
Abstract
Purpose
Spinal abnormalities frequently occur in patients with mucopolysaccharidosis (MPS) types I, II, IV, and VI. The symptoms are manifold, which sometimes prolongs the diagnostic process and delays therapy. Spinal stenosis (SS) with spinal cord compression due to bone malformations and an accumulation of storage material in soft tissue are serious complications of MPS disease. Data on optimal perioperative therapeutic care of SS is limited.
Methods
A retrospective chart analysis of patients with MPS and SS for the time period 01/1998 to 03/2021 was performed. Demographics, clinical data, neurological status, diagnostic evaluations (radiography, MRI, electrophysiology), and treatment modalities were documented. The extent of the SS and spinal canal diameter were analyzed. A Cox regression analysis was performed to identify prognostic factors for neurological outcomes.
Results
Out of 209 MPS patients, 15 were included in this study. The most dominant type of MPS was I (–H) (n = 7; 46.7%). Preoperative neurological deterioration was the most frequent indication for further diagnostics (n = 12; 80%). The surgical procedure of choice was dorsal instrumentation with microsurgical decompression (n = 14; 93.3%). A univariate Cox regression analysis showed MPS type I (–H) to be associated with favorable neurological outcomes.
Conclusion
Early detection of spinal stenosis is highly relevant in patients with MPS. Detailed neurological assessment during follow-up is crucial for timeous detection of patients at risk. The surgical intervention of choice is dorsal instrumentation with microsurgical decompression and resection of thickened intraspinal tissue. Patients with MPS type I (–H) demonstrated the best neurological course.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mucopolysaccharidosis (MPS) is the generic term for a heterogenic group of autosomal recessive lysosomal disorders, mainly diagnosed in pediatric patients. Underlying pathophysiological mechanisms include malfunction of enzymes, which degrades cell products in the lysosomes, leading to an accumulation of glycosaminoglycans (GAGs) and subsequent breakdown of regular cellular functions. Today, seven types of MPS with further subgroups have been identified and classified according to the malfunctioning enzyme [1]. Linked phenotype characteristics can guide the diagnosis of MPS. Nevertheless, in nonspecific clinical presentations, diagnosis might be prolonged [2, 3]. However, prompt diagnosis and early treatment is the only way to prevent organ complications due to disease progression and to ensure stable clinical status and quality of life. Therapeutic approaches include hematopoietic stem cell transplantation in MPS I–H or enzyme replacement therapy in MPS I, II, IVA, VI, and VII [4,5,6]. Additional therapeutic approaches are mainly symptomatic therapies to address clinical symptoms.
In addition to frequent and severe cardiovascular and pulmonary complications, spinal abnormalities are also frequently diagnosed in MPS patients [2, 7, 8]. Clinical knowledge and data on some of these conditions have increased over recent years, including for the relevant, but up to now underestimated, spinal stenosis (SS) with secondary spinal cord compression (SCC) caused by GAG accumulation in the dura mater and surrounding spinal ligaments [9]. SS should be considered a progressive disease that may cause neurological impairment due to myelopathy. Careful neurological assessment, electrophysiological investigation, and spinal MR imaging are important methods to monitor these patients [10]. The therapeutic approach of choice is microsurgical decompression of the spinal canal [1, 8, 11]. Dorsal instrumentation is required if there is instability due to extensive stenosis. Hitherto, systematic data on clinical-neurological presentation and postoperative recovery are scarce and the optimal timing of surgical therapy is still controversial [11,12,13].
In this study, we analyzed our MPS cohort, focusing on patients with spinal stenosis and spinal cord compression to highlight this spinal disease. Patient charts were reviewed, with the focus on their preoperative neurological status, surgical therapy, and postoperative course, to gain insight into the optimal therapeutic regime.
Materials and methods
This retrospective study was conducted in accordance with the ethical guidelines of the Ethical Review Board in Hamburg, Germany (2021–300,037-WF) and the Declaration of Helsinki.
Patients
The patient’s records at the Center for Lysosomal Disorders for the time period from 01/1998 to 03/2021 were reviewed to identify patients with the diagnosis of MPS and SS who had surgical interventions. Data on demographics, MPS type, and additional health signs and symptoms were collected (Table 1). Data on initial neurological symptoms and preoperative neurological status were documented; motor functional impairment was classified as paresis; ataxia, fine motor impairment, and coordination and gait disorders were categorized as clinical myelopathy. If available, electrophysiological data were reviewed (Table 2). Surgical reports were screened for surgical technique and the interval between the onset of the symptoms and surgical intervention (Table 3).
Imaging studies
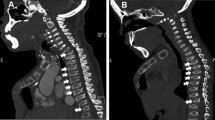
CT (computed tomography) or MRI (magnetic resonance imaging) images were analyzed using the Centricity Universal Viewer Zero Footprint Client® (GE Healthcare, Boston, United States of America). The localization and extent of the SS were determined on sagittal CT and MRI slides. T2-hyperintense intramedullary signals were regarded as radiological indicators of myelopathy (Fig. 1a, b). The spinal canal diameter was determined in axial CT and MRI slides, for both bony and soft tissue structures on pre- and postoperative studies, and measured in millimeters (mm).

Overview of pathogenesis of SS with SCC at the cervical spine. a Sagittal T2-weighted MRI scan with proof of high-grade SS and SCC, with corresponding spinal cord edema (C1–C3). b Corresponding axial T2-weighted MRI scan showing circular SCC by thickened soft tissue. c Exemplary presentation of the laminectomy corridor for microsurgical decompression (dashed line)
Follow-up regime
MPS patients are routinely followed up at least once every 12 months and assessed through a standardized protocol that includes electrocardiogram (ECG); echocardiography (echo); pulmonary function diagnostics; MR imaging and ultrasonography of the abdomen; audiometry; eye examination; X-ray examination of the hip and spine; whole-spine and cranial MR imaging; electrophysiological assessment, including sensory and motor evoked potentials (SEPs and MEPS); nerve conduction velocity (EMG); and neurocognitive examination. If emerging clinical symptoms were detected, patients were referred to the appropriate medical specialists for examination, and, if the disease course was stable, the follow-up intervals were extended.
Surgical procedure
The surgical procedure of choice is microsurgical decompression, often with dorsal instrumentation. Electrophysiological monitoring is done prior to positioning the patient. Intubation is regularly performed with a fiberoptic scope to avoid hyperextension of the neck. If the cervical spine is involved, patients are positioned prone with their heads fixed in the Mayfield skull clamp. The point of the skin incision is determined by X-ray fluoroscopy, and the muscles are then detached from the midline until the bilateral facet joints are exposed. Bleeding is controlled simultaneously to minimize blood loss. Pedicel screws are placed at the cervical and thoracic spine, using a spinal navigation system (Brainlab™, Munich, Germany). Pedicel screw placement at the lumbar spine is performed along the standardized trajectories. Once instrumentation is complete, decompression of the affected segments is performed, and, thereafter, thickened membranes of the dura mater and hypertrophic surrounding spine ligaments are resected. When apparent, meningeal injury with liquorrhea is controlled with microsurgical sutures. After extensive resection of the thickened ligaments and thickened outer dura sheets (Fig. 2c), the spinal dura mater begins to move again in synchrony with the pulse, which is a sign of decompression. Finally, the site is flushed with an antiseptic and sterile saline solution, two subfascial drains are placed, and the wound is closed. Patients are routinely transferred to an intensive care unit for postoperative monitoring.

Overview of surgical procedure in patients with spinal stenosis. a and b Postoperative lateral control X-ray after microsurgical decompression and dorsal instrumentation C2–C6 with pedicle screws. c Resected thickened ligaments and thickened outer dura sheets after surgery
Statistical analysis
Data are displayed as mean ± standard deviation (SD) for continuous variables or absolute and relative numbers for categorical variables. Differences in continuous variables were analyzed with the Mann–Whitney U test, and differences in proportions were analyzed with the chi-square test or Fisher’s exact test. A two-sided p-value less than 0.05 was considered statistically significant. To detect independent risk factors for unfavorable clinical courses, a univariable and multivariable Cox regression analysis was performed. All analyses were performed with SPSS Inc. (Chicago, IL, USA).
Results
Patient cohort
During the period from 01/1998 to 03/2021, 209 patients with the diagnosis of mucopolysaccharidosis were treated in our Center for Lysosomal Disorders. A total of 24 patients with SS were identified. Of these, 18 patients underwent surgical intervention and were assigned for further investigation. All the required information was available for 15 of these patients, and they were included in the study, resulting in a 7.2% overall incidence of SS in our cohort. The most dominant type of MPS was I (–H), with seven patients affected (46.7%). Most patients were diagnosed with MPS within the first 5 years of life (n = 12; 80%). Three patients were diagnosed in adulthood (#1: 17 years, #2: 28 years, and #3: 45 years), which increased the mean overall age of diagnosis to 8.5 ± 11.2 years. Meantime of documented follow-up was 145.8 ± 77 months. All the patients had MPS-linked co-morbidities, of which cardiovascular and orthopedic (excluding spine pathologies) complications were the most common (Table 1).
Neurological assessment
Neurological deterioration preoperatively was the most frequent reason for further diagnostics (n = 12; 80%). Motor deficits affected both the upper and lower extremities of eight patients (53.3%). Preoperative electrophysiological assessment revealed pathological impairment of evoked potentials (EPs) of the upper and lower extremities in eight cases (53.3%), and one patient was diagnosed with electrophysiological impairment of the upper extremities only (6.7%). Clinical myelopathy was evident in nine (60%) cases. During follow-up, five patients had persistent combined motor deficits (33.3%) with accompanying pathological impairment of the EPs (33.3%). Clinical myelopathy improved in four patients (Table 2).
Spine pathologies, surgical procedure, and radiographic findings
The most frequent location of the SS was the upper cervical spine (n = 8; 53.3%), followed by the thoracolumbar spine (n = 3; 20%). The time interval between the onset of symptoms and surgical intervention was 2.43 ± 1.8 months. No statistically significant difference was detected in patients with or without postoperative neurological improvement (p = 0.238). The extent of spinal stenosis was multisegmental, with at least three affected spinal segments in the majority of the patients (n = 14; 93.3%). The surgical procedure of choice in 14 cases was microsurgical decompression with dorsal instrumentation (93.3%) (Fig. 2a, b). The mean preoperative spinal canal diameter measured for bony structures was 11.8 ± 1.7 mm, and it increased to 15.3 ± 1.9 mm (p < 0.05) post-surgery. Consistent mean spinal canal diameter for soft tissue structures expanded from 6.8 ± 1 mm to 10.8 ± 2.2 mm (p < 0.05), as measured in postoperative radiographic controls (Table 3).
Factors associated with spinal cord compression and persisting neurological impairment
In the univariate Cox regression analysis, a diagnosis of MPS type I (–H) (OR: 10; 95% CI: 0.78–128.8; P = 0.047) was associated with favorable neurological outcomes after surgical intervention for SS with secondary SCC.
Histopathological findings
Histology revealed degenerative changes in the ligamentum flavum, with chondroid metaplasia and diffuse, infiltrates of macrophages (Fig. 3). The macrophages were highlighted by the immunohistochemical marker CD68, a member of the lysosomal-associated membrane glycoprotein family (Fig. 4). The pathological findings were consistent with GAG-containing macrophages in MPS type IV.

Ligamentum flavum with chondroid metaplasia (white asterisks) and diffuse infiltrates of macrophages (black asterisks), hematoxylin-eosin staining

Immunohistochemistry for CD68 stain macrophages
Discussion
The main findings of our monocentric and retrospective study are as follows: (1) Spinal stenosis is a frequent spine pathology in patients with MPS and has an impact on short- and long-term neurological outcomes. (2) Most patients diagnosed with spinal stenosis had MPS type I (–H). (3) The univariate Cox regression analysis showed that MPS type I (–H) is an independent factor associated with a favorable neurological outcome.
MPS is an autosomal recessive lysosomal disorder, based on enzymatic malfunction that leads to the accumulation of cell products in lysosomes [2]. MPS is predominantly diagnosed in early childhood, due to an impressive clinical phenotype or because of organ system problems that lead to the initial medical admission [2]. It is important to diagnose patients early to enable early treatment and include them in a systematic follow-up [3, 14, 15]. Skeletal abnormalities, including spinal pathologies, are one of the most frequent co-morbidities in MPS patients, with differing occurrences depending on the type of MPS [16]. Knowledge of and guidance for spine pathologies, such as craniocervical instability or thoracolumbar scoliosis, has improved in recent years [7], but there is still little known about SS with secondary SCC, which is why we focused on this condition [16]. Our overall MPS cohort (n = 209) had a 7.2% (n = 15) incidence of SS. It has been reported in the literature that spinal stenosis occurs predominantly in MPS types IV (–A) and IV, but our findings deviated from this, as type I (–H) predominated in our study. This can be explained by the fact that we, as a transplant center, increasingly treat these patients. There is limited evidence in the literature on MPS type I (–H) and spinal disorders, although Schmidt et al. reported on these disorders and found them to be a relevant problem in MPS type I (–H) patients, which aligns with our findings [9].
Neurological deterioration was a frequent precursor of diagnosis of SS in our cohort, which is why it is important to investigate neurological deterioration, even if other somatic manifestations are more likely to cause clinical deterioration [10]. In line with the complexity of the initial SS diagnosis, regular follow-up is crucial. Current literature reports that detailed and repetitive neurological examinations provide the best evidence to detect neurological deterioration [10]. If neurological abnormalities are diagnosed, further diagnostic evaluation is required, including an MRI and electrophysiological studies [10], and the latter studies are important for diagnosis, severity classification, and follow-up. For example, in a study of MPS type IV–A, Horovitz et al. reported that pathological changes are detected with electrophysiology before changes can be detected with an MRI, and the importance of early detection cannot be overemphasized [17]. The work-up required to diagnose MPS (sub)-type IV (–A) and VI is well-described in the literature by Alden et al. [10], but procedures to refer any MPS patient with suspicion of neurological deterioration are emphasized [11, 17]. In our department, we follow the reported diagnostic protocol, including all the above-mentioned tools. However, 12 patients in our cohort presented with neurological symptoms prior to being diagnosed with SS, which again emphasizes the need for accurate diagnostic follow-up.
Once neurological deterioration has been detected and the diagnosis of SS is confirmed, an optimal treatment concept is required. In our department, surgical intervention is indicated in the event of (1) clinical neurological deterioration or (2) if repetitive electrophysiological abnormalities become evident, even if the patient is clinically asymptomatic. We also recommend an intervention when progressive SS is diagnosed in an MRI, even if there is no functional impairment. This approach is consistent with current recommendations in the literature [10, 17, 18]. There is consensus that prompt surgical intervention is critical to prevent further neurologic deterioration or to reduce symptoms, but there is currently no standardized treatment approach [10, 11]. Most authors recommend dorsal instrumentation with microsurgical decompression as the preferred treatment, which also is the most frequently used approach in our department [10, 17]. Because SS is usually multisegmental, it is necessary to plan the dorsal instrumentation to allow for adequate decompression while still maintaining spinal stability [10]. Based on the pathophysiology of SS with the thickened ligaments and dura sheets and encased cerebral spinal fluid (CSF), only an additive dorsal resection or longitudinal splitting of the thickened structures leads to the relief of the spinal cord (Fig. 2c). To minimize the risk for intraoperative neurological deterioration, there is continuous electrophysiological monitoring while surgery is performed, which is routine in our department.
The ultimate goal of the surgical intervention in these patients is to reverse neurological impairment or to prevent further progression. Six of the patients with preoperative motor deficits and/or neurophysiological abnormalities did profit from the surgery and showed functional improvement at follow-up. It is also important to note that no patient exhibited neurological deterioration associated with the surgery; therefore, we conclude that, despite the invasiveness of surgery, this therapeutic approach is safe if performed by experienced surgeons.
Of the patients who did not profit from surgery, three had been diagnosed with MPS type VI, two with type IV (–A), and one with type I (–H). These results are consistent with the literature, where an unfavorable clinical course is described for MPS IV (–A) and VI [10]. A further univariate Cox regression analysis identified MPS type I (–H) as an independent factor associated with a favorable neurological course after surgery.
Finally, there are some limitations to this study. We present a monocentric and retrospective study with a small number of cases due to the rarity of the underlying disease. Furthermore, we studied a heterogeneous cohort, consisting of children and adults, which means that there are individual differences. However, a strength of this study is that it is the first to systematically investigate the outcome of spinal stenosis in MPS patients.
Conclusion
Spinal stenosis is a relevant concomitant disease in MPS patients. The treatment of choice is prompt surgical intervention in the form of microsurgical decompression with possible concomitant fusion. Regular follow-up examinations are essential to identify (at-risk) patients in a timely manner. Despite the rarity of this disease, every spinal surgeon should be familiar with it.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request (TP).
Code availability
Not applicable.
References
Peck SH, Casal ML, Malhotra NR, Ficicioglu C, Smith LJ (2016) Pathogenesis and treatment of spine disease in the mucopolysaccharidoses. Mol Genet Metab 118(4):232–243. https://doi.org/10.1016/j.ymgme.2016.06.002
Muenzer J (2011) Overview of the mucopolysaccharidoses. Rheumatology (Oxford) 50(Suppl 5):v4-12. https://doi.org/10.1093/rheumatology/ker394
Wood TC, Harvey K, Beck M, Burin MG, Chien YH, Church HJ, D’Almeida V, van Diggelen OP, Fietz M, Giugliani R, Harmatz P, Hawley SM, Hwu WL, Ketteridge D, Lukacs Z, Miller N, Pasquali M, Schenone A, Thompson JN, Tylee K, Yu C, Hendriksz CJ (2013) Diagnosing mucopolysaccharidosis IVA. J Inherit Metab Dis 36(2):293–307. https://doi.org/10.1007/s10545-013-9587-1
Schulze-Frenking G, Jones SA, Roberts J, Beck M, Wraith JE (2011) Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis 34(1):203–208. https://doi.org/10.1007/s10545-010-9215-2
Valayannopoulos V, Wijburg FA (2011) Therapy for the mucopolysaccharidoses. Rheumatology (Oxford) 50(Suppl 5):v49-59. https://doi.org/10.1093/rheumatology/ker396
Noh H, Lee JI (2014) Current and potential therapeutic strategies for mucopolysaccharidoses. J Clin Pharm Ther 39(3):215–224. https://doi.org/10.1111/jcpt.12136
Crostelli M, Mazza O, Mariani M, Mascello D, Iorio C (2019) Spine challenges in mucopolysaccharidosis. Int Orthop 43(1):159–167. https://doi.org/10.1007/s00264-018-4143-0
Terai H, Nakamura H (2020) Surgical management of spinal disorders in people with mucopolysaccharidoses. Int J Mol Sci. https://doi.org/10.3390/ijms21031171
Schmidt M, Breyer S, Lobel U, Yarar S, Stucker R, Ullrich K, Muller I, Muschol N (2016) Musculoskeletal manifestations in mucopolysaccharidosis type I (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J Rare Dis 11(1):93. https://doi.org/10.1186/s13023-016-0470-7
Alden TD, Amartino H, Dalla Corte A, Lampe C, Harmatz PR, Vedolin L (2017) Surgical management of neurological manifestations of mucopolysaccharidosis disorders. Mol Genet Metab 122S:41–48. https://doi.org/10.1016/j.ymgme.2017.09.011
Solanki GA, Alden TD, Burton BK, Giugliani R, Horovitz DD, Jones SA, Lampe C, Martin KW, Ryan ME, Schaefer MK, Siddiqui A, White KK, Harmatz P (2012) A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab 107(1–2):15–24. https://doi.org/10.1016/j.ymgme.2012.07.018
Mut M, Cila A, Varli K, Akalan N (2005) Multilevel myelopathy in Maroteaux-Lamy syndrome and review of the literature. Clin Neurol Neurosurg 107(3):230–235. https://doi.org/10.1016/j.clineuro.2004.05.003
Jurecka A, Opoka-Winiarska V, Jurkiewicz E, Marucha J, Tylki-Szymanska A (2012) Spinal cord compression in Maroteaux-Lamy syndrome: case report and review of the literature with effects of enzyme replacement therapy. Pediatr Neurosurg 48(3):191–198. https://doi.org/10.1159/000345635
Tylki-Szymanska A, Czartoryska B, Bunge S, van Diggelen OP, Kleijer WJ, Poorthuis BJ, Huijmans JG, Gorska D (1998) Clinical, biochemical and molecular findings in a two-generation Morquio A family. Clin Genet 53(5):369–374. https://doi.org/10.1111/j.1399-0004.1998.tb02747.x
Gosele S, Dithmar S, Holz FG, Volcker HE (2000) Late diagnosis of Morquio syndrome. Clinical histopathological findings in a rare mucopolysaccharidosis. Klin Monbl Augenheilkd 217(2):114–117. https://doi.org/10.1055/s-2000-10394
Baccouche H, Maunz M, Beck T, Fogarassy P, Beyer M (2009) Echocardiographic assessment and monitoring of the clinical course in a patient with Tako-Tsubo cardiomyopathy by a novel 3D-speckle-tracking-strain analysis. Eur J Echocardiogr 10(5):729–731. https://doi.org/10.1093/ejechocard/jep064
Horovitz DD, Magalhaes Tde S, Pena e Costa A, Carelli LE, Souza e Silva D, de Linhares e Riello AP, Llerena JC, Jr. (2011) Spinal cord compression in young children with type VI mucopolysaccharidosis. Mol Genet Metab 104(3):295–300. https://doi.org/10.1016/j.ymgme.2011.07.019
White KK, Sousa T (2013) Mucopolysaccharide disorders in orthopaedic surgery. J Am Acad Orthop Surg 21(1):12–22. https://doi.org/10.5435/JAAOS-21-01-12
Funding
Open Access funding enabled and organized by Projekt DEAL. Not applicable.
Author information
Authors and Affiliations
Contributions
T.P., N.M., and S.E. contributed to the design and implementation of the research. T.P. and M.L. performed the data collection and analysis of the results. T.P. and S.E. took the lead in writing the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript. S.E. supervised the project.
Corresponding author
Ethics declarations
Conflict of interest
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pantel, T., Lindschau, M., Luebke, A.M. et al. Spinal cord compression in patients with mucopolysaccharidosis. Eur Spine J 31, 1693–1699 (2022). https://doi.org/10.1007/s00586-022-07168-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00586-022-07168-0