
Abstract
Gastro-oesophageal reflux disease (GORD) is a chronic condition characterised by visceral pain in the distal oesophagus. The current first-line treatment for GORD is proton pump inhibitors (PPIs), however, PPIs are ineffective in a large cohort of patients and long-term use may have adverse effects. Emerging evidence suggests that nerve fibre number and location are likely to play interrelated roles in nociception in the oesophagus of GORD patients. Simultaneously, alterations in cells of the oesophageal mucosa, namely epithelial cells, mast cells, dendritic cells, and T lymphocytes, have been a focus of GORD research for several years. The oesophagus of GORD patients exhibits both macro- and micro-inflammation as a response to chronic acidic reflux at the epithelium. In other conditions of the GI tract, such as IBS and IBD, well-characterised bidirectional processes between immune cells and mucosal nerve fibres contribute to pathogenesis and symptom generation. Sensory alterations in these conditions such as nerve fibre outgrowth and hypersensitivity can be driven by inflammatory processes, which promote visceral pain signalling. This review will examine what is currently known of the molecular pathways linking inflammation and sensory perception leading to the development of GORD symptoms and explore potentially relevant mechanisms in other GI regions which may indicate new areas in GORD research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastro-oesophageal reflux disease (GORD) is a condition that affects approximately 20–25% of UK adults, and its prevalence continues to increase globally [1]. The primary symptom of GORD is heartburn, described as the sensation of pain or burning in the distal oesophagus or epigastric region [2]. Heartburn is a type of visceral pain, resulting from the activation of afferent sensory nerve fibres in the oesophageal mucosa. Activation of oesophageal sensory afferents is likely to result from several factors, including impaired barrier function, inflammation or microinflammation, and afferent sensitisation [3,4,5,6]. These factors indicate communication between noxious refluxate, epithelial cells, innate and adaptive immune systems, and mucosal sensory nerves.
GORD patients suffer chronic, long-term pain symptoms, which arises partly from the sensitisation of sensory nerves, both peripherally and centrally [7]. Innervation of the oesophagus is provided by spinal and vagal nerve branches that relay action potentials generated in afferent nerve fibres of the oesophageal mucosa to the central nervous system (CNS) [8,9,10]. In the vagal pathway, signals are transmitted to the nucleus tractus solitarius in the brainstem via nodose and jugular ganglia [8]. In the spinal pathway, afferent nerves synapse with cell bodies in the thoracic and cervical dorsal root ganglia (DRG) [8]. Sensory innervation to the oesophageal mucosa is provided by both vagal and spinal pathways, as shown by calcitonin gene-related peptide (CGRP) immunostaining in afferent fibres [11]. Spinal afferents are considered critical in nociception in response to acid reflux and sensitisation due to inflammation [10]. The molecular mechanisms underlying this sensitisation are beginning to be elucidated, with neuro-immune mechanisms likely to contribute significantly, similar to other visceral pain conditions [12,13,14,15].
In many peripheral organs such as the lungs, skin, and proximal and distal gastrointestinal (GI) tract, characterised neuroimmune interactions, communication between cells of the nervous and immune systems, are known to play a role in pain perception in various pathologies [12]. In the GI tract, these processes contribute to homeostatic and pathological functions [16,17,18]. In irritable bowel syndrome (IBS), a condition marked by visceral pain and microinflammation of the distal GI tract, several neuroimmune pathways are well defined, including sensitisation of nociceptive neurones by immune mediators such as histamine [13]. In inflammatory bowel disease (IBD), a chronic inflammatory condition of the large intestine, similar neuroimmune networks have been identified that promote visceral pain, as well as inflammation through interactions between neuropeptides and immune cells [14, 15]. Visceral pain signalling in the lower gut is mediated primarily via extrinsic networks, specifically sensory nociceptive neurones with cell bodies in the DRG innervating all layers of the lower GI tract [19]. Therefore, insights from lower GI tract pathologies are suggestive of potential oesophageal neuroimmune mechanisms relevant to GORD.
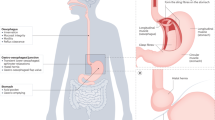
There is a wealth of evidence describing chronic inflammation in the oesophageal mucosa of GORD patients, regardless of macroscopic inflammation. Emerging evidence also points to the role of alterations in mucosal sensory innervation in GORD, specifically in driving heartburn symptoms. The aim of this review is to summarise current evidence of neuroimmune interactions in the oesophageal mucosa in GORD, as well as draw parallels with other GI mucosal barrier sites, both proximal and distal, which have established evidence of neuroimmune interactions in pathology. Possible neuroimmune interactions in GORD are summarised in Fig. 1.

Potential neuro-immune interactions in the oesophagus of GORD patients, resulting in pain and inflammation. a In the healthy oesophagus, microbial products, epithelial cells, and mucosal immune cells may act symbiotically to promote mucosal tolerance and barrier function. b In GORD oesophagus, bacterial dysbiosis may lead to immune cell activation and direct activation of sensory afferent fibres. High levels of reflux as well as increased epithelial permeability allow exposure of superficial sensory nerves, immune cells, and epithelial cells to acid and bile salts, resulting in the production of inflammatory mediators, as well as TRPV1 activation of CGRP + sensory nerves. Degranulating mast cells release histamine and prostaglandins, including prostaglandin D2 (PGD2) which may activate H1 receptor and Prostaglandin D2 receptor 1 (DP1) on sensory afferent nerve fibres, respectively. Substance P released from afferent fibres in response to neuronal activation may bind NK1R on mast cells, T lymphocytes, and epithelial cells to promote the expression of inflammatory genes. Created with BioRender.com
Neuroanatomy of the oesophagus
Both vagal and spinal afferents are likely to be important in pain signalling and extra-oesophageal symptoms such as anxiety and depression in GORD [20]. The anatomical location of these afferent fibres in the distal oesophagus, as well as their mucosal pattern could prescribe a role in the sensing of refluxed stomach contents.
Spinal nerves with ganglia in the spinal cord and which signal to the CNS, are implicated in causing, for example, heartburn [21]. Spinal afferent innervation of the oesophagus is provided via the DRG [8]. In the oesophagus of the cat and dog, retrograde tracing of DRG projections in the oesophagus has demonstrated spinal afferents localised at all levels of the oesophagus [22, 23]. CGRP has been used as a marker for spinal sensory afferents in the oesophagus due to its abundance in nerve fibres originating from the DRG and its paucity in vagal nerve fibres, as has been confirmed in mammals through combined retrograde labelling and immunohistochemistry [24, 25]. In the rat, CGRP + spinal nerve fibres are present throughout the length of the oesophagus, with many projections terminating in the mucosa [26]. In humans, CGRP + nerve fibres have been detected in all layers of the oesophagus, including the lamina propria and epithelium, throughout the length of the oesophagus [4, 5, 27]. These mucosal spinal afferents are likely involved in pain processing, and in the context of GORD, their proximity to the lumen could allow for the detection of noxious contents.
The oesophagus receives vagal innervation from C-fibres originating from nodose and jugular ganglia [28]. The vagus nerve contains sensory and motor nerve fibres with functions including gastro-oesophageal junction (GOJ) function and mechanosensation in the smooth muscle layer of the human oesophagus [29]. Calretinin, a neuronal calcium-binding protein, has been used as a putative immunohistochemical marker for vagal afferents in the oesophagus as it is present in retrograde labelled afferent neurones originating from the nodose and jugular ganglia but not the DRG [25]. Calretinin + nerve fibres have been found to be densely expressed in all layers of the upper and lower third of the rat oesophagus [25]. In this study, DRG neurones did not positively stain for calretinin, suggesting a high specificity for vagal neurones [25]. In mouse oesophagus, AAV-GFP labelling of vagal fibres from jugular/nodose ganglia revealed vagal innervation throughout the oesophageal mucosa [30]. In guinea pig, both jugular and nodose vagal afferents have been found throughout the length of the oesophagus, although their localisation within the mucosa has not been thoroughly investigated [28]. In humans, neurones containing neuropeptide Y (NPY) and vasoactive intestinal peptide (VIP), which are found in vagal nodose fibres, have been detected in the oesophageal mucosa [31,32,33]. These vagal afferents within the oesophageal mucosa have the potential for chemosensation or detection of inflammatory mediators due to their proximity to the epithelium.
Classically, vagal afferents have been thought to be involved in sensing physiological stimuli, However, evidence points towards the activation of peripheral afferent vagal fibres in the control of mood and pain modulation, as well as peripherally released neuropeptides from vagal afferents and efferents in immune homeostasis [34, 35]. Studies in guinea pig oesophagus show vagal afferents, specifically jugular C fibres, are activated by acid via TRPV1 and can also respond to pathological distension, suggesting a role in nociception [36]. Although a population of oesophageal vagal afferents have nociceptive properties, knowledge of contribution to pain perception is limited. Current evidence suggests vagal afferents may have a role in pain signal modulation [35], likely via projections from the nucleus tractus solitarius (NTS) which is involved in central pain processing [37]. The role of vagal activation in visceral pain is only partially understood, however, signalling through vagal afferents may also contribute to affective-emotional disorder in visceral pain conditions [20]. Vagal afferent activation also appears to produce an analgesic effect in some conditions. For example, high intensity electrical activation (≥ 150 μA) of capsaicin-sensitive vagal afferents in rats inhibits spinal nociception in response to thermal and mechanical stimuli at the skin, likely via descending vagal pathways [38].
Understanding the nociceptive capabilities of vagal afferents and alterations in GORD could reveal a role for this pathway in pain perception and affective symptoms. In an immunohistochemical study of the human oesophagus, percentage of papillae containing VIP + nerve fibres was increased in inflamed mucosa compared to healthy controls [32]. Further, in an ex vivo study, almost all guinea pig oesophageal vagal nodose neurones were found to be responsive to bradykinin via B2R [39]. Bradykinin is released by cell types including epithelial cells and immune cells in response to acid exposure [40, 41]. Therefore, some vagal afferents in the oesophageal mucosa have chemosensory ability and may be involved in pain modulation in response to noxious stimuli in GORD.
Neurochemistry of the oesophagus in GORD
CGRP
As described previously, CGRP is a key neuropeptide in nociception in the GI tract due to it being localised mainly to spinal nerve fibres originating from the DRG [19]. For this reason, CGRP is often used to characterise sensory neurones likely to be involved in pain perception. CGRP acts as a neurotransmitter in the DRG via a G-protein coupled receptor (GPCR) containing the protein subunit RAMP1 [42]. In rat oesophagus, CGRP is expressed in almost all neurones (99%) originating from levels C2-T12 of the DRG, but only a small proportion (2%) of vagal nodose and jugular neurones innervating the distal oesophagus [24, 26]. Therefore, neuronal CGRP reactivity in the distal oesophagus, where heartburn symptoms of GORD originate, is found predominantly in spinal afferent fibres. Activation of these spinal afferents by luminal contents such as refluxate or by inflammatory products results in the perception of pain, for example, oesophageal perfusion of pH ~ 1 pepsin solution in rats results in potent activation of CGRP + nerves in the DRG [11, 43]. In healthy human distal oesophageal mucosa, CGRP + nerve fibres are expressed in the lamina propria as well as some projections into the epithelium [11]. However, in the oesophageal mucosa of NERD patients, CGRP + nerve fibres are significantly more dense compared to healthy volunteers, and this CGRP + nerve fibre density was negatively correlated with oesophageal distention pain threshold [44]. However, in an immunohistochemical study of ERD oesophageal mucosa, no difference was found in the percentage of papillae which contained a CGRP + nerve fibre [32], a result that may be explained by the criteria used by this study that excluded the epithelium and subepithelial lamina propria. Increased density of CGRP + sensory nerve fibres may be important in heartburn symptoms specifically in NERD patients.
Notably, CGRP-containing nerve fibres lay closer to the luminal surface of the oesophageal epithelium in NERD patients compared to healthy controls [4]. This difference is not seen in functional heartburn (FH), ERD, or Barrett’s Oesophagus (BE) [4, 45]. In NERD oesophagus, a significantly greater proportion of epithelial CGRP + nerve fibres are positive for the proton receptor, TRPV1, compared to ERD patients [5]. Taken together, these CGRP + TRPV1 + nerve fibres are likely to be involved in sensing acidic refluxate and the perception of pain in GORD. NERD patients characteristically retain an intact epithelial barrier compared to ERD, and therefore the superficiality of sensory afferents could be critical in their sensing of noxious luminal contents compared to ERD, which is typified by epithelial erosions [46].
Given current evidence, it is unlikely that CGRP has direct effects on epithelial and immune cells in the oesophageal mucosa. However, it is probable that the alterations in the location and receptor expression of CGRP + nerve fibres in NERD augments nociception, likely partly via immune interactions. For example, NGF secreted by intestinal mast cells can contribute to nerve growth under stress conditions, resulting in hypersensitivity [47, 48]. Furthermore, increased sensitisation of TRPV1 on CGRP + neurones in GORD could potentially contribute to nociception. This may be attributed to inflammatory or bacterial products, including ATP, and TLR agonists such as HMGB1 and LPS, which has been demonstrated in DRG neurones in vitro and in vivo [49]. Endogenous mediators of TRPV1 sensitisation, such as ATP may contribute to neuronal hypersensitivity in GORD [40]. In a rat model of oesophagitis, mRNA expression of ATP receptor P2X3 in vagal and spinal afferents was higher compared to controls [50]. Further research is required to determine the expression of immune product receptors on sensory afferents in the human GORD oesophagus, as this could elucidate mechanisms of neuronal sensitisation leading to heartburn.
Substance P
Substance P (SP) is a neuropeptide contained in a subtype of nociceptive neurones and is found in both spinal and vagal nerves. In the oesophagus of mammals, including the opossum and the cat, SP plays a role in oesophageal peristalsis and the function of the lower oesophageal sphincter (LES), where it is co-released with acetylcholine (ACh) by vagal efferent nerves [24, 51, 52]. In pain signalling of the spinal pathway, SP released from afferent sensory nerve fibres binds preferentially to neurokinin 1 receptor (NK1R) within the dorsal horn [53]. In the opossum oesophagus, SP-containing nerve fibres are present in all layers of the oesophagus, including the mucosa [54, 55]. SP + nerve fibres are present in healthy human oesophageal mucosa [31] and the percentage of papillae containing SP + neurones have been observed to be unchanged between healthy controls and ERD patients [32]. However, the mucosal protein level of SP is elevated in NERD patients [56], with a negative correlation between SP + neurone density and oesophageal distention pain threshold [44].
More research is needed to determine alterations in SP innervation in GORD phenotypes, as this may contribute to visceral hypersensitivity. However, it’s possible that elevated levels of sensory SP + neurones play a role in pain perception in NERD and not ERD as SP-ergic innervation appears to be altered specifically in NERD oesophagus. As SP is found in both vagal and spinal fibres, activation of SP + afferents could contribute to visceral pain as well as anxiety/depression symptoms in GORD. An image of a CGRP + SP + (spinal) nerve fibre in the oesophageal mucosa is shown in Fig. 2.

CGRP + Substance P + nerve fibres in a papilla of a non-erosive reflux disease (NERD) patient. The basal layer of the epithelium is indicated with a ‘B’. The papilla is marked with an arrowhead. Scale bar = 20 µm
Neurogenic inflammation in GORD
Neurogenic inflammation is a conserved process, occurring in the skin, trachea, bladder, and GI tract [57, 58]. Sensory afferent nerve fibres at barrier sites participate in neurogenic inflammation which requires sensing of noxious components such as acid (via TRPV1), bacterial/viral components (via TLRs), or ATP released from damaged epithelial cells (via P2X receptors) [59]. Subsequent neuronal activation results in rapid local release of neuropeptides such as SP and CGRP from afferent endings which act on cell types including epithelial, endothelial, and leukocytes, invoking a rapid inflammatory response [19, 53, 59]. SP binding to NK1R on endothelial cells leads to reduced endothelial integrity and upregulation of leukocyte adhesion molecules, facilitating plasma and leukocyte extravasation [58, 60]. SP + neurone density is altered in gut pathologies including in IBS patients where density in colonic mucosa is threefold higher compared to healthy controls [61] with similar findings in IBS subtypes [62, 63]. High SP + nerve fibre density is also reported in inflammatory skin conditions prurigo nodularis, psoriasis, and atopic dermatitis [64].
There is emerging evidence of SP-mediated neurogenic inflammation in the oesophagus. An ex vivo study of cat oesophageal tissue showed pH2 incubation-induced neuronal SP release via TRPV1 [65]. Interestingly, NK1R mRNA has been reported to be increased in both NERD and ERD mucosa [56, 66]. This is consistent with inflammatory conditions of the skin with a neurogenic inflammatory component, such as chronic prurigo [67]
CGRP, a potent vasodilator at local blood vessels [68], promotes leukocyte infiltration to the injury site during acute inflammation but also acts as an anti-inflammatory mediator [14, 69]. In the skin, local release of CGRP upon neuronal activation contributes to oedema and leukocyte infiltration [70]. However, CGRP may also have an anti-inflammatory function in the skin and on endothelial cells by downregulation of NF-κB activation [71]. Indeed, CGRP knockout mice with DSS-induced colitis suffer worse symptoms driven by decreased activation of TGF-β-expressing CD4 + Tim4 + intestinal macrophages [72]. This data suggests CGRP may have an anti-inflammatory role at barrier sites, particularly during long-term high-grade inflammatory conditions such as IBD, but its function as a vasodilator facilitates acute inflammation. RAMP1 mRNA expression is low in the oesophageal mucosa of healthy individuals and is not altered in NERD, suggesting mucosal CGRP does not act locally [56].
Neuro-immune crosstalk in GORD
It is widely accepted that GORD is likely a condition driven by mucosal inflammation rather than direct acid damage [73]. Several cell types, comprising epithelial, immune, and neuronal cells are involved in the inflammatory pathogenesis of GORD. In vitro studies have indicated that the components of refluxate, acid and bile salts, induce the production of inflammatory mediators such as COX-2, IL-1β, and IL-8 in oesophageal epithelial cell lines [74, 75]. In patients with ERD and NERD, there is chronic upregulation of inflammatory cytokines including IL-1β, IL-8, IL-33, TNF-⍺, and IFN-γ [74, 76,77,78]. Among the immune cells implicated in GORD pathogenesis and likely to release the aforementioned cytokines, are mast cells, eosinophils, dendritic cells, and T/B lymphocytes.
Mast cells
Mast cells have been a focus of lower GI pathology research for many years, and have the most evidence supporting a neuro-immune function. In NERD patients, there are a significantly higher number of mast cells in oesophageal mucosa compared to healthy volunteers, as well as a greater proportion of degranulated mast cells [79]. Zhong et al. reported a greater number of intraepithelial mast cells in patients with reflux chest pain syndromes (RCS), which includes patients with retrosternal and epigastric pain as main symptoms, rather than typical reflux symptoms such as regurgitation [80]. Therefore, oesophageal mast cell degranulation may contribute to heartburn symptoms in GORD patients. However, it is necessary to be cautious when comparing immune cell counts between oesophageal biopsies due to inherent limitations. For example, as immune cells are present in greater numbers in the lamina propria compared to the epithelium, the proportion of the biopsy containing these two layers can drive alterations in mucosal immune cell counts if not controlled for. Other limitations include sloughing off of the most luminal epithelial layers during biopsy preparation, which can inflate cell counts. Finally, endoscopic oesophageal biopsies typically contain almost entirely epithelium which, among other factors, can make the biopsy difficult to orientate and papillae difficult to visualise when counting intrapapillary immune cells.
In IBS, a relationship between mast cells and sensory nerves has been described [81]. One study found the number of mast cells within 5 μm of a nerve fibre to be greatly increased in the mucosa of the descending colon in IBS patients compared to healthy controls, as well as a higher proportion of degranulated mast cells in the descending colon mucosa [82]. The number of mucosal mast cells within 5 μm of a nerve fibre, determined with electron microscopy, was correlated with the severity and frequency of abdominal pain [82]. In eosinophilic oesophagitis (EoE), a high density of mucosal mast cells within the oesophagus is associated with heartburn perception, and the mast cells are in close proximity to TRPV1 + nerve fibres [83]. Although, this pattern of proximity is not as clear as in the lower gut, as mast cells are not as close to nerve fibres.
Mast cell products can sensitise neurones within the GI tract, for example, in guinea pig oesophagus, mast cell-derived histamine and prostaglandin D2 have been found to sensitise vagal nodose C-fibres to distension via H1 and DP1 receptors, respectively [84, 85]. Activation of these receptors increases firing of guinea pig airway vagal afferents, partly due to sensitisation of TRPV1 via intracellular GPCR signalling cascades [86, 87]. In IBS, the degranulation state of mast cells, rather than just density, is important in symptom generation [88]. More research is required to investigate the degranulation states of mast cells in GORD and downstream impacts of mast cell products, including histamine and NGF, on mucosal nociceptors.
Mast cells may contribute to neuronal hypersensitivity in GORD, but neuropeptides released in neurogenic inflammation may also, in turn, promote mast cell degranulation, driving symptom generation. Cultured human mast cells express receptors for SP and VIP, and incubation of mast cells with SP or VIP can lead to degranulation [89]. The number of mast cells expressing VPAC-1, a VIP receptor, was found to be increased in EoE [90]. Precise interactions between oesophageal VIP + neurones and mast cells have not been elucidated but it is possible that increased vagal activation in response to reflux could promote mast cell degranulation. Research is required to determine the nature of neurone-mast cell signalling in GORD. A likely interaction, which requires further study, may be SP signalling via NK1R to promote mast cell degranulation, driving neuronal hypersensitivity and epithelial permeability. The possible functions of mast cells in GORD are summarised in Fig. 1.
Eosinophils
Eosinophils are also innate immune cells involved in inflammatory diseases of the GI tract. Epithelial eosinophils are very rare in healthy controls, as well as a very low mRNA level of eosinophil chemotaxins such as eotaxin1-3 and eosinophil maturation mediators such as IL-5 [91,92,93]. As the main diagnostic criterion for EoE is ≥ 15 eosinophils/ HPF, epithelial eosinophil infiltration is expectedly increased in these patients [94]. Epithelial mediators released during allergen challenge induce a type 2 inflammatory response, leading to the production of eosinophil chemotactic and maturation mediators including eotaxin1-3, which are upregulated in EoE [93]. Pathological functions of infiltrating eosinophils include barrier disruption by granule proteins such as major basic protein (MBP), mast cell recruitment and activation by IL-9, as well as tissue remodelling by IL-13 [95]. Eosinophils have also been shown to express neurotrophic factors such as NGF as well as various mediators that may promote neuronal activation such as MBP [96]. However, EoE therapeutics, including monoclonal antibodies, targeting eosinophils have been unsuccessful in improving clinical outcomes, suggesting that eosinophils may be symptomatic, rather than drivers, of disease progression in EoE [95, 96].
Most studies investigating epithelial eosinophil infiltration in GORD have described a low number of intraepithelial eosinophils, particularly in comparison with EoE [91, 92, 97,98,99]. However, there is a moderate increase in mucosal mRNA levels of eotaxin1-3 between healthy controls and GORD patients [93]. Also, one study applying a lower threshold of eosinophil infiltration (≥ 5 eosinophils/ HPF) found eosinophil infiltration in 26.1% of GORD patients with oesophagitis and 35.7% of GORD patients without oesophagitis studied, compared to 0% of healthy controls [92]. Eosinophil infiltration in the oesophagus may be related to dysphagia symptoms [100]. GORD patients listing dysphagia as their primary symptom display a significantly greater number of eosinophils/HPF compared to those listing dysphagia as a secondary or tertiary symptoms, as well as patients without dysphagia [100]. This could be related to fibrosis induced by eosinophil-derived mediators. Overall, there is disagreement surrounding the extent of eosinophil infiltration in GORD, which could be prescribed to different pathologists, counting, and biopsy collection methods as described earlier. Although eosinophils may be an important factor in some GORD symptoms such as dysphagia, they are unlikely to be an effective treatment target in this disorder.
Dendritic cells
Dendritic cells (DCs) are antigen-presenting cells (APCs) that link innate and adaptive immune systems. DCs in the oesophagus are not well characterised during homeostatic or pathological conditions. In Barrett’s oesophagus, a condition defined by intestinal metaplasia of the distal oesophagus due in part to chronic acid reflux, CD83 + DCs are present in the lamina propria and form clusters with T and B lymphocytes [101]. DCs of the human intestine are phenotyped based on expression of protein markers, primarily the presence of CD103 [102]. CD103 expression defines the conventional DC1 subset, which has a role in cross-presentation of exogenous antigens to lymphocytes [102]. Similar categorisations exist for DCs in blood as well as at other barrier sites such as the gastric mucosa and skin [103]. Although there is a limited characterisation of oesophageal DCs when compared to those of the intestine, there is evidence of a CD103 + CD11c + DC population in healthy oesophageal mucosa. However, most oesophageal DC research relates specifically to adenocarcinoma and tumour infiltrating DCs.
In contrast to the intestinal mucosa, it is known that Langerhans cells (LC), a subset of immature DCs that express CD1a, are commonly found in the oesophageal epithelium, in particular the suprabasal region [104, 105]. LCs are powerful antigen-presenting cells (APCs) and, therefore, play a role in maintaining antigenic tolerance or promoting immunity at barrier sites [106]. In the skin, LC function has been characterised. During steady state, when no exogenous antigen is detected, LCs promote the proliferation of CD4 + Tregs, dependent on contact-mediated interactions via MHC class II and CD80-86, as well as the release of the cytokines IL-2 and IL-15, leading to peripheral tolerance [106,107,108]. LCs also prime antigen-specific T-lymphocytes during an inflammatory response [109]. The microenvironment of the epidermis is critical in driving LC function. Indeed, TGF-β, a mediator released by keratinocytes during homeostatic conditions is important in inhibiting LC maturity and their retention in the epidermis [110]. Whereas, intradermal injection of TNF-α, which is released by epidermal keratinocytes during inflammation, promotes LC migration from the epidermis to the dermis [111]. LCs in the oesophagus are likely to respond in similar ways to the epithelial microenvironment, which may contribute to the inflammatory epithelial T cell response which is observed early in GORD onset.
Due to the paucity of studies regarding DC quantification and localisation in the oesophagus, it remains difficult to theorise potential neuroimmune interactions between DCs and afferent nerve fibres in the oesophagus. However, in the human colonic mucosa, CD103 + (“conventional DC1”) DCs were found to be in close apposition to CGRP + sensory nerve fibres [112]. No such relationship has been demonstrated in the oesophagus. In murine-derived DCs, NK1R agonism in vitro increases expression of co-stimulatory and activation markers CD80, CD83, CD86, CD40, and MHC class II [113]. NK1R agonism also inhibits DC production of IL-10, but not IL-1β, IL-6, or TNF-⍺. In vitro challenging of murine CD11c + DCs with SP provokes NF-κB activation [114]. Whereas, in vitro treatment of cultured LC-like cells with VIP reduces their capacity to present antigen to T cells whilst downregulating IL-12 and IL-1β production [115]. Therefore, there could be bi-directional communication via sensory pathways which may be altered in GORD.
Lymphocytes
T lymphocytes have a demonstrated role in the onset of GORD, and their activity is regulated by interactions with tissue DCs. In an oesophagoduodenostomy model of reflux oesophagitis, T-cell infiltration into the oesophageal mucosa is an initial event preceding the development of oesophagitis [116]. Similarly, in GORD patients, 1 week after discontinuing PPI treatment, intraepithelial lymphocyte infiltration is significantly increased [117]. A study by Osman et al. showed epithelial T lymphocyte counts are reduced following 1 month of PPI treatment [118]. Taken together, these studies suggest a rapid and reversible increase in mucosal lymphocyte infiltration in response to acid reflux. This finding has been consistently noted in cross-sectional studies, describing an T lymphocyte epithelial infiltration in GORD patients in various patient cohorts [80, 92, 119, 120]. An in vitro study by Huo et al. showed condition media from the oesophageal epithelial cell line NES-B10T exposed to acidic bile salt promoted T cell migration [75]. Therefore, soluble mediators released from epithelial cells in response to reflux may be responsible for driving T-cell infiltration into the epithelium. The exact function of these infiltrating T lymphocytes, however, as prescribed by the method of their activation and subsequent molecular expression, remains largely unknown.
The most comprehensive study of T cell subtypes in GORD investigated cell surface marker expression of ex vivo cultured T cells from healthy control, Barrett’s Oesophagus, and reflux oesophagitis patients [121]. T lymphocytes isolated from reflux oesophagitis patients displayed an inflammatory phenotype, with a high proportion of cytotoxic Granzyme B + cells [121]. Two cross-sectional studies found intraepithelial T lymphocytes to be predominantly CD8 + in the healthy and inflamed oesophagus [120, 122]. A study of 200 reflux oesophagitis patients found the ratio of CD4 + :CD8 + T cells to be approximately 2:1, which is within the healthy range for adults [118, 123]. However, the focus of this study on circulating T cells rather than T cells in the oesophageal mucosa severely limits its application in mucosal pathogenesis. Lastly, a study of paediatric patients found an increased proportion of Treg cells (FOXP3 +) in the oesophageal mucosa in EoE and GORD [119]. However, due to the young age of recruited patients, this may not be directly translatable to adult GORD, as paediatric and adult GORD can differ in clinical presentation, and T cell populations alter dramatically with age [124, 125]. Overall, T cells appear to be involved in the pathogenesis of oesophagitis in GORD. The inflammatory microenvironment of the GORD oesophagus may be responsible for promoting T cell maintenance and altered gene expression. Further characterisation of T cell populations in GORD is required, including NERD and FH, the pathogenesis of which is still unclear.
In vitro studies have established a role of SP in T cell activity. We have demonstrated the expression of NK1R on a population of T lymphocytes in the oesophageal mucosa of GORD patients, as shown in Fig. 3. NK1R expression on T cells can be induced by the mucosal microenvironment. For example, in a model of NSAID-induced colitis, NK1R expression was induced in T lymphocytes of IL-10 knockout animals [126]. Further in vitro experimentation revealed that Th1 cytokine IL-12 promoted and IL-10 inhibited NK1R expression in isolated mouse CD4 + T cells [126]. IL-12 also promoted NK1R expression in a human T cell line [127], possibly via an NF-κB-mediated pathway [128]. NK1R activation on human T lymphocytes induces expression of macrophage inflammatory protein-1β (MIP-1β), enhancing T cell chemotaxis [129]. Additionally, NK1R agonism may play a role in augmenting T cell cytokine production, as in T cells isolated from IL-10 knockout mice with NSAID-induced colitis, TGF-β and SP were required for upregulated production of IL-17 and IFN-γ [130]. Therefore, SP may specifically augment migration, activation, and subsequent cytokine release of mucosal T cells in GORD patients. NK1R expression on T cells appears highly dependent on the microenvironment and more research is required to determine how this may affect NK1R + T cells in GORD.

NK1R + T lymphocytes in the oesophageal epithelium of two non-erosive reflux disease (NERD) patients. A Interepithelial T lymphocytes positive for the substance P receptor, NK1R. B A T lymphocyte in an oesophageal papilla expressing NK1R. NK1R + T lymphocytes are marked with an arrowhead. Scale bar = 20 µm
Unlike T lymphocytes, B lymphocytes are not well researched in the oesophagus. In contrast to the intestinal wall, the oesophagus does not contain Peyer’s patches, organised lymphoid follicles containing clusters of T and B cells. However, B cells have been observed in the papillae and lamina propria, but not the epithelium, of the healthy oesophagus [131]. Epithelial and papillary CD20 + B cell infiltration is higher in EoE compared to healthy controls [131, 132]. However, EoE is an allergic-mediated disease and is associated with other Mendelian atopic conditions [133]. In EoE, exposure of the epithelium to allergens induces the release of Th2-priming mediators including IL-33 [133]. These Th2 cells then secrete IL-13, promoting B cell chemotaxis and proliferation [133]. Therefore, EoE pathogenesis is markedly different from GORD. Indeed, epithelial B lymphocyte infiltration during GORD pathogenesis has been shown to be minimal [117]. Although, one study has described a high number (700 ± 30/10 HPFs) of clustered B cells in the oesophageal mucosa of GORD patients which is markedly reduced (10 ± 2/10 HPFs) by 1 month of PPI treatment [118]. However, these figures and observations are outliers and can possibly be attributed to the aforementioned issues of cell counting from oesophageal biopsies, particularly the depth of the biopsies.
The oesophageal microbiome and its potential role in neuroimmune pathways
The gut microbiome has received increasing attention as a regulator of mucosal inflammation, permeability, and neuronal sensitivity [134, 135]. Throughout the intestine, commensal bacterial products interact directly with cells of the immune system, including DCs, T lymphocytes, and epithelial cells to promote tolerance and maintain epithelial function [134]. These interactions are facilitated by microbial products, including short-chain fatty acids (SCFAs) and polysaccharides, released by intestinal flora which resides in the mucus layer [134]. Antimicrobial peptides including β-defensins and secretory leukocyte protease inhibitor (SLPI) produced by epithelial cells limit host exposure to commensal bacteria [134]. The particular species which make up the microbiome vary greatly through the length of the GI tract, and depend on the luminal environment, including pH and nutrient levels, however, 90% of bacterial species in healthy human faecal samples are of Firmicutes and Bacteroidetes phyla [136, 137].
Disruption in the GI microbiome has been implicated in the pathogenesis and progression of several conditions including infection, metabolic syndromes, IBD, and IBS [134]. As well as its role in epithelial permeability and inflammation, accumulating evidence suggests that alterations in the microbiome is also involved in the onset of pain symptoms in GI conditions [135]. Mice treated with antibiotics (bacitracin/neomycin) for 10 days caused hypersensitivity to colorectal distension compared to placebo-treated mice [138], with a higher concentration of myeloperoxidase in tissue and higher SP innervation in the submucosa [138], both of which were normalised following co-administration of live Lactobacillus paracasei and its metabolic products [138]. In a study of zymosan-induced colitis in neonate rats, supplementation with Lactobacillus rhamnosus GG for 39 days rescued hypersensitivity to colonic distension [139], coinciding with altered levels of 5-HT, noradrenaline, and dopamine in various CNS regions [139]. This suggests the role of the microbiome in regulating hypersensitivity to painful stimuli, potentially involving central sensitisation. Commensal bacteria may also exert anti-nociceptive functions directly on sensory neurones, as Lactobacillus reuteri inhibited TRPV1-mediated activation of DRG neurones [140]. Other bacterial products such as LPS, β-glycan, or SCFAs may also bind directly to receptors on peripheral neurones, including TLRs and bile acid receptors, to induce peripheral sensitisation of sensory neurones [135]. Therefore, dysbiosis in the microbiome of the GI tract can result in bacteria acting directly, or indirectly via immune cells, on afferent neurones to induce or protect from hyperalgesia. Possible impacts of the oesophageal microbiome and bacterial dysbiosis in GORD pathogenesis are summarised in Fig. 1.
The oesophagus supports a distinct resident microbiome [141, 142], and lacks a permanent mucous layer, allowing bacteria to directly adhere to squamous cells [141, 143]. The majority of bacteria populating the distal oesophagus are of Firmicutes (70%) followed by Bacteroidetes (20%) phylum [141]. Similar bacterial proportions have been reported using 16S rRNA analysis of oesophageal biopsies as well as oesophageal secretions collected by a string test [144,145,146].
Several studies have found altered oesophageal microbiomes in oesophageal disease patients [146,147,148,149]. Unsupervised clustering analysis of 16S rDNA from distal oesophageal biopsies of healthy volunteers and ERD patients revealed two clusters based on the combined genetic distance of microbiome samples [148]. The ‘Type I’ microbiome contained predominantly Firmicutes phylum, specifically Streptococcus genus, with 11/12 healthy oesophageal samples belonging to this group [148]. However, 7/12 of oesophagitis samples had a ‘Type II’ microbiome, characterised by a lower proportion of Streptococcus genus and a higher proportion of gram-negative anaerobes/microaerophiles [148]. Bacterial load in the distal oesophagus is higher in GORD patients compared to healthy controls, with no change in number of taxa present (α-diversity) between groups [146]. GORD patients treated with PPIs had a lower proportion of Firmicutes and a greater proportion of Proteobacteria bacteria compared to healthy patients, perhaps as a result of low-acid reflux altering the oesophageal microenvironment [146], although paediatric and adult samples were analysed together; age is a key variable in oesophageal microbiome composition [146, 149]. In a study by Deshpande et al., α-diversity was unchanged between healthy controls and GORD patients [149]. However, gram-negative bacteria were present in greater numbers in GORD oesophagus and pathway analysis revealed differences in microbial pathways including lactic acid production, hexitol degradation, and heme biosynthesis [149]. Microbial dysbiosis could result in the production of products that act directly on afferent sensory nerves or alter the epithelial-immune homeostasis, resulting in the production of inflammatory mediators which sensitise or activate these nerves.
Conclusion
Neuroimmune pathways are key mediators of both physiological and pathological processes. Several cell types are involved in these processes, including spinal and vagal afferent neurones, epithelial cells, bacteria, and immune cells. Visceral pain and chronic inflammation are major symptoms of GORD, and there are similarities between the cell types involved in other GI conditions. Emerging evidence describes sensory changes in the oesophageal mucosa of GORD patients in the presence of macro- and micro-inflammation. As described in the lower gut and other organs, these sensory alterations are likely to be driven partly by inflammatory processes, which has subsequent effects promoting visceral pain. Investigation of these interactions in the oesophagus could elucidate pathways leading to heartburn, particularly in NERD and FH patients. Further research is required to determine the exact mechanisms which are relevant in the oesophagus and develop future treatments which meet the needs of PPI-refractory GORD patients.
References
El-Serag HB, Sweet S, Winchester CC, et al. Update on the epidemiology of gastro-oesophageal reflux disease: a systematic review. Gut. 2014;63:871–80.
Clarrett DM, Hachem C. Gastroesophageal reflux disease (GERD). Mo Med. 2018;115:214–8.
Weijenborg PW, Smout AJ, Verseijden C, et al. Hypersensitivity to acid is associated with impaired esophageal mucosal integrity in patients with gastroesophageal reflux disease with and without esophagitis. Am J Physiol Gastrointest Liver Physiol. 2014;307:G323–9.
Woodland P, Shen Ooi JL, Grassi F, et al. Superficial esophageal mucosal afferent nerves may contribute to reflux hypersensitivity in nonerosive reflux disease. Gastroenterology. 2017;153:1230–9.
Ustaoglu A, Sawada A, Lee C, et al. Heartburn sensation in nonerosive reflux disease: pattern of superficial sensory nerves expressing TRPV1 and epithelial cells expressing ASIC3 receptors. Am J Physiol Gastrointest Liver Physiol. 2021;320:G804–15.
Yoshida N. Inflammation and oxidative stress in gastroesophageal reflux disease. J Clin Biochem Nutr. 2007;40:13–23.
Hungin APS, Molloy-Bland M, Scarpignato C. Revisiting montreal: new insights into symptoms and their causes, and implications for the future of GERD. Am J Gastroenterol. 2019;114:414–21.
Kuo B, Urma D. Esophagus-anatomy and development. GI Motility online. 2006. https://doi.org/10.1038/gimo6.
Brierley SM, Hughes P, Harrington A, et al. Chapter 24—innervation of the gastrointestinal tract by spinal and vagal afferent nerves. In: Johnson LR, Ghishan FK, Kaunitz JD, et al., editors. Physiology of the gastrointestinal tract. 5th ed. Boston: Academic Press; 2012. p. 703–31.
Sengupta JN. Esophageal sensory physiology. GI Motil Online, 2006. https://doi.org/10.1038/gimo6.s
Ustaoglu A, Woodland P. Esophageal afferent innervation and its role in gastro-esophageal reflux disease symptoms. Curr Opin Gastroenterol. 2021;37:372–7.
Klein Wolterink RGJ, Wu GS, Chiu IM, et al. Neuroimmune interactions in peripheral organs. Annu Rev Neurosci. 2022;45:339–60.
Vanuytsel T, Bercik P, Boeckxstaens G. Understanding neuroimmune interactions in disorders of gut-brain interaction: from functional to immune-mediated disorders. Gut. 2023;72:787–98.
Khalil M, Zhang Z, Engel MA. Neuro-immune networks in gastrointestinal disorders. Visc Med. 2019;35:52–60.
Wang H, Foong JPP, Harris NL, et al. Enteric neuroimmune interactions coordinate intestinal responses in health and disease. Mucosal Immunol. 2022;15:27–39.
Populin L, Stebbing MJ, Furness JB. Neuronal regulation of the gut immune system and neuromodulation for treating inflammatory bowel disease. FASEB Bioadv. 2021;3:953–66.
Stakenborg N, Viola MF, Boeckxstaens GE. Intestinal neuro-immune interactions: focus on macrophages, mast cells and innate lymphoid cells. Curr Opin Neurobiol. 2020;62:68–75.
Jacobson A, Yang D, Vella M, et al. The intestinal neuro-immune axis: crosstalk between neurons, immune cells, and microbes. Mucosal Immunol. 2021;14:555–65.
Phillips RJ, Powley TL. Innervation of the gastrointestinal tract: patterns of aging. Auton Neurosci. 2007;136:1–19.
Bonaz B, Sinniger V, Pellissier S. Vagal tone: effects on sensitivity, motility, and inflammation. Neurogastroenterol Motil. 2016;28:455–62.
Sengupta JN. Visceral pain: the neurophysiological mechanism. Handb Exp Pharmacol. 2009;194:31–74.
Collman PI, Tremblay L, Diamant NE. The distribution of spinal and vagal sensory neurons that innervate the esophagus of the cat. Gastroenterology. 1992;103:817–22.
Khurana RK, Petras JM. Sensory innervation of the canine esophagus, stomach, and duodenum. Am J Anatomy. 1991;192:293–306.
Neuhuber WL, Raab M, Berthoud HR, et al. Innervation of the mammalian esophagus. In: Rallison S, editor., et al., Advances in anatomy embryology and cell biology. Heidelberg: Springer; 2006.
Dütsch M, Eichhorn U, Wörl J, et al. Vagal and spinal afferent innervation of the rat esophagus: a combined retrograde tracing and immunocytochemical study with special emphasis on calcium-binding proteins. J Comp Neurol. 1998;398:289–307.
Wank M, Neuhuber WL. Local differences in vagal afferent innervation of the rat esophagus are reflected by neurochemical differences at the level of the sensory ganglia and by different brainstem projections. J Comp Neurol. 2001;435:41–59.
Singaram C, Sengupta A, Sweet MA, et al. Nitrinergic and peptidergic innervation of the human oesophagus. Gut. 1994;35:1690.
Yu M, Chang C, Undem BJ, et al. Capsaicin-sensitive vagal afferent nerve-mediated interoceptive signals in the esophagus. Molecules. 2021;26:3929.
Wang YB, de Lartigue G, Page AJ. Dissecting the role of subtypes of gastrointestinal vagal afferents. Front Physiol. 2020;11:643.
Harsanyiova J, Ru F, Zatko T, et al. Vagus nerves provide a robust afferent innervation of the mucosa throughout the body of the esophagus in the mouse. Dysphagia. 2020;35:471–8.
Wattchow DA, Furness JB, Costa M, et al. Distributions of neuropeptides in the human esophagus. Gastroenterology. 1987;93:1363–71.
Newton M, Kamm MA, Soediono PO, et al. Oesophageal epithelial innervation in health and reflux oesophagitis. Gut. 1999;44:317–22.
Zhuo H, Ichikawa H, Helke CJ. Neurochemistry of the nodose ganglioN. Prog Neurobiol. 1997;52:79–107.
Breit S, Kupferberg A, Rogler G, et al. Vagus nerve as modulator of the brain-gut axis in psychiatric and inflammatory disorders. Front Psychiatry. 2018;9:44.
Gottfried-Blackmore A, Habtezion A, Nguyen L. Noninvasive vagal nerve stimulation for gastroenterology pain disorders. Pain Manag. 2020;11:89–96.
Yu X, Hu Y, Yu S. Effects of acid on vagal nociceptive afferent subtypes in guinea pig esophagus. Am J Physiol Gastrointest Liver Physiol. 2014;307:G471–8.
Page AJ, Li H. Meal-sensing signaling pathways in functional dyspepsia. Front Syst Neurosci. 2018;12:10.
Ren K, Zhuo M, Randich A, et al. Vagal afferent stimulation-produced effects on nociception in capsaicin-treated rats. J Neurophysiol. 1993;69:1530–40.
Yu S, Ouyang A. TRPA1 in bradykinin-induced mechanical hypersensitivity of vagal C fibers in guinea pig esophagus. Am J Physiol Gastrointest Liver Physiol. 2009;296:G255–65.
Morales-Lazaro SL, Simon SA, Rosenbaum T. The role of endogenous molecules in modulating pain through transient receptor potential vanilloid 1 (TRPV1). J Physiol. 2013;591:3109–21.
Ma J, Altomare A, Rieder F, et al. ATP: a mediator for HCl-induced TRPV1 activation in esophageal mucosa. Am J Physiol Gastrointest Liver Physiol. 2011;301:G1075–82.
Cottrell GS, Roosterman D, Marvizon J-C, et al. Localization of calcitonin receptor-like receptor and receptor activity modifying protein 1 in enteric neurons, dorsal root ganglia, and the spinal cord of the rat. J Comp Neurol. 2005;490:239–55.
Harrington AM, Brierley SM, Isaacs NJ, et al. Identifying spinal sensory pathways activated by noxious esophageal acid. Neurogastroenterol Motil. 2013;25:e660–8.
Xu X, Li Z, Zou D, et al. High expression of calcitonin gene-related peptide and substance P in esophageal mucosa of patients with non-erosive reflux disease. Dig Dis Sci. 2013;58:53–60.
Nikaki K, Woodland P, Lee C, et al. Esophageal mucosal innervation in functional heartburn: closer to healthy asymptomatic subjects than to non-erosive reflux disease patients. Neurogastroenterol Motil. 2019;31: e13667.
Woodland P, Al-Zinaty M, Yazaki E, et al. In vivo evaluation of acid-induced changes in oesophageal mucosa integrity and sensitivity in non-erosive reflux disease. Gut. 2013;62:1256–61.
Zhang L, Song J, Bai T, et al. Sustained pain hypersensitivity in the stressed colon: role of mast cell-derived nerve growth factor-mediated enteric synaptic plasticity. Neurogastroenterol Motil. 2018;30: e13430.
Barreau F, Cartier C, Ferrier L, et al. Nerve growth factor mediates alterations of colonic sensitivity and mucosal barrier induced by neonatal stress in rats. Gastroenterology. 2004;127:524–34.
Wu Y, Wang Y, Wang J, et al. TLR4 mediates upregulation and sensitization of TRPV1 in primary afferent neurons in 2,4,6-trinitrobenzene sulfate-induced colitis. Mol Pain. 2019;15:1744806919830018.
Banerjee B, Medda BK, Schmidt J, et al. Altered expression of P2X3 in vagal and spinal afferents following esophagitis in rats. Histochem Cell Biol. 2009;132:585–97.
Reynolds JC, Ouyang A, Cohen S. A lower esophageal sphincter reflex involving substance P. Am J Physiol Gastrointest Liver Physiol. 1984;246:G346–54.
Mukhopadhyay AK. Effect of substance P on the lower esophageal sphincter of the opossum. Gastroenterology. 1978;75:278–82.
Chang CT, Jiang BY, Chen CC. Ion channels involved in substance P-mediated nociception and antinociception. Int J Mol Sci. 2019;20:1596.
Singaram C, Sengupta A, Spechler SJ, et al. Mucosal peptidergic innervation of the opossum esophagus and anal canal: a comparison with snout skin. J Auton Nerv Syst. 1990;29:231–40.
Christensen J, Williams TH, Jew J, et al. Distribution of immunoreactive substance P in opossum esophagus. Dig Dis Sci. 1989;34:513–20.
Yoshida N, Kuroda M, Suzuki T, et al. Role of nociceptors/neuropeptides in the pathogenesis of visceral hypersensitivity of nonerosive reflux disease. Dig Dis Sci. 2013;58:2237–43.
Figini M, Emanueli C, Grady EF, et al. Substance P and bradykinin stimulate plasma extravasation in the mouse gastrointestinal tract and pancreas. Am J Physiol Gastrointest Liver Physiol. 1997;272:G785–93.
Gonzalez HL, Carmichael N, Dostrovsky JO, et al. Evaluation of the time course of plasma extravasation in the skin by digital image analysis. J Pain. 2005;6:681–8.
Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. 2012;15:1063–7.
Miyazaki Y, Satoh T, Nishioka K, et al. STAT-6-mediated control of P-selectin by substance P and interleukin-4 in human dermal endothelial cells. Am J Pathol. 2006;169:697–707.
Akbar A, Yiangou Y, Facer P, et al. Increased capsaicin receptor TRPV1-expressing sensory fibres in irritable bowel syndrome and their correlation with abdominal pain. Gut. 2008;57:923.
Sohn W, Lee OY, Lee SP, et al. Mast cell number, substance P and vasoactive intestinal peptide in irritable bowel syndrome with diarrhea. Scand J Gastroenterol. 2013;49:43–51.
Keszthelyi D, Troost FJ, Jonkers DM, et al. Alterations in mucosal neuropeptides in patients with irritable bowel syndrome and ulcerative colitis in remission: a role in pain symptom generation? Eur J Pain. 2013;17:1299–306.
Ständer S, Yosipovitch G. Substance P and neurokinin 1 receptor are new targets for the treatment of chronic pruritus. Br J Dermatol. 2019;181:932–8.
Harnett KM, Rieder F, Behar J, et al. Viewpoints on Acid-induced inflammatory mediators in esophageal mucosa. J Neurogastroenterol Motil. 2010;16:374–88.
Kim JJ, Kim N, Park JH, et al. Comparison of tight junction protein-related gene mRNA expression levels between male and female gastroesophageal reflux disease patients. Gut and Liver. 2018;12:411–9.
Ohanyan T, Schoepke N, Eirefelt S, et al. Role of substance P and its receptor neurokinin 1 in chronic prurigo: a randomized, proof-of-concept, controlled trial with topical aprepitant. Acta Derm Venereol. 2018;98:26–31.
Brain SD, Tippins JR, Morris HR, et al. Potent vasodilator activity of calcitonin gene-related peptide in human skin. J Investig Dermatol. 1986;87:533–6.
Ray JC, Allen P, Bacsi A, et al. Inflammatory complications of CGRP monoclonal antibodies: a case series. J Headache Pain. 2021;22:121.
Zarban AA, Chaudhry H, de Sousa VJ, et al. Elucidating the ability of CGRP to modulate microvascular events in mouse skin. Int J Mol Sci. 2022;23:12246.
Granstein RD, Wagner JA, Stohl LL, et al. Calcitonin gene-related peptide: key regulator of cutaneous immunity. Acta Physiol (Oxf). 2015;213:586–94.
Wu W, Feng B, Liu J, et al. The CGRP/macrophage axis signal facilitates inflammation recovery in the intestine. Clin Immunol. 2022;245: 109154.
Souza RF, Bayeh L, Spechler SJ, et al. A new paradigm for GERD pathogenesis. Not acid injury, but cytokine-mediated inflammation driven by HIF-2alpha: a potential role for targeting HIF-2alpha to prevent and treat reflux esophagitis. Curr Opin Pharmacol. 2017;37:93–9.
Ma J, Altomare A, Guarino M, et al. HCl-induced and ATP-dependent upregulation of TRPV1 receptor expression and cytokine production by human esophageal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2012;303:G635–45.
Huo X, Agoston AT, Dunbar KB, et al. Hypoxia-inducible factor-2alpha plays a role in mediating oesophagitis in GORD. Gut. 2017;66:1542–54.
Zavala-Solares MR, Fonseca-Camarillo G, Valdovinos M, et al. Gene expression profiling of inflammatory cytokines in esophageal biopsies of different phenotypes of gastroesophageal reflux disease: a cross-sectional study. BMC Gastroenterol. 2021;21:201.
Monkemuller K, Wex T, Kuester D, et al. Interleukin-1beta and interleukin-8 expression correlate with the histomorphological changes in esophageal mucosa of patients with erosive and non-erosive reflux disease. Digestion. 2009;79:186–95.
Sei H, Oshima T, Shan J, et al. Esophageal epithelial-derived IL-33 Is upregulated in patients with heartburn. PLoS ONE. 2016;11: e0154234.
Yu Y, Ding X, Wang Q, et al. Alterations of mast cells in the esophageal mucosa of the patients with non-erosive reflux disease. Gastroenterology Res. 2011;4:70–5.
Zhong C, Liu K, Wang K, et al. Developing a diagnostic understanding of GERD phenotypes through the analysis of levels of mucosal injury, immune activation, and psychological comorbidity. Dis Esophagus. 2018;31:doy039.
Hasler WL, Grabauskas G, Singh P, et al. Mast cell mediation of visceral sensation and permeability in irritable bowel syndrome. Neurogastroenterol Motil. 2022;34: e14339.
Barbara G, Stanghellini V, De Giorgio R, et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702.
Zhang S, Shoda T, Aceves SS, et al. Mast cell-pain connection in eosinophilic esophagitis. Allergy. 2022;77:1895–9.
Yu S, Kollarik M, Ouyang A, et al. Mast cell-mediated long-lasting increases in excitability of vagal C fibers in guinea pig esophagus. Am J Physiol Gastrointest Liver Physiol. 2007;293:G850–6.
Zhang S, Grabauskas G, Wu X, et al. Role of prostaglandin D2 in mast cell activation-induced sensitization of esophageal vagal afferents. Am J Physiol Gastrointest Liver Physiol. 2013;304:G908–16.
Maher SA, Birrell MA, Adcock JJ, et al. Prostaglandin D2 and the role of the DP1, DP2 and TP receptors in the control of airway reflex events. Eur Respir J. 2015;45:1108–18.
Fabisiak A, Wlodarczyk J, Fabisiak N, et al. Targeting histamine receptors in irritable bowel syndrome: a critical appraisal. J Neurogastroenterol Motil. 2017;23:341–8.
Dothel G, Barbaro MR, Boudin H, et al. Nerve fiber outgrowth is increased in the intestinal mucosa of patients with irritable bowel syndrome. Gastroenterology. 2015;148(1002–11): e4.
Kulka M, Sheen CH, Tancowny BP, et al. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123:398–410.
Kanamori A, Tanaka F, Takashima S, et al. Esophageal mast cells may be associated with the perception of symptoms in patients with eosinophilic esophagitis. Esophagus. 2023;20:333–41.
Jamali E, Kazeminegad B, Ahadi M, et al. Quantity and distribution of eosinophils in esophageal specimens of adults: an Iranian population-based study. Iran J Pathol. 2022;17:136–42.
Zand Irani M, Talley NJ, Ronkainen J, et al. Neutrophils, eosinophils, and intraepithelial lymphocytes in the squamous esophagus in subjects with and without gastroesophageal reflux symptoms. Hum Pathol. 2021;115:112–22.
Bhattacharya B, Carlsten J, Sabo E, et al. Increased expression of eotaxin-3 distinguishes between eosinophilic esophagitis and gastroesophageal reflux disease. Hum Pathol. 2007;38:1744–53.
Gonsalves NP, Aceves SS. Diagnosis and treatment of eosinophilic esophagitis. J Allergy Clin Immunol. 2020;145:1–7.
Doyle AD, Masuda MY, Kita H, et al. Eosinophils in eosinophilic esophagitis: the road to fibrostenosis is paved with good intentions. Front Immunol. 2020;11: 603295.
Salvo-Romero E, Rodino-Janeiro BK, Albert-Bayo M, et al. Eosinophils in the gastrointestinal tract: key contributors to neuro-immune crosstalk and potential implications in disorders of brain-gut interaction. Cells. 2022;11:1644.
Wong S, Tippett M, Zobel J, et al. Distal esophageal wall thickness correlates with dysphagia in adult patients with eosinophilic esophagitis. Esophagus. 2022;19:554–9.
Basseri B, Levy M, Wang HL, et al. Redefining the role of lymphocytes in gastroesophageal reflux disease and eosinophilic esophagitis. Dis Esophagus. 2010;23:368–76.
Mueller S, Neureiter D, Aigner T, et al. Comparison of histological parameters for the diagnosis of eosinophilic oesophagitis versus gastro-oesophageal reflux disease on oesophageal biopsy material. Histopathology. 2008;53:676–84.
Ayazi S, Hagen JA, Chandrasoma P, et al. Esophageal intraepithelial eosinophils in dysphagic patients with gastroesophageal reflux disease. Dig Dis Sci. 2009;55:967–72.
Bobryshev YV, Tran D, Killingsworth MC, et al. Dendritic cells in Barrett’s esophagus and esophageal adenocarcinoma. J Gastrointest Surg. 2009;13:44–53.
Stagg AJ. Intestinal dendritic cells in health and gut inflammation. Front Immunol. 2018;9:2883.
Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154:3–20.
Lucendo AJ, Navarro M, Comas C, et al. Immunophenotypic characterization and quantification of the epithelial inflammatory infiltrate in eosinophilic esophagitis through stereology: an analysis of the cellular mechanisms of the disease and the immunologic capacity of the esophagus. The Am J Surg Pathol. 2007;31:598–606.
Terris B, Potet F. Structure and role of langerhans’ cells in the human oesophageal epithelium. Digestion. 1995;56:9–14.
Yan B, Liu N, Li J, et al. The role of Langerhans cells in epidermal homeostasis and pathogenesis of psoriasis. J Cell Mol Med. 2020;24:11646–55.
Idoyaga J, Fiorese C, Zbytnuik L, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest. 2013;123:844–54.
Seneschal J, Clark Rachael A, Gehad A, et al. Human epidermal langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity. 2012;36:873–84.
Cumberbatch M, Dearman RJ, Kimber I. Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin-1 beta for migration. Immunology. 1997;92:388–95.
Kel JM, Girard-Madoux MJ, Reizis B, et al. TGF-beta is required to maintain the pool of immature Langerhans cells in the epidermis. J Immunol. 2010;185:3248–55.
Groves RW, Allen MH, Ross EL, et al. Tumour necrosis factor alpha is pro-inflammatory in normal human skin and modulates cutaneous adhesion molecule expression. Br J Dermatol. 1995;132:345–52.
Peiris M, Weerts Z, Aktar R, et al. A putative anti-inflammatory role for TRPM8 in irritable bowel syndrome-an exploratory study. Neurogastroenterol Motil. 2021;33: e14170.
Janelsins BM, Sumpter TL, Tkacheva OA, et al. Neurokinin-1 receptor agonists bias therapeutic dendritic cells to induce type 1 immunity by licensing host dendritic cells to produce IL-12. Blood. 2013;121:2923–33.
Marriott I, Mason MJ, Elhofy A, et al. Substance P activates NF-κB independent of elevations in intracellular calcium in murine macrophages and dendritic cells. J Neuroimmunol. 2000;102:163–71.
Kodali S, Ding W, Huang J, et al. Vasoactive intestinal peptide modulates Langerhans cell immune function. J Immunol. 2004;173:6082–8.
Souza RF, Huo X, Mittal V, et al. Gastroesophageal reflux might cause esophagitis through a cytokine-mediated mechanism rather than caustic acid injury. Gastroenterology. 2009;137:1776–84.
Dunbar KB, Agoston AT, Odze RD, et al. Association of acute gastroesophageal reflux disease with esophageal histologic changes. JAMA. 2016;315:2104.
Ahmed Osman H, Aly SS, Mahmoud HS, et al. Effect of acid suppression on peripheral T-lymphocyte subsets and immunohistochemical esophageal mucosal changes in patients with gastroesophageal reflux disease. J Clin Gastroenterol. 2019;53:e362–70.
Mousavinasab F, Babaie D, Nilipour Y, et al. Increased number of regulatory T cells in esophageal tissue of patients with eosinophilic esophagitis in comparison to gastro esophageal reflux disease and control groups. Allergol Immunopathol (Madr). 2019;47:431–6.
Resnick MB, Finkelstein Y, Weissler A, et al. Assessment and diagnostic utility of the cytotoxic T-lymphocyte phenotype using the specific markers granzyme-B and TIA-1 in esophageal mucosal biopsies. Hum Pathol. 1999;30:397–402.
Lind A, Koenderman L, Kusters JG, et al. Squamous tissue lymphocytes in the esophagus of controls and patients with reflux esophagitis and Barrett’s esophagus are characterized by a non-inflammatory phenotype. PLoS ONE. 2014;9: e106261.
Moiseff R, Olson N, Suriawinata AA, et al. CD8 T-Cell-predominant lymphocytic esophagitis is one of the major patterns of lymphocytic inflammation in gastroesophageal reflux disease. Arch Pathol Lab Med. 2021;145:1138–43.
McBride JA, Striker R. Imbalance in the game of T cells: what can the CD4/CD8 T-cell ratio tell us about HIV and health? PLoS Pathog. 2017;13: e1006624.
Rybak A, Pesce M, Thapar N, et al. Gastro-esophageal reflux in children. Int J Mol Sci. 2017;18:1671.
Jagger A, Shimojima Y, Goronzy JJ, et al. Regulatory T cells and the immune aging process: a mini-review. Gerontology. 2013;60:130–7.
Weinstock JV, Blum A, Metwali A, et al. Substance P regulates Th1-type colitis in IL-10 knockout mice1. J Immunol. 2003;171:3762–7.
Blum AM, Metwali A, Crawford C, et al. Interleukin 12 and antigen independently induce substance P receptor expression in T cells in murine schistosomiasis mansoni. FASEB J. 2001;15:950–7.
Weinstock JV, Blum A, Metwali A, et al. IL-18 and IL-12 signal through the NF-κB pathway to induce NK-1R expression on T cells1. J Immunol. 2003;170:5003–7.
Guo C-J, Lai J-P, Luo H-M, et al. Substance P up-regulates macrophage inflammatory protein-1β expression in human T lymphocytes. J Neuroimmunol. 2002;131:160–7.
Beinborn M, Blum A, Hang L, et al. TGF-beta regulates T-cell neurokinin-1 receptor internalization and function. Proc Natl Acad Sci U S A. 2010;107:4293–8.
Vicario M, Blanchard C, Stringer KF, et al. Local B cells and IgE production in the oesophageal mucosa in eosinophilic oesophagitis. Gut. 2010;59:12–20.
Straumann A, Bauer M, Fischer B, et al. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J Allergy Clin Immunol. 2001;108:954–61.
Ryu S, Lee KH, Tizaoui K, et al. Pathogenesis of eosinophilic esophagitis: a comprehensive review of the genetic and molecular aspects. Int J Mol Sci. 2020;21:7253.
Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014;157:121–41.
Guo R, Chen LH, Xing C, et al. Pain regulation by gut microbiota: molecular mechanisms and therapeutic potential. Br J Anaesth. 2019;123:637–54.
Vich Vila A, Imhann F, Collij V, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci Transl Med. 2018;10:8914.
Qiu P, Ishimoto T, Fu L, et al. The gut microbiota in inflammatory bowel disease. Front Cell Infect Microbiol. 2022;12:33992.
Verdu EF, Bercik P, Verma-Gandhu M, et al. Specific probiotic therapy attenuates antibiotic induced visceral hypersensitivity in mice. Gut. 2006;55:182–90.
Kannampalli P, Pochiraju S, Chichlowski M, et al. Probiotic Lactobacillus rhamnosus GG (LGG) and prebiotic prevent neonatal inflammation-induced visceral hypersensitivity in adult rats. Neurogastroenterol Motil. 2014;26:1694–704.
Perez-Burgos A, Wang L, McVey Neufeld KA, et al. The TRPV1 channel in rodents is a major target for antinociceptive effect of the probiotic Lactobacillus reuteri DSM 17938. J Physiol. 2015;593:3943–57.
Pei Z, Bini EJ, Yang L, et al. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci U S A. 2004;101:4250–5.
Kaymak T, Hruz P, Niess JH. Immune system and microbiome in the esophagus: implications for understanding inflammatory diseases. FEBS J. 2022;289:4758–72.
Dixon J, Strugala V, Griffin SM, et al. Esophageal mucin: an adherent mucus gel barrier is absent in the normal esophagus but present in columnar-lined Barrett’s esophagus. Am J Gastroenterol. 2001;96:2575–83.
Fillon SA, Harris JK, Wagner BD, et al. Novel device to sample the esophageal microbiome–the esophageal string test. PLoS ONE. 2012;7: e42938.
Facchin S, Calgaro M, Pandolfo M, et al. Salivary microbiota composition may discriminate between patients with eosinophilic oesophagitis (EoE) and non-EoE subjects. Aliment Pharmacol Ther. 2022;56:450–62.
Harris JK, Fang R, Wagner BD, et al. Esophageal microbiome in eosinophilic esophagitis. PLoS ONE. 2015;10: e0128346.
Pei Z, Yang L, Peek RM, et al. Bacterial biota in reflux esophagitis and Barrett’s esophagus. World J Gastroenterol. 2005;11:7277–83.
Yang L, Lu X, Nossa CW, et al. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137:588–97.
Deshpande NP, Riordan SM, Castano-Rodriguez N, et al. Signatures within the esophageal microbiome are associated with host genetics, age, and disease. Microbiome. 2018;6:227.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
T.L. received an unrestricted, investigator-initiated research grant from Reckitt Benckiser UK. M.P. has no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leech, T., Peiris, M. Mucosal neuroimmune mechanisms in gastro-oesophageal reflux disease (GORD) pathogenesis. J Gastroenterol 59, 165–178 (2024). https://doi.org/10.1007/s00535-023-02065-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00535-023-02065-9