal ganglia: possible involvement of G(olf) in the coupling of dopamine D1 receptor with adenylyl cyclase. J Neurosci 13:2237–2248
Herve D, Le Moine C, Corvol JC, Belluscio L, Ledent C, Fienberg AA, Jaber M, Studler JM, Girault JA (2001) Galpha(olf) levels are regulated by receptor usage and control dopamine and adenosine action in the striatum. J Neurosci 21:4390–4399
Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ (1994) Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron 13:1235–1244
Hurley MJ, Mash DC, Jenner P (2001) Dopamine D(1) receptor expression in human basal ganglia and changes in Parkinson’s disease. Brain Res Mol Brain Res 87:271–279
Jones MW, Errington ML, French PJ, Fine A, Bliss TV, Garel S, Charnay P, Bozon B, Laroche S, Davis S (2001) A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat Neurosci 4:289–296. https://doi.org/10.1038/85138
Joyce JN (1991) Differential response of striatal dopamine and muscarinic cholinergic receptor subtypes to the loss of dopamine. I. Effects of intranigral or intracerebroventricular 6-hydroxydopamine lesions of the mesostriatal dopamine system. Exp Neurol 113:261–276
Ko WK, Martin-Negrier ML, Bezard E, Crossman AR, Ravenscroft P (2014) RGS4 is involved in the generation of abnormal involuntary movements in the unilateral 6-OHDA-lesioned rat model of Parkinson’s disease. Neurobiol Dis 70:138–148. https://doi.org/10.1016/j.nbd.2014.06.013
Konradi C, Westin JE, Carta M, Eaton ME, Kuter K, Dekundy A, Lundblad M, Cenci MA (2004) Transcriptome analysis in a rat model of l-DOPA-induced dyskinesia. Neurobiol Dis 17:219–236. https://doi.org/10.1016/j.nbd.2004.07.005
Kumar R, Riddle L, Griffin SA, Grundt P, Newman AH, Luedtke RR (2009) Evaluation of the D3 dopamine receptor selective antagonist PG01037 on l-dopa-dependent abnormal involuntary movements in rats. Neuropharmacology 56:944–955. https://doi.org/10.1016/j.neuropharm.2009.01.020
LaPlant Q, Vialou V, Covington HE 3rd, Dumitriu D, Feng J, Warren BL, Maze I, Dietz DM, Watts EL, Iniguez SD, Koo JW, Mouzon E, Renthal W, Hollis F, Wang H, Noonan MA, Ren Y, Eisch AJ, Bolanos CA, Kabbaj M, Xiao G, Neve RL, Hurd YL, Oosting RS, Fan G, Morrison JH, Nestler EJ (2010) Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci 13:1137–1143. https://doi.org/10.1038/nn.2619
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. https://doi.org/10.1016/j.cell.2012.03.017
Lebel M, Chagniel L, Bureau G, Cyr M (2010) Striatal inhibition of PKA prevents levodopa-induced behavioural and molecular changes in the hemiparkinsonian rat. Neurobiol Dis 38:59–67. https://doi.org/10.1016/j.nbd.2009.12.027
Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD (2006) Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem 281:15763–15773. https://doi.org/10.1074/jbc.M511767200
Li L, Carter J, Gao X, Whitehead J, Tourtellotte WG (2005a) The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol Cell Biol 25:10286–10300. https://doi.org/10.1128/mcb.25.23.10286-10300.2005
Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ (2005b) The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 15:1961–1967. https://doi.org/10.1016/j.cub.2005.09.043
Li L, Homan KT, Vishnivetskiy SA, Manglik A, Tesmer JJ, Gurevich VV, Gurevich EV (2015) G protein-coupled receptor kinases of the GRK4 protein subfamily phosphorylate inactive G protein-coupled receptors (GPCRs). J Biol Chem 290:10775–10790. https://doi.org/10.1074/jbc.M115.644773
Lim SA, Xia R, Ding Y, Won L, Ray WJ, Hitchcock SA, McGehee DS, Kang UJ (2015) Enhanced histamine H2 excitation of striatal cholinergic interneurons in l-DOPA-induced dyskinesia. Neurobiol Dis 76:67–76. https://doi.org/10.1016/j.nbd.2015.01.003
Lindgren HS, Rylander D, Iderberg H, Andersson M, O’Sullivan SS, Williams DR, Lees AJ, Cenci MA (2011) Putaminal upregulation of FosB/DeltaFosB-like immunoreactivity in Parkinson’s disease patients with dyskinesia. J Parkinsons Dis 1:347–357
Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J (2005) Rheb binds and regulates the mTOR kinase. Curr Biol 15:702–713. https://doi.org/10.1016/j.cub.2005.02.053
Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121:179–193. https://doi.org/10.1016/j.cell.2005.02.031
Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, Greenberg ME (2014) Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci 17:1330–1339. https://doi.org/10.1038/nn.3808
Mandal R, Becker S, Strebhardt K (2016) Stamping out RAF and MEK1/2 to inhibit the ERK1/2 pathway: an emerging threat to anticancer therapy. Oncogene 35:2547–2561. https://doi.org/10.1038/onc.2015.329
Marcellino D, Ferre S, Casado V, Cortes A, Le Foll B, Mazzola C, Drago F, Saur O, Stark H, Soriano A, Barnes C, Goldberg SR, Lluis C, Fuxe K, Franco R (2008) Identification of dopamine D1–D3 receptor heteromers: indications for a role of synergistic D1–D3 receptor interactions in the striatum. J Biol Chem 283:26016–26025. https://doi.org/10.1074/jbc.m710349200
Marcotte ER, Sullivan RM, Mishra RK (1994) Striatal G-proteins: effects of unilateral 6-hydroxydopamine lesions. Neurosci Lett 169:195–198
Marshall JF, Navarrete R, Joyce JN (1989) Decreased striatal D1 binding density following mesotelencephalic 6-hydroxydopamine injections: an autoradiographic analysis. Brain Res 493:247–257
Mela F, Millan MJ, Brocco M, Morari M (2010) The selective D(3) receptor antagonist, S33084, improves parkinsonian-like motor dysfunction but does not affect l-DOPA-induced dyskinesia in 6-hydroxydopamine hemi-lesioned rats. Neuropharmacology 58:528–536. https://doi.org/10.1016/j.neuropharm.2009.08.017
Miller CA, Campbell SL, Sweatt JD (2008) DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol Learn Mem 89:599–603. https://doi.org/10.1016/j.nlm.2007.07.016
Mons N, Cooper DM (1994) Selective expression of one Ca(2+)-inhibitable adenylyl cyclase in dopaminergically innervated rat brain regions. Brain Res Mol Brain Res 22:236–244
Morigaki R, Okita S, Goto S (2017) Dopamine-induced changes in Galphaolf protein levels in striatonigral and striatopallidal medium spiny neurons underlie the genesis of l-DOPA-induced dyskinesia in parkinsonian mice. Front Cell Neurosci 11:26. https://doi.org/10.3389/fncel.2017.00026
Nash JE, Johnston TH, Collingridge GL, Garner CC, Brotchie JM (2005) Subcellular redistribution of the synapse-associated proteins PSD-95 and SAP97 in animal models of Parkinson’s disease and l-DOPA-induced dyskinesia. FASEB J 19:583–585. https://doi.org/10.1096/fj.04-1854fje
Nestler EJ, Barrot M, Self DW (2001) DeltaFosB: a sustained molecular switch for addiction. Proc Natl Acad Sci USA 98:11042–11046. https://doi.org/10.1073/pnas.191352698
Park HY, Kang YM, Kang Y, Park TS, Ryu YK, Hwang JH, Kim YH, Chung BH, Nam KH, Kim MR, Lee CH, Han PL, Kim KS (2014) Inhibition of adenylyl cyclase type 5 prevents l-DOPA-induced dyskinesia in an animal model of Parkinson’s disease. J Neurosci 34:11744–11753. https://doi.org/10.1523/JNEUROSCI.0864-14.2014
Park HY, Ryu YK, Kim YH, Park TS, Go J, Hwang JH, Choi DH, Rhee M, Lee CH, Kim KS (2016) Gadd45beta ameliorates l-DOPA-induced dyskinesia in a Parkinson’s disease mouse model. Neurobiol Dis 89:169–179. https://doi.org/10.1016/j.nbd.2016.02.013
Paul S, Nairn AC, Wang P, Lombroso PJ (2003) NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat Neurosci 6:34–42
Pavon N, Martin AB, Mendialdua A, Moratalla R (2006) ERK phosphorylation and FosB expression are associated with l-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry 59:64–74
Peebles CL, Yoo J, Thwin MT, Palop JJ, Noebels JL, Finkbeiner S (2010) Arc regulates spine morphology and maintains network stability in vivo. Proc Natl Acad Sci USA 107:18173–18178. https://doi.org/10.1073/pnas.1006546107
Penit-Soria J, Durand C, Besson MJ, Herve D (1997) Levels of stimulatory G protein are increased in the rat striatum after neonatal lesion of dopamine neurons. NeuroReport 8:829–833
Picconi B, Centonze D, Hakansson K, Bernardi G, Greengard P, Fisone G, Cenci MA, Calabresi P (2003) Loss of bidirectional striatal synaptic plasticity in l-DOPA-induced dyskinesia. Nat Neurosci 6:501–506
Pimoule C, Schoemaker H, Reynolds GP, Langer SZ (1985) [3H]SCH 23390 labeled D1 dopamine receptors are unchanged in schizophrenia and Parkinson’s disease. Eur J Pharmacol 114:235–237
Plath N, Ohana O, Dammermann B, Errington ML, Schmitz D, Gross C, Mao X, Engelsberg A, Mahlke C, Welzl H, Kobalz U, Stawrakakis A, Fernandez E, Waltereit R, Bick-Sander A, Therstappen E, Cooke SF, Blanquet V, Wurst W, Salmen B, Bosl MR, Lipp HP, Grant SG, Bliss TV, Wolfer DP, Kuhl D (2006) Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52:437–444. https://doi.org/10.1016/j.neuron.2006.08.024
Porras G, Berthet A, Dehay B, Li Q, Ladepeche L, Normand E, Dovero S, Martinez A, Doudnikoff E, Martin-Negrier ML, Chuan Q, Bloch B, Choquet D, Boue-Grabot E, Groc L, Bezard E (2012) PSD-95 expression controls l-DOPA dyskinesia through dopamine D1 receptor trafficking. J Clin Investig 122:3977–3989. https://doi.org/10.1172/JCI59426
Price DL, Rockenstein E, Ubhi K, Phung V, MacLean-Lewis N, Askay D, Cartier A, Spencer B, Patrick C, Desplats P, Ellisman MH, Masliah E (2010) Alterations in mGluR5 expression and signaling in Lewy body disease and in transgenic models of alpha-synucleinopathy—implications for excitotoxicity. PLoS One 5:e14020. https://doi.org/10.1371/journal.pone.0014020
Rahman Z, Gold SJ, Potenza MN, Cowan CW, Ni YG, He W, Wensel TG, Nestler EJ (1999) Cloning and characterization of RGS9-2: a striatal-enriched alternatively spliced product of the RGS9 gene. J Neurosci 19:2016–2026
Rahman Z, Schwarz J, Gold SJ, Zachariou V, Wein MN, Choi KH, Kovoor A, Chen CK, DiLeone RJ, Schwarz SC, Selley DE, Sim-Selley LJ, Barrot M, Luedtke RR, Self D, Neve RL, Lester HA, Simon MI, Nestler EJ (2003) RGS9 modulates dopamine signaling in the basal ganglia. Neuron 38:941–952
Ramamoorthi K, Fropf R, Belfort GM, Fitzmaurice HL, McKinney RM, Neve RL, Otto T, Lin Y (2011) Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science 334:1669–1675. https://doi.org/10.1126/science.1208049
Rangel-Barajas C, Silva I, Lopez-Santiago LM, Aceves J, Erlij D, Floran B (2011) l-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol Dis 41:51–61. https://doi.org/10.1016/j.nbd.20
Ross EM, Wilkie TM (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem 69:795–827. https://doi.org/10.1146/annurev.biochem.69.1.795
Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 101:13489–13494. https://doi.org/10.1073/pnas.0405659101
Ruiz-DeDiego I, Mellstrom B, Vallejo M, Naranjo JR, Moratalla R (2015a) Activation of DREAM (downstream regulatory element antagonistic modulator), a calcium-binding protein, reduces l-DOPA-induced dyskinesias in mice. Biol Psychiatry 77:95–105. https://doi.org/10.1016/j.biopsych.2014.03.023
Ruiz-DeDiego I, Naranjo JR, Herve D, Moratalla R (2015b) Dopaminergic regulation of olfactory type G-protein alpha subunit expression in the striatum. Mov Disord 30:1039–1049. https://doi.org/10.1002/mds.26197
Ruvinsky I, Meyuhas O (2006) Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem Sci 31:342–348
Rylander D, Recchia A, Mela F, Dekundy A, Danysz W, Cenci MA (2009) Pharmacological modulation of glutamate transmission in a rat model of l-DOPA-induced dyskinesia: effects on motor behavior and striatal nuclear signaling. J Pharmacol Exp Ther 330:227–235. https://doi.org/10.1124/jpet.108.150425
Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Herve D, Greengard P, Fisone G (2007) Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in l-DOPA-induced dyskinesia. J Neurosci 27:6995–7005. https://doi.org/10.1523/jneurosci.0852-07.2007
Santini E, Alcacer C, Cacciatore S, Heiman M, Herve D, Greengard P, Girault JA, Valjent E, Fisone G (2009a) l-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem 108:621–633. https://doi.org/10.1111/j.1471-4159.2008.05831.x
Santini E, Heiman M, Greengard P, Valjent E, Fisone G (2009b) Inhibition of mTOR signaling in Parkinson’s disease prevents l-DOPA-induced dyskinesia. Sci Signal 2:ra36. https://doi.org/10.1126/scisignal.2000308
Santini E, Sgambato-Faure V, Li Q, Savasta M, Dovero S, Fisone G, Bezard E (2010) Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in l-DOPA-induced dyskinesia. PLoS One 5:e12322. https://doi.org/10.1371/journal.pone.0012322
Santini E, Feyder M, Gangarossa G, Bateup HS, Greengard P, Fisone G (2012) Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism. J Biol Chem 287:27806–27812. https://doi.org/10.1074/jbc.m112.388413
Savasta M, Dubois A, Benavides J, Scatton B (1988) Different plasticity changes in D1 and D2 receptors in rat striatal subregions following impairment of dopaminergic transmission. Neurosci Lett 85:119–124
Schiffmann SN, Fisone G, Moresco R, Cunha RA, Ferre S (2007) Adenosine A2A receptors and basal ganglia physiology. Prog Neurobiol 83:277–292. https://doi.org/10.1016/j.pneurobio.2007.05.001
Schuster S, Nadjar A, Guo JT, Li Q, Ittrich C, Hengerer B, Bezard E (2008) The 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor lovastatin reduces severity of l-DOPA-induced abnormal involuntary movements in experimental Parkinson’s disease. J Neurosci 28:4311–4316
Sgambato-Faure V, Buggia V, Gilbert F, Levesque D, Benabid AL, Berger F (2005) Coordinated and spatial upregulation of arc in striatonigral neurons correlates with l-dopa-induced behavioral sensitization in dyskinetic rats. J Neuropathol Exp Neurol 64:936–947
Shinotoh H, Inoue O, Hirayama K, Aotsuka A, Asahina M, Suhara T, Yamazaki T, Tateno Y (1993) Dopamine D1 receptors in Parkinson’s disease and striatonigral degeneration: a positron emission tomography study. J Neurol Neurosurg Psychiatry 56:467–472
Sodersten E, Feyder M, Lerdrup M, Gomes AL, Kryh H, Spigolon G, Caboche J, Fisone G, Hansen K (2014) Dopamine signaling leads to loss of Polycomb repression and aberrant gene activation in experimental Parkinsonism. PLoS Genet 10:e1004574. https://doi.org/10.1371/journal.pgen.1004574
Spigolon G, Cavaccini A, Trusel M, Tonini R, Fisone G (2018) cJun N-terminal kinase (JNK) mediates cortico-striatal signaling in a model of Parkinson’s disease. Neurobiol Dis 110:37–46. https://doi.org/10.1016/j.nbd.2017.10.015
Stoof JC, Kebabian JW (1981) Opposing roles for D-1 and D-2 dopamine receptors in efflux of cyclic AMP from rat neostriatum. Nature 294:366–368
Suarez LM, Solis O, Carames JM, Taravini IR, Solis JM, Murer MG, Moratalla R (2014) l-DOPA treatment selectively restores spine density in dopamine receptor D2-expressing projection neurons in dyskinetic mice. Biol Psychiatry 75:711–722. https://doi.org/10.1016/j.biopsych.2013.05.006
Suarez LM, Solis O, Aguado C, Lujan R, Moratalla R (2016) l-DOPA oppositely regulates synaptic strength and spine morphology in D1 and D2 striatal projection neurons in dyskinesia. Cereb Cortex 26:4253–4264. https://doi.org/10.1093/cercor/bhw263
Subramaniam S, Napolitano F, Mealer RG, Kim S, Errico F, Barrow R, Shahani N, Tyagi R, Snyder SH, Usiello A (2011) Rhes, a striatal-enriched small G protein, mediates mTOR signaling and l-DOPA-induced dyskinesia. Nat Neurosci 15:191–193. https://doi.org/10.1038/nn.2994
Taymans JM, Kia HK, Claes R, Cruz C, Leysen J, Langlois X (2004) Dopamine receptor-mediated regulation of RGS2 and RGS4 mRNA differentially depends on ascending dopamine projections and time. Eur J Neurosci 19:2249–2260. https://doi.org/10.1111/j.0953-816X.2004.03336.x
Tekumalla PK, Calon F, Rahman Z, Birdi S, Rajput AH, Hornykiewicz O, Di Paolo T, Bedard PJ, Nestler EJ (2001) Elevated levels of DeltaFosB and RGS9 in striatum in Parkinson’s disease. Biol Psychiatry 50:813–816
Thiele SL, Chen B, Lo C, Gertler TS, Warre R, Surmeier JD, Brotchie JM, Nash JE (2014) Selective loss of bi-directional synaptic plasticity in the direct and indirect striatal output pathways accompanies generation of parkinsonism and l-DOPA induced dyskinesia in mouse models. Neurobiol Dis 71:334–344. https://doi.org/10.1016/j.nbd.2014.08.006
Thomas G, Siegmann M, Gordon J (1979) Multiple phosphorylation of ribosomal protein S6 during transition of quiescent 3T3 cells into early G1, and cellular compartmentalization of the phosphate donor. Proc Natl Acad Sci USA 76:3952–3956
Toki S, Kawasaki H, Tashiro N, Housman DE, Graybiel AM (2001) Guanine nucleotide exchange factors CalDAG-GEFI and CalDAG-GEFII are colocalized in striatal projection neurons. J Comp Neurol 437:398–407
Treisman R (1996) Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol 8:205–215
Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD (2007) Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci 27:5885–5894
Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, Bezard E, Caron MG (2015) Targeting beta-arrestin2 in the treatment of l-DOPA-induced dyskinesia in Parkinson’s disease. Proc Natl Acad Sci USA 112:E2517–E2526. https://doi.org/10.1073/pnas.1502740112
Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol JC, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Herve D, Girault JA (2005) Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. Proc Natl Acad Sci USA 102:491–496
Valjent E, Aubier B, Corbille AG, Brami-Cherrier K, Caboche J, Topilko P, Girault JA, Herve D (2006) Plasticity-associated gene Krox24/Zif268 is required for long-lasting behavioral effects of cocaine. J Neurosci 26:4956–4960
Visanji NP, Fox SH, Johnston T, Reyes G, Millan MJ, Brotchie JM (2009) Dopamine D3 receptor stimulation underlies the development of l-DOPA-induced dyskinesia in animal models of Parkinson’s disease. Neurobiol Dis 35:184–192. https://doi.org/10.1016/j.nbd.2008.11.010
Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA (2007) Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of l-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry 62:800–810
Whitmarsh AJ (2007) Regulation of gene transcription by mitogen-activated protein kinase signaling pathways. Biochim Biophys Acta 1773:1285–1298. https://doi.org/10.1016/j.bbamcr.2006.11.011
Won L, Ding Y, Singh P, Kang UJ (2014) Striatal cholinergic cell ablation attenuates l-DOPA induced dyskinesia in Parkinsonian mice. J Neurosci 34:3090–3094. https://doi.org/10.1523/JNEUROSCI.2888-13.2014
Xie J, Wang X, Proud CG (2016) mTOR inhibitors in cancer therapy. F1000Research 5:2078. https://doi.org/10.12688/f1000research.9207.1
Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O’Brien RJ, Worley P (2003) Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron 39:513–528
Yang W, Klaman LD, Chen B, Araki T, Harada H, Thomas SM, George EL, Neel BG (2006) An Shp2/SFK/Ras/Erk signaling pathway controls trophoblast stem cell survival. Dev Cell 10:317–327. https://doi.org/10.1016/j.devcel.2006.01.002
Zhang J, Xu TX, Hallett PJ, Watanabe M, Grant SG, Isacson O, Yao WD (2009) PSD-95 uncouples dopamine-glutamate interaction in the D1/PSD-95/NMDA receptor complex. J Neurosci 29:2948–2960. https://doi.org/10.1523/JNEUROSCI.4424-08.2009
Zhuang X, Belluscio L, Hen R (2000) G(olf)alpha mediates dopamine D1 receptor signaling. J Neurosci 20:RC91
Acknowledgements
GF is supported by the Swedish Research Council, Parkinson Foundation in Sweden and Swedish Brain Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Spigolon, G., Fisone, G. Signal transduction in l-DOPA-induced dyskinesia: from receptor sensitization to abnormal gene expression. J Neural Transm 125, 1171–1186 (2018). https://doi.org/10.1007/s00702-018-1847-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-018-1847-7