
Abstract
Purpose
The study aims to assess the feasibility, safety, and tolerability of CareMin650, a new photobiomodulation device, in patients treated by radiotherapy (RT) and to collect preliminary data on efficacy for prevention and treatment of oral mucositis (OM) and radiation dermatitis (RD).
Methods
Safe PBM is a French, multicentric, prospective, non-comparative study which include patients with head and neck cancer (H&NC, cohort A) or breast cancer (BC, cohort B) treated in prophylactic (cohorts A1 and B1) or curative setting (cohort A2 and B2). Prophylactic treatment was administered from D1 to end of RT, at a dose of 3 J/cm2. Curative treatment started when a grade 1 to grade 3 lesion had occurred and was pursued until end of RT. Primary endpoint was incidence of device-related adverse events (AEs). OM and RD lesions were graded according to CTCAE V3.
Results
Overall, 72 patients were included (22, 9, 23, and 18 in cohorts A1, A2, B1, and B2, respectively). No device-related AE was reported after 1312 CareMin650 sessions. In cohorts A1 and B1, median time to first OM or RD lesion was 20 days. One BC patient developed G3 RD after completion of RT and discontinuation of CareMin650. Four H&NC patients developed G3 OM. In cohorts A2 and B2, lesions improved or stabilized in 71% of patients. Rates of satisfaction were high among patients and users.
Conclusion
CareMin650 is feasible, safe, and well tolerated for preventive or curative treatment of OM and RD in cancer patients treated with RT. Preliminary efficacy results are promising.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Oral mucositis (OM) and radiation dermatitis (RD) are among the most frequent and disabling side effects of radiotherapy (RT). OM affects nearly all patients treated with RT ± chemotherapy (CT) for head and neck cancer (H&NC) [1], resulting in dysphagia and pain, weight loss, and necessity of enteral alimentation in some cases, as well as increased risk of infections with potentially life-threatening sepsis [2,3,4,5]. OM may jeopardize planned anticancer treatment, potentially leading to decreased efficacy [3]. OM-related consequences significantly increase the cost of patients’ management [4] and impair quality of life (QoL). Very few options showed efficacy for prophylaxis and/or treatment of OM, despite a huge variety of treatments experimented. However, low-level laser therapy (LLLT), now referred to as photobiomodulation (PBM), demonstrated significant benefits in several randomized clinical trials, in particular in patients with H&NC [6,7,8,9,10,11]. According to recent guidelines, PBM is recommended to prevent OM in patients receiving high-dose CT as a conditioning regimen for stem cell transplantation (SCT) and in patients undergoing RT for H&NC [12, 13].
RD affects approximately 95% of patients who receive RT [14], ranging from mild erythema to dry or moist desquamation and ulceration [15]. Nearly all women treated with RT for breast cancer (BC) experience some degree of RD [16]. High-quality data are still insufficient to support specific strategies in the management of RD [15]. However, there is increasing evidence on the benefits of PBM in this setting [17].
PBM involves absorption of red and near-infrared light by mitochondrial chromophores. The electron transfer rate of the respiratory chain is increased, driving upward the production of adenosine triphosphate, reactive oxygen species, and nitric oxide, thus simulating genes involved in tissue repair. Factors involved in inflammation and immunity are recruited to act at a tissue level [18,19,20]. Thus, PBM impacts all the stages of wound repair and tissue regeneration. It also prevents fibrosis, reduces pain (absorption of energy by nociceptors), and prevents tissue death [19, 21].
CareMin650 has been developed to improve practical use of PBM. Delivery of light from LED, emitted by a flexible surface (fabric made of woven optic fibers) in contact with the skin or mucosa, is accurately controlled, reproducible, and operator independent. The device consists of an electronic box generating the light, and pads connected to the box through an optic fiber cable: either oral pads measuring 2.6 × 5.5 cm2 emitting light on both sides or derma pads measuring 15.6 × 5.5cm2, emitting light on one side (Fig. 1). The dose in J/cm2 is selected on the light box that calculates automatically the length of the session to reach the selected dose. Average irradiation times are 1 min 47 s for oral pads and 2 min 23 s for derma pads at a dose of 3 J/cm2. At a dose of 6 J/cm2, they are 3 min 34 s and 4 min 46 s, respectively. Disposable single-use sleeves are placed on the pads before applying them to the skin or the mucosa. The main objectives of this study were to show feasibility, safety, and tolerability of CareMin650 and provide preliminary data on efficacy.

CareMin650. A Light box and oral pads. B Light box and derma pad. C Oral pads placed in the mouth. D Derma pad placed on the neck
Methods
Design and patients
Safe PBM was a French multicentric prospective non-comparative study, designed to assess CareMin650 in patients with H&NC (cohort A) or BC (cohort B), in prophylaxis (cohorts A1 and B1) or cure (cohorts A2 and B2) (Supplementary Fig. 1). Eligible patients were aged 18 years or above, had histologically proven cancer (BC in cohorts B1 and B2, squamous cell carcinoma of oropharynx, nasopharynx, hypopharynx, larynx, or oral cavity in cohorts A1 or A2), and ECOG performance status ≤ 2. In preventive cohorts, patients were scheduled to receive RT (on at least 50% of the oral mucosa, at a total dose of at least 40 Gy for H&NC) and had no lesions at inclusion. In cohorts A2 and B2, patients had previously started RT and presented with OM and/or RD lesions of grade 1 to 3. BC patients were required to have undergone tumor resection (breast conservative surgery or mastectomy) while prior surgery was not mandatory in cohorts A1 or A2. In all cohorts, concomitant treatment with CT and/or targeted therapies was permitted. Patients were excluded if they had known allergy to polyurethane. In H&NC patients, specific exclusion criteria were active bleeding or high risk of bleeding, Hb < 8 g/dL, neutrophils < 1000 mm3, or platelets < 50 000/mm3. In cohorts B1 and B2, patients were excluded if they had received prior irradiation to the same breast. Consecutive patients were included in each subgroup, until the target number had been reached. All study procedures were in accordance with the ethical standards and with the 1964 Helsinki Declaration. All patients signed informed consent before any study procedure was implemented. The study was approved by the Comité de Protection des Personnes (CPP) CPP Sud-Est VI and by the ANSM (Agence Nationale du Médicament et des Produits de Santé).
Treatment
CareMin650 started at inclusion in the study and was pursued until the end of RT. The minimal number of sessions per week was 3, performed immediately before or after RT; however, 5 sessions/week were recommended. Any healthcare professional could administer the treatment after appropriate training. Oral pads and derma pads delivered red light with wavelength of 650 nm, and irradiance of 28 mW/cm2 for oral pads and 21 mW/cm2 for derma pads. Doses for prophylactic and curative treatments were 3 J/cm2 and 6 J/cm2, respectively. If OM or RD occurred in a patient from a prophylactic cohort, the dose was increased to 6 J/cm2. Conversely, if lesions in patients from curative cohorts resolved before the end of RT, the dose was decreased to 3 J/cm2. In cohorts A1 and B1, pads were applied on irradiated areas presenting a risk of RT-related complications. In cohorts A2 and B2, pads were applied on each lesion.
Standard OM and RD prophylaxis, including oral hygiene using soft toothbrush and bicarbonate mouthwashes for OM, was implemented according to sites’ habits. In case of lesions, usual local care, analgesics, and corticosteroids were allowed and therapies considered necessary for the subject’s well‐being could be administered at the discretion of the investigator. Keratinocyte growth factors (palifermin) and other PBM treatments were not allowed during the study.
Assessments
Safety was assessed throughout the study, and adverse events (AEs) were graded according to NCI CTCAE v4. Examination of skin and oral mucosa was performed at each CareMin650 session to assess local tolerance and detect any new lesion. In case of lesion, time of occurrence, size, location, grade according to NCI CTCAE v3, and time to resolution (defined as lesion not requiring further treatment) were reported. Once a week, data were collected on pain, using a Visual Analogic Scale (VAS) graded from 0 (no pain) to 100 (intolerable pain), analgesic consumption, xerostomia, and in case of OM, consequences on food intake. Quality of life was assessed using the SF-12 questionnaire, filled in at baseline and at the end of RT. Patients’ and users’ satisfaction questionnaires were filled in at the end of RT. A follow-up visit was performed 10 ± days after the end of treatment visit.
Statistics
The safety and modified intention-to-treat (mITT) sets comprised all included subjects who had at least one CareMin650 session and at least one safety or efficacy evaluation, respectively. The per protocol (PP) set comprised all subjects without major protocol violation, defined by (i) less than 10 CareMin650 sessions in cohorts A1 or B1 or less than 5 sessions in cohorts A2 or B2 or (ii) start of CareMin650 more than 3 days after start of RT in cohorts A1 or B1. The primary endpoint of the study was the rate of device-related AEs. Secondary endpoints included incidence and grade of lesions, pain, and patients’ and users’ satisfaction.
The number of observations needed was estimated at 300 to allow detecting an undesirable effect occurring at a frequency of 1% with a probability of 95% [22]. Assuming that patients would undergo at least 10 sessions on average, the total number of patients to analyze was set at 60. With an estimation of 20% not eligible for analysis, the total number of subjects to include was 72. Analyses were only descriptive. Quantitative variables were described by mean, standard deviation, median, minimum, and maximum. Qualitative variables were described by counts and percentages. No statistical test was performed. Missing data were not replaced.
The study is registered with ClinicalTrials.gov, number NCT03988556.
Results
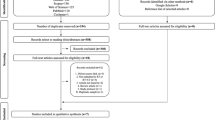
Patients were recruited from July 2019 to November 2020. In total, 74 were screened, and 72 were included and analyzed in the mITT set (22, 9, 23, and 18 patients in cohorts A1, A2, B1, and B2, respectively), while 58 were analyzed in the PP population (17, 8, 19, and 14 patients, respectively) (Fig. 2). Baseline characteristics are summarized in Table 1. Median age was 61.4 years. Relevant comorbidities were more frequent in cohorts A than in cohorts B, cardiovascular disorders and diabetes mellitus being the most prevalent. H&NC were located mainly in oropharynx (41.9%) and oral cavity (32.3%) followed by nasopharynx (9.7%), larynx (9.7%), and hypopharynx (6.5%); 71% of patients had surgery before starting RT and 11 patients received concomitant cisplatin-based CT. In patients with BC, 70.7% of tumors were located in the upper outer quadrant and 92% had stage I or II tumors. Median time from diagnosis to inclusion was 3.5 months. All patients in cohorts A were treated with IMRT and/or VMAT while in cohorts B, 51.2% were treated by RT-3D. The median number of CareMin650 sessions per week was 3.88 and the percentage of weeks with at least 3 sessions was 87.5%, 100%, 83.3%, and 100% in cohorts A1, A2, B1, and B2 respectively.

Disposition of patients
Safety
In total, 1312 sessions of CareMin650 were performed during the study, including 530, 156, 455, and 171 in cohorts A1, A2, B1, and B2, respectively. Nine patients reported 14 treatment-emergent adverse events, none of which being related to the device.
OM and RD lesions, preventive setting
The PP population comprised 36 patients (17 and 19 in cohorts A1 and B1, respectively). The median number of areas requiring application of pads was 3 in cohort A1 and 2 in cohort B1. All patients developed some degree of OM and/or RD lesion. Median time from start of CareMin650 to first lesion was 20 days in both cohorts, ranging from 7 to 34 days in cohort A1, from 3 to 49 days in cohort B1 (Supplementary Figs. 2 and 3). Most lesions were grade 1 or 2 (Table 2). Only 1 patient developed G3 RD in cohort B1 (left axilla), diagnosed after the end of RT at the follow-up visit, G3 upfront. Four patients had G3 OM in cohort A1, after 7, 14, 19, and 25 days, mean 16.25 ± 7.63 days, median: 16.5 days. One lesion, located on hard palate, was diagnosed at grade 3 upfront. Other lesions were diagnosed at grade 1 (one case, tongue and cheek) or at grade 2 (two cases, tongue and cheek in one patient, lips in another patient). In the mITT population (n = 45), the maximal grade of lesions throughout the study was 0 in one patient (cohort B1), 1 in 12 patients [3 (13.6%) in A1, 9 (39.2%) in B1], 2 in 25 patients [13 (59.1%) in A1, 12 (52.2%) in B1], and 3 in 7 patients [6 (27.3%) in A1 and 1 (4.3%) in B1]. The 2 additional patients with G3 OM had started CareMin650 on day 7 and day 5 of RT, respectively.
OM and RD lesions, curative setting
At inclusion, the median number of lesions was 2 (1–3) in cohort A2 and 1 (1–4) in cohort B2. The median time from start of RT to inclusion was 27 days (9–40) and 25 days (11–41), respectively. In the PP population (n = 22), the maximal grade of lesions at inclusion in cohorts A2 and B2 was 1 in 62.5% and 78.6% of cases, respectively, 2 in 25.0% and 21.4% of cases, and 3 in 12.5% and 0% of cases. At the end of radiotherapy visit, 15/21 patients (71.4%) had a maximal grade equal to or lower than that of inclusion (Table 3). At the follow-up visit, lesions had disappeared in 3 patients from cohort B2 (one with G1 and 2 with G2 lesion at inclusion). In the mITT population (n = 27), results were similar with 69% of patients showing stabilization or improvement at the end of RT and disappearance of all lesions in 4 cases at the follow-up visit (cohort B2).
Other criteria
In 57 patients data on pain were available at inclusion and at least once during the study. Mean maximal pain during the study was the highest in cohorts A2 (42.9 ± 26.3) and A1 (35.8 ± 28.1) while it remained low in cohorts B1 (11.6 ± 15.5 and 11.1 ± 17.8, respectively). Data on pain are shown in Supplementary Table 1.
Data on patient’s satisfaction were available for 64 patients. A majority of patients declared that the application of the device was not burdensome (81.3%), provoked no discomfort (76.6%), and that the duration of sessions was acceptable (68.8%). Overall, 87.5% reported no pain at all during applications, while 5 (7.8%) reported slight but tolerable pain and only 3 (15.8%), all in cohort A1, found the application quite painful. The patient preferred maintaining the device in contact with skin or mucosa himself during the session in 89.1% of cases. Overall, most patients were very satisfied (60.3%) or satisfied (33.3%) with the device. The application of CareMin650 was performed by physicians, nurses, residents, radiologic technologists, or clinical research assistants, depending on sites decision and organization. The installation of the device was considered easy in all cases (very easy: 79.4%; rather easy: 20.6%). The device was considered very handy, rather handy, and rather unhandy in 69.1%, 22.1%, and 8.8% of cases, respectively. Duration of sessions was assessed as rather short, acceptable, and rather long in 4.4%, 73.5%, and 22.1% of cases, respectively. Overall, users found the device rather satisfactory (71.6%) or very satisfactory (23.9%) and 87.5% declared that they would like to use it in routine practice.
Discussion
Although PBM demonstrated efficacy and safety in a number of randomized clinical trials and meta-analyses [23,24,25,26,27], it is rarely used in routine practice. Indeed, treatment is time consuming, and equipment is cumbersome and unwieldy; lasers are set with various parameters (wavelength, irradiance, pulse structure, coherence, polarization, energy, fluence) resulting in lack of standardization. Finally, the procedure is not fully reproducible and operator dependent as the distance from the skin or mucosa is difficult to assess accurately. Thus, the amount of energy delivered cannot be exactly known. CareMin650 is a small and handy device allowing reproducible and accurately controlled delivery of light thanks to direct application of lightning tissue on skin and mucosa. This study aimed at evaluating its feasibility and safety in as many situations as possible, leading to inclusion of 4 subgroups of patients, who underwent applications of pads on mucosa and/or skin, on intact or damaged tissues. The study was performed at highly experienced radiotherapy sites. Characteristics of radiotherapy reflect usual practice with IMRT/VMAT being used in all H&NC patients while one half of patients with BC received RT-3D. Duration, total dose, and dose per fraction are consistent with current recommendations. The percentage of patients receiving concomitant chemotherapy was quite low (33%) but not unusual.
The study protocol recommended at least 3 sessions per week, ideally 5. Compliance to treatment was good with a median number of 3.88 sessions per week, which suggests that CareMin650 therapy is feasible even in sites with high patient flow. Indeed, the device is easy to use as the operator only needs to select a dose and the lightbox automatically calculates the duration of application required to deliver this dose. Therefore, application can be performed by any healthcare professional who has been properly trained. In this study, physicians, residents, nurses, radiologic technologists, or CRAs were in charge of device utilization.
PBM is known to have good local tolerance [6, 23]. However, a possible concern with CareMin650 was that direct contact of a pad on skin or mucosa could provoke pain or irritation, especially in case of pre-existing lesion. Our results show that local tolerance was very good as no device-related adverse event relating to local pain, irritation, or unpleasant feelings has been reported during 1312 sessions. In particular, all patients from cohort A2 reported that application was not painful, and overall, only 3 patients (4.7%) declared that the application was rather painful and provoked discomfort. Concerns on risks of PBM-related proliferation of tumor cells have been raised and extensively debated due to conflicting in vitro data. However, the use of PBM for more than 30 years and the increasing number of published data in OM prevention in H&NC and SCT patients suggest that PBM does not influence tumor or treatment outcomes and overall survival [28]. Data in H&NC patients treated with LLLT during RT without prior surgery and long-term follow-up are very reassuring [7]. Specific evaluation of CareMin650 in an in vitro study showed that irradiated cancer cells do not proliferate when illuminated [29].
The study was not designed to demonstrate efficacy. However, preliminary findings can be observed. Only one case of grade 3 RD occurred in the preventive cohorts, and it was diagnosed after the end of RT, therefore did not occur during CareMin650 treatment.
In H&NC patients, grade 3 OM occurred in 4 cases (23.5%), which is lower than reported in the literature, as incidence is usually around 50% and in any case, always exceeds 30% in the absence of efficacious preventive treatment [1, 3]. Importantly, these 4 cases occurred at the same investigational site, among 5 patients included. This discrepancy between sites could be explained by several hypotheses. First, a high variability has been shown in the grading of OM lesions using CTCAEv3 criteria, with discordance rates of 34% between local investigators and central review [30]. In our study, lesions were graded locally, and no training had been implemented at the beginning of the study to standardize grading. Thus, a very likely explanation lies in the variable analysis of lesions across sites, although grading was to be performed by a physician. The choice of CTCAEv3 for grading had been made because it was considered more accurate than WHO grading scale or CTCAEv5. However, it is also more difficult to use and the distinction between grade 2 and 3 appears very difficult and subject to investigator’s interpretation. Moreover, the choice of grading scale can influence the results. In our study, among patients with food intake limitations, only 3 had G3 OM; therefore, using WHO grading scale, the number of patients with G3 OM lesions would probably have been 3. Second, it has been observed that some lesions were reported late, although symptoms suggesting OM had been described days or weeks earlier, leading to delay in dose increase. This highlights the importance of early detection of lesions with dose increase to 6 J/cm2 as soon as a G1 lesion appears. Finally, differences in patients’ population might partly explain a higher incidence of severe lesions, for example, different exposures to tobacco and alcohol in one site (North of France) compared to others. Unfortunately, alcohol consumption and smoking status were not recorded in this study. In the ITT population, 2 additional patients developed G3 OM lesions. Both had started CareMin650 several days after the start of RT, suggesting that starting PBM on day 1 of RT is probably key for prevention of OM. No conclusion can be drawn on the effects of CareMin650 in curative settings at this point, due to the small sample size. However, it seems that lesions were stabilized or improved in most cases, although treatment started at grade 2 or 3 in 27% of cases. Finally, safety and efficacy are not the only criteria for a therapy to be implemented in clinical practice. It has to be acceptable for patients and healthcare professionals. In this study, patients’ and users’ questionnaires showed the high rate of satisfaction towards the device.
This study has limitations. It was not a randomized controlled trial, so that no conclusion can be drawn on efficacy. As already mentioned, no standardization training for grading had been performed which raises questions on consistency across investigators. Some data are lacking to better interpret the results, such as smoking habits or alcohol consumption. Finally, the size of each cohort was low, especially in curative settings. Recruitment turned out to be easier in cohorts A1 and B1 that were almost completed when the COVID-19 pandemic started. At this time, inclusions were put on hold and ongoing treatments were interrupted, leading to a high number of early study discontinuations. Inclusions resumed after 3 months, in some but not all sites, at a very low rate and it was eventually decided to stop the study as the overall target had been reached, despite the imbalance between cohorts.
Conclusion
OM and RD are frequent and disabling adverse effects of RT. Every effort should be made to reduce their incidence and severity in order to improve patients’ quality of life and optimize supportive care. PBM is not routinely used despite proven efficacy and clear recommendations, because of the lack of easy-to-use and reliable equipment. The new CareMin650 device has shown very good safety and tolerance as well as promising efficacy results that will require confirmation in a larger prospective trial to allow wider utilization in daily practice.
Data Availability
A clinical study report has been sent to ANSM.
Code availability
Not applicable.
References
Lalla RV, Bowen J, Barasch A et al (2014) MASCC/ISOO clinical practice guidelines for the management of mucositis secondary to cancer therapy. Cancer 120(10):1453–1461
Scully C, Sonis S, Diz PD (2006) Oral mucositis. Oral Dis 12(3):229–241
Trotti A, Bellm LA, Epstein JB et al (2003) Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol 66(3):253–262
Elting LS, Cooksley CD, Chambers MS, Garden AS (2007) Risk, outcomes, and costs of radiation-induced oral mucositis among patients with head-and-neck malignancies. Int J Radiat Oncol Biol Phys 68(4):1110–1120
Murphy BA, Beaumont JL, Isitt J et al (2009) Mucositis-related morbidity and resource utilization in head and neck cancer patients receiving radiation therapy with or without chemotherapy. J Pain Symptom Manage 38(4):522–532
Antunes HS, Herchenhorn D, Small IA et al (2013) Phase III trial of low-level laser therapy to prevent oral mucositis in head and neck cancer patients treated with concurrent chemoradiation. Radiother Oncol 109(2):297–302
Antunes HS, Herchenhorn D, Small IA et al (2017) Long-term survival of a randomized phase III trial of head and neck cancer patients receiving concurrent chemoradiation therapy with or without low-level laser therapy (LLLT) to prevent oral mucositis. Oral Oncol 71:11–15
Bensadoun RJ, Franquin JC, Ciais G et al (1999) Low-energy He/Ne laser in the prevention of radiation-induced mucositis. A multicenter phase III randomized study in patients with head and neck cancer. Support Care Cancer 7(4):244–52
Gautam AP, Fernandes DJ, Vidyasagar MS, Maiya AG, Vadhiraja BM (2012) Low level laser therapy for concurrent chemoradiotherapy induced oral mucositis in head and neck cancer patients — a triple blinded randomized controlled trial. Radiother Oncol 104(3):349–354
Oton-Leite AF, Elias LS, Morais MO et al (2013) Effect of low level laser therapy in the reduction of oral complications in patients with cancer of the head and neck submitted to radiotherapy. Spec Care Dentist 33(6):294–300
Gautam AP, Fernandes DJ, Vidyasagar MS, Maiya AG, Guddattu V (2015) Low level laser therapy against radiation induced oral mucositis in elderly head and neck cancer patients—a randomized placebo controlled trial. J Photochem Photobiol B 144:51–56
Elad S, Cheng KKF, Lalla RV et al (2020) MASCC/ISOO clinical practice guidelines for the management of mucositis secondary to cancer therapy. Cancer 126(19):4423–4431
Zadik Y, Arany PR, Fregnani ER et al (2019) Systematic review of photobiomodulation for the management of oral mucositis in cancer patients and clinical practice guidelines. Support Care Cancer 27(10):3969–3983
Seite S, Bensadoun RJ, Mazer JM (2017) Prevention and treatment of acute and chronic radiodermatitis. Breast Cancer (Dove Med Press) 9:551–557
Wong RK, Bensadoun RJ, Boers-Doets CB et al (2013) Clinical practice guidelines for the prevention and treatment of acute and late radiation reactions from the MASCC Skin Toxicity Study Group. Support Care Cancer 21(10):2933–2948
Kole AJ, Kole L, Moran MS (2017) Acute radiation dermatitis in breast cancer patients: challenges and solutions. Breast Cancer (Dove Med Press) 9:313–323
Robijns J, Censabella S, Claes S et al (2018) Prevention of acute radiodermatitis by photobiomodulation: a randomized, placebo-controlled trial in breast cancer patients (TRANSDERMIS trial). Lasers Surg Med 50(7):763–771
Hamblin MR, Victor Pires de Sousa M, Agrawal T (2016) Handbook of low-level laser therapy: Pan Stanford Publishing, pp. 1133
Huang YY, Sharma SK, Carroll J, Hamblin MR (2011) Biphasic dose response in low level light therapy — an update. Dose Response 9(4):602–618
Courtois E, Bouleftour W, Guy JB et al (2021) Mechanisms of PhotoBioModulation (PBM) focused on oral mucositis prevention and treatment: a scoping review. BMC Oral Health 21(1):220
Fekrazad R, Chiniforush N (2014) Oral mucositis prevention and management by therapeutic laser in head and neck cancers. J Lasers Med Sci 5(1):1–7
Begaud B (1998) Dictionnaire de Pharmaco-épidémiologie. 3e édition: Bordeaux
Bjordal JM, Bensadoun RJ, Tuner J, Frigo L, Gjerde K, Lopes-Martins RA (2011) A systematic review with meta-analysis of the effect of low-level laser therapy (LLLT) in cancer therapy-induced oral mucositis. Support Care Cancer 19(8):1069–1077
Oberoi S, Zamperlini-Netto G, Beyene J, Treister NS, Sung L (2014) Effect of prophylactic low level laser therapy on oral mucositis: a systematic review and meta-analysis. PLoS ONE 9(9):e107418
Clarkson JE, Worthington HV, Furness S, McCabe M, Khalid T, Meyer S (2010) Interventions for treating oral mucositis for patients with cancer receiving treatment. Cochrane Database Syst Rev (8):CD001973
Migliorati C, Hewson I, Lalla RV et al (2013) Systematic review of laser and other light therapy for the management of oral mucositis in cancer patients. Support Care Cancer 21(1):333–341
Bensadoun RJ, Nair RG (2012) Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol 24(4):363–370
Bensadoun RJ, Epstein JB, Nair RG et al (2020) Safety and efficacy of photobiomodulation therapy in oncology: a systematic review. Cancer Med 9(22):8279–8300
Courtois E, Guy JB, Axisa F, et al (2021) Photobiomodulation by a new optical fiber device: analysis of the in vitro impact on proliferation/migration of keratinocytes and squamous cell carcinomas cells stressed by X-rays. Lasers Med Sci 36(7):1445–1454
Ueno T, Zenda S, Konishi T et al (2019) The post hoc analysis comparing the severity grades of chemoradiotherapy-induced oral mucositis scored between the central and local assessors in a multicenter, randomized controlled trial of rebamipide for head and neck cancer. Int J Clin Oncol 24(3):241–247
Funding
The research has been funded by NeoMedLight, 90 rue Frédéric Fays, 69 100 Villeurbanne, sponsor of the clinical trial.
Author information
Authors and Affiliations
Contributions
All authors included patients in the study and contributed to writing and reviewing the article.
Corresponding author
Ethics declarations
Ethics approval
The study was approved by the Comité de Protection des Personnes (CPP) CPP Sud-Est VI on April 11, 2019.
Consent to participate
All patients included in the study signed an informed consent form prior to any study procedure.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bensadoun, RJ., Bollet, M.A., Liem, X. et al. New photobiomodulation device for prevention and cure of radiotherapy-induced oral mucositis and dermatitis: results of the prospective Safe PBM study. Support Care Cancer 30, 1569–1577 (2022). https://doi.org/10.1007/s00520-021-06574-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00520-021-06574-2