
Abstract
Renal ciliopathies are a common cause of kidney failure in children and adults, and this study reviewed their ocular associations. Genes affected in renal ciliopathies were identified from the Genomics England Panels. Ocular associations were identified from Medline and OMIM, and the genes additionally examined for expression in the human retina (https://www.proteinatlas.org/humanproteome/tissue) and for an ocular phenotype in mouse models (http://www.informatics.jax.org/). Eighty-two of the 86 pediatric-onset renal ciliopathies (95%) have an ocular phenotype, including inherited retinal degeneration, oculomotor disorders, and coloboma. Diseases associated with pathogenic variants in ANK6, MAPKBP1, NEK8, and TCTN1 have no reported ocular manifestations, as well as low retinal expression and no ocular features in mouse models. Ocular abnormalities are not associated with the most common adult-onset "cystic" kidney diseases, namely, autosomal dominant (AD) polycystic kidney disease and the AD tubulointerstitial kidney diseases (ADTKD). However, other kidney syndromes with cysts have ocular features including papillorenal syndrome (optic disc dysplasia), Hereditary Angiopathy Nephropathy, Aneurysms and muscle Cramps (HANAC) (tortuous retinal vessels), tuberous sclerosis (retinal hamartomas), von Hippel-Lindau syndrome (retinal hemangiomas), and Alport syndrome (lenticonus, fleck retinopathy). Ocular abnormalities are associated with many pediatric-onset renal ciliopathies but are uncommon in adult-onset cystic kidney disease. However the demonstration of ocular manifestations may be helpful diagnostically and the features may require monitoring or treatment.
Similar content being viewed by others


Introduction
Renal ciliopathies account for at least 15% of children and 5% of adults with kidney failure [1,2,3,4]. It is important to identify these diseases because treatments that slow disease progression are increasingly available and because other organs may also be affected [5].
Renal ciliopathies result from pathogenic variants in more than 100 genes (https://panelapp.genomicsengland.co.uk/). The cysts arise mainly from abnormal cilium signaling in the tubules and collecting ducts [2]. The primary cilia on the surface of the tubular epithelial cells function as antennae that sense mechanical, osmotic, and chemical stimuli and signal through pathways that are responsible for maintaining normal tubular structure. These pathways control cell proliferation, differentiation, and orientation, with disruption resulting in cyst development.
The clinical features of renal ciliopathies manifest in organs where the affected genes are expressed and ciliary signaling is important, such as the liver, heart, lungs, skeleton, nerves, and eyes [6,7,8]. The pattern of extra-renal involvement may be helpful diagnostically.
Renal ciliopathies in children have mainly autosomal recessive (AR) inheritance and include the various forms of nephronophthisis, as well as Senior-Loken, Joubert, Bardet-Biedl, Meckel, and Jeune syndromes, cranioectodermal dysplasia, and the orofaciodigital syndromes (Table 1). These are often associated with ocular abnormalities.
The most common adult-onset diseases included here are autosomal dominant (AD) polycystic kidney disease (ADPKD) and AD tubulointerstitial kidney disease (ADTKD), previously known as “medullary cystic kidney disease”) due to pathogenic variants in MUC1, UMOD, REN, or HNF1B. ADTKD is characterised by microcysts rather than cysts. Tuberous sclerosis is common but results less often in kidney failure. Other diseases associated with kidney cysts include ARPKD, the rare von Hippel-Lindau syndrome, papillorenal syndrome, and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps or HANAC. X-linked (XL) and AD Alport syndrome are also associated with kidney cysts, but their pathogenesis is unclear [16, 17]. In addition, acquired cystic kidneys are common in individuals undergoing dialysis [18]. Cystic liver disease without significant kidney cysts usually results from variants in other genes [19].
The eye is often involved in genetic kidney disease because, despite their different functions, the eye and kidney share developmental pathways and some specialized structural features [20]. Both the kidney and the eye undergo organogenesis in the 4th to 7th embryonic week, and perturbations during this period result in kidney and ocular phenotypes [21]. Primary cilia on the surface of the tubule epithelium are critical in maintaining tubular structure and orientation, and disruption leads to cyst formation. Cilia in the photoreceptor outer segment relay sensory stimuli to the brain via the visual pathway, and defects result in retinal degeneration [22]. The retinal pigment cells—Bruch’s membrane—choriocapillaris and glomerular filtration barrier also share structural features [23].
While genetic testing has become more widely available, ocular phenotyping is a simple, non-invasive method that may suggest the diagnosis. Genetic testing also indicates the mode of inheritance and other at-risk family members. In addition, ophthalmic examination identifies vision-threatening conditions that require regular monitoring and, in some cases, treatment.
The aim of this study was to review the reported ophthalmic associations of renal ciliopathies in children and cystic diseases in adults in order to identify features that might help the nephrologist recognize the disease’s genetic basis. The study also examined whether individual genes were expressed in the retina or associated with ocular features in mouse models, which suggested ocular abnormalities might yet be found in the corresponding human disease.
Strategy
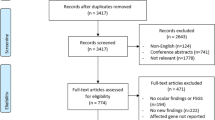
The major genes affected in the renal ciliopathies and other forms of cystic kidney disease were identified from the Genomics England Renal ciliopathy panel (v1.64; https://panelapp.genomicsengland.co.uk/) and from reviews [16, 24]. Gene names were then searched in OVID Medline together with (“AND”) the following search terms: “ocular,” “ophthal*,” “optic*,” “eye,” “vision*,” “cornea,” “iris”, “pupil,” “lens,” “retina,” “choroid,” “fovea,” “macula,” and “optic nerve” to identify relevant manuscripts. Abstracts were reviewed, and relevant manuscripts examined for ocular abnormalities. Inclusion criteria were studies describing a renal ciliopathy or cystic kidney disease, associated ocular phenotypes, and publication in a peer-reviewed journal. Articles were excluded if they were not written in English or if the full text was not available online. The search was conducted between August and September 2020 and reviewed in August–October 2022. Gene names and corresponding diseases were also searched for “eye” features in OMIM in October 2022.
Expression of the genes was examined in the retina in the Human Protein Atlas (https://www.proteinatlas.org/humanproteome/tissue); and the effects of the corresponding genes on ocular manifestations examined in mouse models in the Mouse Genome Informatics website (http://www.informatics.jax.org/) in October 2022.
Renal ciliopathies with a mainly pediatric presentation
Eighty-six genes associated with a mainly pediatric presentation were examined from the renal ciliopathy green and amber lists after PKD1, 2, PKHD1, MUC1, UMOD, and HNF1B had been excluded (Table 2).
Eighty-two of these genes (95%) had a reported ocular phenotype in human disease. Four renal ciliopathy genes (ANK6, MAPKBP1, NEK8, TCTN1) had no ocular phenotype in human disease and were associated with low retinal expression (< 10 transcripts per million, TPM) and no mouse ocular phenotype.
Eighty-five of the 86 genes (99%) were expressed in the retina. The exception was ICK which is associated with endocrine cerebro-osteodysplasia, but its kidney phenotype is unclear. Fifty-eight of these genes (67%) were expressed at > 10 TPM in the retina. Sixty genes (70%) had an ocular phenotype in the corresponding mouse model. Thirteen genes were expressed both at low levels in the retina and not associated with an ocular phenotype in the mouse model, but nine of these had a reported phenotype in human disease.
Ocular phenotypes in nephronophthisis and other renal ciliopathies
The descriptions for the clinical features of these diseases often overlap, and almost all of these genes have an ocular phenotype. The most common ocular abnormalities are inherited retinal degeneration, coloboma, and oculomotor abnormalities (Tables 1, 2, 3; Figs. 1, 2).

Inherited retinal degeneration in renal ciliopathies demonstrating a a waxy optic disc, attenuated arterioles, and widespread mottling of the retinal pigment epithelium in nephronophthisis; with OCT scan demonstrating outer retinal atrophy with a small area of retained subfoveal photoreceptors (below); b infrared fundus image demonstrating a ring of retained normal retina; c automated visual field test demonstrating constricted visual field; d inferonasal retinal pigmentation in an adult with Bardet-Biedl syndrome; and e retinal thinning and intraretinal pigment migration with OCT scan demonstrating associated cystoid macular edema (below)

Optic disc anomalies and coloboma in renal ciliopathies demonstrating a iris coloboma and b large chorioretinal coloboma
Senior-Loken syndrome is diagnosed when there is retinal disease as well as nephronophthisis, which occurs in at least 15% of cases [27]. Many gene variants have been reported [28,29,30]. Pathogenic variants in IQCB1 (NPHP5) are always associated with retinal abnormalities.
Joubert syndrome is characterized by nephronophthisis with neurological disease resulting in hypotonia and developmental delay. The “molar tooth sign” on MRI is pathognomonic. Pathogenic variants affect up to 30 different genes. Variants in the AHI1, CPLANE1, CC2D2A, CEP290, and TMEM67 genes represent 40% of all cases [31,32,33]. Neuro-ophthalmic abnormalities are common, with retinal disease and coloboma occurring less frequently [34].
Bardet-Biedl syndrome includes nephronophthisis, polydactyly, obesity, diabetes, genital anomalies, and inherited retinal degeneration. More than 20 genes are implicated, most of which are found in the “BBSome,” a protein complex involved in ciliary trafficking [35]. Retinal disease occurs in 90% of cases [36] but coloboma and oculomotor disorders are uncommon.
Meckel syndrome is a severe multi-system illness that is often fatal in the perinatal period. Pathogenic variants in the same genes may also result in milder disease, such as Joubert syndrome, with inherited retinal degeneration, coloboma, microphthalmia, or oculomotor disorders [12, 37].
Jeune syndrome (“asphyxiating thoracic dystrophy”) and cranioectodermal dysplasia (Sensenbrenner syndrome) are rare skeletal dystrophies that occur because of disrupted intraflagellar transport. Nephronophthisis is associated with high myopia, inherited retinal degeneration, and neuro-ophthalmic features [14, 38, 39].
Orofaciodigital syndrome type 1 includes malformations of the face, mouth, and digits and is often associated with kidney cysts, fibrocystic liver disease, and neurological, skeletal, and cardiac anomalies [40]. The less common types 2–13 do not appear to have kidney cysts. Ocular abnormalities are limited to hypertelorism and epicanthal folds [41].
Common ocular phenotypes in pediatric ciliopathies (Table 3; Figs. 1, 2)
Inherited retinal degeneration
Inherited retinal degeneration (dystrophy, disease) encompasses a heterogeneous group of diseases characterized by photoreceptor degeneration and impaired vision. It is categorized by the photoreceptor type affected, with disease predominantly affecting the rods, cones, or both. Retinitis pigmentosa is the most common form [42]. Rods are affected early with subsequent progression involving the cones. Night blindness due to rod dysfunction is an early feature. Peripheral visual field deficits may progress to tunnel vision. The retina has thinned vessels, abnormal pigmentation, retinal pigment atrophy, and waxy pallor of the optic disc [43].
The diagnosis of Senior-Loken syndrome (nephronophthisis with inherited retinal degeneration) depends on the use of retinal imaging, fundus autofluorescence, optical coherence tomography, and electroretinography [44]. The clinical outcome is highly variable, even among individuals with the same gene variants [45]. Gene therapy is currently being explored as treatment [42, 46,47,48,49], and intravitreal antisense oligonucleotide injections have improved visual acuity and oculomotor stability for at least the CEP290 variants [50].
Leber congenital amaurosis is a severe form of congenital or early-onset inherited retinal degeneration affecting both rods and cones. Presentation is typically in the first few months of life with impaired vision, nystagmus, sluggish pupillary light reflexes, and increased eye rubbing (the “oculo-digital sign”) that may increase the risk of keratoconus [51, 52]. Fundus imaging may be normal early, but vascular attenuation, optic disc pallor, and abnormal pigmentation occur with time [53]. Pathogenic variants in CEP290 and RPGRIP1L are responsible for 20% and 5% of cases, respectively [54].
Coloboma
Overall coloboma are normally found in one in 2000–10,000 births, and one or both eyes may be affected [55, 56]. Coloboma are common in genetic kidney disease but also occur as an isolated phenomenon, with CAKUT, some genetic glomerulopathies or tubulopathies and with other genetic diseases that do not affect the kidneys.
Coloboma occur from abnormal closure of the embryonic fissure during the sixth to seventh week of gestation. Coloboma of the iris and ciliary body result from abnormal anterior closure, and coloboma of the optic nerve, retina, and choroid from failed posterior closure [57].
Chorioretinal coloboma are detected on fundus imaging as discrete, white lesions commonly affecting the inferonasal quadrant with overlying pigmentation. Complications include a 30% increased risk of retinal detachment possibly with visual field defect [58], and amblyopia [59], cataract, glaucoma, and increased refractive error [60,61,62].
Optic nerve coloboma vary in size with the largest resembling the “morning glory” anomaly with enlargement, dysplasia, and funneling of the optic disc, radial orientation of retinal vessels, and peripapillary pigmentation [63].
Neuro-ophthalmological disorders
These are particularly common in nephronophthisis and related ciliopathies, especially Joubert syndrome. Congenital oculomotor apraxia occurs in 80% of individuals with Joubert syndrome [64] and is characterized by the inability to initiate voluntary eye movements, most often horizontal saccades. In order to compensate, individuals use “head thrusting” to bring an object of interest into their field of vision after head control is established at 6 months of age [65, 66]. Head thrusting tends to diminish after the first year of life, and no specific intervention alters disease progression. Various forms of nystagmus have been described including “pendular” and “seesaw” types [67]. Strabismus occurs in most patients with Joubert syndrome and often requires surgical correction [34].
Renal ciliopathies and cystic kidney disease with a mainly adult-onset presentation
Ten genes associated with cystic kidney disease that include the commonest genetic causes of cystic kidney disease, ADPKD and ADTKD, were examined (Table 4; Fig. 3). Ocular features are not described in these diseases. In addition, PKD1 and all ADTKD genes are expressed at very low levels in the human retina. Only PKD2, GANAB, and DNAJB11 are found at levels > 10 transcripts per million (TPM) but still have no reported ocular phenotype in human disease nor in mouse models.

Ocular abnormalities in cystic kidney disease demonstrating a tuberous sclerosis with retinal astrocytoma and OCT with intraretinal location (below); b von Hippel-Lindau syndrome with treated hemangioma; c congenital vascular tortuosity typical of HANAC; d X-linked Alport syndrome with fleck dystrophy that spares the macula, and OCT that demonstrates the temporal retinal thinning and characteristic macular profile (below); and e renal coloboma syndrome with retinal vessels emerging from the side rather than the center of the optic disc
AD polycystic kidney disease
At least 90% of all individuals with ADPKD have a pathogenic variant in PKD1 or PKD2 which code for a transmembrane protein complex that regulates the transport of cations in the tubule and are not associated with ocular features. These genes are expressed at low levels in the human retina, and mouse models have no ocular phenotype. Pathogenic variants in GANAB and DNAJB11 are rare causes of polycystic kidney disease and while expressed in the retina do not result in an ocular phenotype in mouse models.
AR polycystic kidney disease
AR polycystic kidney disease (ARPKD) is caused by pathogenic variants in PKHD1 which encodes fibrocystin, a cell membrane ciliary receptor. ARPKD is not associated with an ocular phenotype, but genome-wide association studies have recently suggested PKHD1 as a marker for myopia [77], raised intraocular pressure, and primary open-angle glaucoma [78]. The mouse models have no ocular phenotype. A rare cause of ARPKD, DZIP1L, is also not expressed in the retina, and again the mouse models have no ocular phenotype.
AD tubulointerstitial kidney disease
Pathogenic variants in UMOD, MUC1, and REN have no reported ocular associations. There is an isolated report of a unilateral coloboma and visual loss associated with a pathogenic variant in HNF1B (renal cysts and diabetes syndrome), a regulator of gene expression during nephron development [79], but this has not been reproduced and may be coincidental. None of these four proteins is expressed to any extent in the human retina, and none of the mouse models has ocular features.
Ocular phenotypes in syndromic forms of cystic kidney disease
In contrast, all 6 genes associated with syndromic cystic kidney disease have ocular manifestations. These include the renal coloboma syndrome (“morning glory anomaly”); von Hippel-Lindau syndrome (retinal hemangioblastoma); tuberous sclerosis (iris and retinal hamartoma); HANAC (tortuous retinal vessels); and X-linked Alport syndrome (anterior lenticonus, corneal dystrophy, fleck retinopathy). The ocular features are distinctive and generally diagnostic.
Renal coloboma syndrome
Inheritance is AD, and about 80% of individuals with a pathogenic variant in PAX2, a transcriptional regulator, have renal coloboma syndrome [80]. Variants in this gene result typically in focal and segmental glomerulosclerosis (FSGS), but congenital anomalies of the kidney and urinary tract (CAKUT) and kidney cysts also occur. Ocular associations include optic nerve dysplasia, including both optic nerve coloboma and the “morning glory” anomaly [71]. Subtle forms may be overlooked. Scleral staphyloma, chorioretinal coloboma, optic nerve cyst, microcornea, microphthalmia, macular dysplasia [81,82,83], nystagmus and retinal detachment [84] also occur. Iris coloboma are not a feature. The ocular abnormality is present at birth, may be bilateral, but varies in each eye and does not necessarily affect the visual fields. However, complications such as retinal detachment may occur and patients require monitoring.
Von Hippel-Lindau disease
Von Hippel Lindau disease is an AD “tumor syndrome” that results from pathogenic variants in the VHL tumor suppressor gene [85]. Retinal capillary hemangiomas are present in 50 to 85% of affected individuals by the age of 25 years [85, 86, 87]. Most retinal capillary hemangiomas are due to Von Hippel-Lindau disease, and their presence prompts genetic testing [88]. Small hemangiomas do not affect vision, but larger lesions result in complications including subretinal exudation, fibrovascular proliferation, neovascular glaucoma, and retinal detachment. Regular monitoring is important to avoid loss of vision, with treatments including laser photocoagulation, photodynamic therapy, anti-VEGF injections, and vitreoretinal surgery [89,90,91,92].
Tuberous sclerosis complex 1 and 2
Tuberous sclerosis is another AD “tumor syndrome,” caused by pathogenic variants in the TSC1 or TSC2 tumor suppressor genes, tuberin and hamartin, that inhibit the mammalian target of rapamycin (mTOR) pathway. Affected individuals have the characteristic retinal lesions of astrocytic hamartomas which may be flat, nodular, or transitional. Flat lesions occur in 50–70% of patients [93] and are yellow-gray retinal patches that obscure vessels. Nodular or “mulberry” lesions are calcified, elevated defects with well-defined margins found in half of all patients [94]. Transitional lesions are less common and have features of both flat and nodular hamartomas. Hamartomas rarely affect vision, but ongoing monitoring is important because of the risk of subretinal exudates and retinal detachment [95]. Hamartomas affecting the optic nerve may be mistaken for papilledema. Tuberous sclerosis also results in subependymal giant cell astrocytomas in 20% of patients that can cause papilledema [96]. Less common retinal findings include areas of peripheral hypopigmentation and chorioretinal coloboma [93]. Non-retinal findings in the eye are rare but include iris hypopigmentation [97] and juvenile cataract [98].
Alport syndrome
Alport syndrome results from pathogenic variants in the COL4A5, COL4A3, or COL4A4 genes. These contribute to the collagen IV α3α4α5 network in the basement membranes of the lens capsule, cornea, and the inner nuclear layer, and Bruch’s membrane of the retina. Kidney cysts have been reported in the XL and AD forms of Alport syndrome [16, 17]. Cysts typically occur before the age of 50 years but are few in number, and do not distort the kidney size or contribute to kidney failure [99]. With the XL and AR forms of Alport syndrome, lenticonus, central fleck retinopathy, and temporal retinal thinning are common [100], and corneal erosions and macular atrophy may occur [23]. More severe genetic variants (large rearrangements, truncating changes) are associated with more damaging ocular phenotypes [101,102,103]. AD Alport syndrome has no reported ocular features [104].
Kidney cysts are also found in HANAC. This results from pathogenic variants in COL4A1 which codes for the collagen IV a1 chain found in blood vessel basement membranes. The cysts are usually few in number and do not result in kidney failure [105]. Retinal vessels are tortuous on imaging [74] and cerebral small vessel disease may be present [106].
Discussion
The pediatric-onset renal ciliopathies are often associated with ocular abnormalities. These include inherited retinal degeneration, coloboma, and oculomotor disorders, but there is often variable penetrance and expression even in family members with the same disease-causing variant. The greater prevalence of ocular features in renal ciliopathies correlates with the frequent expression of the genes in the retina and the presence of ocular phenotypes in the corresponding mouse models.
In contrast, ophthalmic abnormalities are much less common or do not occur in genetic "cystic" kidney diseases that present in adulthood, namely, ADPKD, and ADTKD. ARPKD also has no ocular features. Ocular abnormalities are more common in the rarer cystic kidney syndromes such as tuberous sclerosis and von Hippel-Lindau syndrome where they affect at least half of all affected individuals. Ocular features are also found in some other cystic kidney diseases such as papillorenal syndrome and Alport syndrome. In general, the genes affected in cystic kidney disease are not expressed in the retina and most mouse models do not have an ocular phenotype.
It was generally difficult to determine how often ocular abnormalities were associated with disease due to individual genes. This was because often few individuals were reported for any gene, only some had undergone a formal ophthalmic examination and many had only been examined at presentation. However, one large well-studied cohort with Joubert syndrome had ocular motor apraxia in 80% of affected individuals, strabismus in 74%, nystagmus in 72%, ptosis in 43%, chorioretinal coloboma in 30%, optic nerve atrophy in 22%, and inherited retinal degeneration in 38% [64].
In summary, ocular features of the inherited renal ciliopathies and cystic kidney diseases may be useful diagnostically. Most forms of nephronophthisis and the other renal ciliopathies with a mainly pediatric onset have ocular features reported. Inherited retinal degeneration is a useful pointer to the diagnosis of nephronophthisis and Bardet-Biedl syndrome. Oculomotor disorders may be a feature of ciliopathies, especially Joubert syndrome. Coloboma suggest a diagnosis of nephronophthisis but also occur in other forms of genetic kidney disease. In contrast, ADPKD, ARPKD, and ADTKD are not associated with ocular features, but retinal hemangioma and astrocytic hamartoma suggest von Hippel-Lindau syndrome and tuberous sclerosis, respectively.
In children a complete ophthalmological examination is helpful especially where a kidney biopsy has non-specific or inconclusive findings and genetic testing is problematic. While genetic testing is increasingly available, ophthalmic examination is a fast, inexpensive, and non-invasive method that also demonstrates potentially vision-threatening disease. Furthermore, sight-threatening conditions associated with kidney disease may require ophthalmological referral for monitoring and treatment, for example, for refractive error, glaucoma, or strabismus. Monitoring and treatment is necessary for retinal hemangiomas in von Hippel-Lindau disease, astrocytic hamartoma in tuberous sclerosis, and complications of coloboma such as retinal detachment [58, 95, 107, 108]. Inherited retinal degeneration is not currently treatable, but monitoring for progression and complications is necessary [45, 109]. In the future, gene-specific therapies may be helpful. Genetic testing should be performed in individuals where these diseases are suspected.
The strengths of this study were use of the Genomics England panel for the list of genes confirmed by experts to be affected in the renal ciliopathies, the inclusion of OMIM in the clinical review, and the screening of a retinal expression database and of mouse models for ocular features. The study’s major limitations were the few reports of some diseases, and the paucity of detailed ophthalmic examinations described; the variable penetrance and expression of the genetic variants; and the lack of examination of gene expression in parts of the eye other than the retina. In addition, some of the ocular associations may have been coincidental and sometimes the ocular phenotypes may have been mild or overlooked. Mouse models do not precisely replicate human disease but the finding of a mouse ocular phenotype suggests that these features should be sought more closely in the corresponding human disease.
In conclusion, the finding of ocular abnormalities in a suspected renal ciliopathy or cystic kidney disease may confirm the disease’s genetic basis, implicate the affected gene, indicate the necessity of monitoring for ocular complications, and suggest future treatments. These ocular associations may also help us better understand the pathogenesis of the corresponding kidney disease.
Data availability
All the data examined here is included in the manuscript, the Supplementary material or the cited databases.
References
Harris PC, Torres VE (2009) Polycystic kidney disease. Annu Rev Med 60:321–337
Guay-Woodford LM (2006) Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatr Nephrol 21:1369–1376
Saunier S, Salomon R, Antignac C (2005) Nephronophthisis. Curr Opin Genet Dev 15:324–331
Luo F, Tao YH (2018) Nephronophthisis: a review of genotype-phenotype correlation. Nephrology (Carlton) 23:904–911
Rastogi A, Ameen KM, Al-Baghdadi M, Shaffer K, Nobakht N, Kamgar M, Lerma EV (2019) Autosomal dominant polycystic kidney disease: updated perspectives. Ther Clin Risk Manag 15:1041–1052
Badano JL, Mitsuma N, Beales PL, Katsanis N (2006) The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 7:125–148
Hildebrandt F, Benzing T, Katsanis N (2011) Ciliopathies. N Engl J Med 364:1533–1543
Lee JE, Gleeson JG (2011) A systems-biology approach to understanding the ciliopathy disorders. Genome Med 3:59
Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, Utsch B, Sayer JA, Lillo C, Jimeno D, Coucke P, De Paepe A, Reinhardt R, Klages S, Tsuda M, Kawakami I, Kusakabe T, Omran H, Imm A, Tippens M, Raymond PA, Hill J, Beales P, He S, Kispert A, Margolis B, Williams DS, Swaroop A, Hildebrandt F (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 37:282–288
Romani M, Micalizzi A, Kraoua I, Dotti MT, Cavallin M, Sztriha L, Ruta R, Mancini F, Mazza T, Castellana S, Hanene B, Carluccio MA, Darra F, Máté A, Zimmermann A, Gouider-Khouja N, Valente EM (2014) Mutations in B9D1 and MKS1 cause mild Joubert syndrome: expanding the genetic overlap with the lethal ciliopathy Meckel syndrome. Orphanet J Rare Dis 9:72
Forsythe E, Beales PL (2013) Bardet-Biedl syndrome. Eur J Hum Genet 21:8–13
Hartill V, Szymanska K, Sharif SM, Wheway G, Johnson CA (2017) Meckel-Gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr 5:244–244
Baujat G, Huber C, El Hokayem J, Caumes R, Do Ngoc Thanh C, David A, Delezoide AL, Dieux-Coeslier A, Estournet B, Francannet C, Kayirangwa H, Lacaille F, Le Bourgeois M, Martinovic J, Salomon R, Sigaudy S, Malan V, Munnich A, Le Merrer M, Le Quan Sang KH, Cormier-Daire V (2013) Asphyxiating thoracic dysplasia: clinical and molecular review of 39 families. J Med Genet 50:91–98
Arts H, Knoers N (2018) Cranioectodermal dysplasia. GeneReviews. University of Washington, Seattle
Toriello HV, Franco B, Bruel AL, Thauvin-Robinet C (1993) Oral-facial-digital syndrome type I. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews. University of Washington, Seattle
Sevillano AM, Gutierrez E, Morales E, Hernandez E, Molina M, Gonzalez E, Praga M (2014) Multiple kidney cysts in thin basement membrane disease with proteinuria and kidney function impairment. Clin Kidney J 7:251–256
Gulati A, Sevillano AM, Praga M, Gutierrez E, Alba I, Dahl NK, Besse W, Choi J, Somlo S (2020) Collagen IV gene mutations in adults with bilateral renal cysts and CKD. Kidney Int Rep 5:103–108
Dunnill MS, Millard PR, Oliver D (1977) Acquired cystic disease of the kidneys: a hazard of long-term intermittent maintenance haemodialysis. J Clin Pathol 30:868–877
Boerrigter MM, Bongers E, Lugtenberg D, Nevens F, Drenth JPH (2021) Polycystic liver disease genes: practical considerations for genetic testing. Eur J Med Genet 64:104160
Izzedine H, Bodaghi B, Launay-Vacher V, Deray G (2003) Eye and kidney: from clinical findings to genetic explanations. J Am Soc Nephrol 14:516–529
Bodaghi B, Massamba N, Izzedine H (2014) The eye: a window on kidney diseases. Clin Kidney J 7:337–338
Yildiz O, Khanna H (2012) Ciliary signaling cascades in photoreceptors. Vision Res 75:112–116
Savige J, Liu J, DeBuc DC, Handa JT, Hageman GS, Wang YY, Parkin JD, Vote B, Fassett R, Sarks S, Colville D (2010) Retinal basement membrane abnormalities and the retinopathy of Alport syndrome. Invest Ophthalmol Vis Sci 51:1621–1627
Kim B, King BF Jr, Vrtiska TJ, Irazabal MV, Torres VE, Harris PC (2016) Inherited renal cystic diseases. Abdom Radiol (NY) 41:1035–1051
Feldhaus B, Weisschuh N, Nasser F, den Hollander AI, Cremers FPM, Zrenner E, Kohl S, Zobor D (2020) CEP290 Mutation spectrum and delineation of the associated phenotype in a large German cohort: a monocentric study. Am J Ophthalmol 211:142–150
Moloney TP, Patel C, Gole GA (2014) Exudative vasculopathy in a child with Leber congenital amaurosis. J AAPOS 18:297–299
Wolf MTF (2015) Nephronophthisis and related syndromes. Curr Opin Pediatr 27:201–211
Srivastava S, Sayer JA (2014) Nephronophthisis. J Pediatr Genet 3:103–114
Hildebrandt F, Zhou W (2007) Nephronophthisis-associated ciliopathies. J Am Soc Nephrol 18:1855–1871
Konig J, Kranz B, Konig S, Schlingmann KP, Titieni A, Tonshoff B, Habbig S, Pape L, Haffner K, Hansen M, Buscher A, Bald M, Billing H, Schild R, Walden U, Hampel T, Staude H, Riedl M, Gretz N, Lablans M, Bergmann C, Hildebrandt F, Omran H, Konrad M, Gesellschaft fur Padiatrische Nephrologie (GPN) (2017) Phenotypic spectrum of children with nephronophthisis and related ciliopathies. Clin J Am Soc Nephrol 12:1974–1983
Parisi M, Glass I (1993) Joubert syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews. University of Washington, Seattle
Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O’Day D, Alswaid A, Ramadevi AR, Lingappa L, Lourenço C, Martorell L, Garcia-Cazorla À, Ozyürek H, Haliloğlu G, Tuysuz B, Topçu M, Chance P, Parisi MA, Glass IA, Shendure J, Doherty D (2015) Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet 52:514–522
Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, Zein W, Brooks BP, Heller T, Soldatos A, Oden NL, Yildirimli D, Vemulapalli M, Mullikin JC, Program NCS, Malicdan MCV, Gahl WA, Gunay-Aygun M (2017) Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med 19:875–882
Brooks BP, Zein WM, Thompson AH, Mokhtarzadeh M, Doherty DA, Parisi M, Glass IA, Malicdan MC, Vilboux T, Vemulapalli M, Mullikin JC, Gahl WA, Gunay-Aygun M (2018) Joubert syndrome: ophthalmological findings in correlation with genotype and hepatorenal disease in 99 patients prospectively evaluated at a single center. Ophthalmology 125:1937–1952
Suspitsin EN, Imyanitov EN (2016) Bardet-Biedl Syndrome. Mol Syndromol 7:62–71
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA (1999) New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet 36:437–446
Alexiev BA, Lin X, Sun CC, Brenner DS (2006) Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med 130:1236–1238
Pilotto E, Midena E, Longhin E, Frizziero L (2022) Retinal dystrophy in Jeune syndrome: a multimodal imaging characterization. Retin Cases Brief Rep 16:183–185
Wilson DJ, Weleber RG, Beals RK (1987) Retinal Dystrophy in Jeune’s Syndrome. Arch Ophthalmol 105:651–657
Feather SA, Winyard PJ, Dodd S, Woolf AS (1997) Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease: clinical, radiological and histopathological features of a new kindred. Nephrol Dial Transplant 12:1354–1361
Larralde de Luna M, Raspa ML, Ibargoyen J (1992) Oral-facial-digital type 1 syndrome of Papillon-Leage and Psaume. Pediatr Dermatol 9:52–56
Ziccardi L, Cordeddu V, Gaddini L, Matteucci A, Parravano M, Malchiodi-Albedi F, Varano M (2019) Gene Therapy in Retinal Dystrophies. Int J Mol Sci 20:5722
Hamel C (2006) Retinitis pigmentosa. Orphanet J Rare Dis 1:40
Nash BM, Wright DC, Grigg JR, Bennetts B, Jamieson RV (2015) Retinal dystrophies, genomic applications in diagnosis and prospects for therapy. Trans Pediatr 4:139–163
Hohman TC (2017) Hereditary Retinal Dystrophy. Handb Exp Pharmacol 242:337–367
Sahel JA, Dalkara D (2019) Gene therapy for retinal dystrophy. Nat Med 25:198–199
Prado DA, Acosta-Acero M, Maldonado RS (2020) Gene therapy beyond luxturna: a new horizon of the treatment for inherited retinal disease. Curr Opin Ophthalmol 31:147–154
Patel U, Boucher M, de Léséleuc L, Visintini S (2018) Voretigene Neparvovec: an emerging gene therapy for the treatment of inherited blindness. In: CADTH Issues in Emerging Health Technologies. Canadian Agency for Drugs and Technologies in Health, Ottawa (ON), 2016(169):1–11
Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, Georgiadis A, Mowat FM, Beattie SG, Gardner PJ, Feathers KL, Luong VA, Yzer S, Balaggan K, Viswanathan A, de Ravel TJ, Casteels I, Holder GE, Tyler N, Fitzke FW, Weleber RG, Nardini M, Moore AT, Thompson DA, Petersen-Jones SM, Michaelides M, van den Born LI, Stockman A, Smith AJ, Rubin G, Ali RR (2015) Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 372:1887–1897
Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, Roman AJ, Sumaroka A, Han IC, Hochstedler MD, Pfeifer WL, Sohn EH, Taiel M, Schwartz MR, Biasutto P, Wit W, Cheetham ME, Adamson P, Rodman DM, Platenburg G, Tome MD, Balikova I, Nerinckx F, Zaeytijd J, Van Cauwenbergh C, Leroy BP, Russell SR (2019) Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med 25:225–228
Chung DC, Traboulsi EI (2009) Leber congenital amaurosis: clinical correlations with genotypes, gene therapy trials update, and future directions. J AAPOS 13:587–592
Elder MJ (1994) Leber congenital amaurosis and its association with keratoconus and keratoglobus. J Pediatr Ophthalmol Strabismus 31:38–40
Kumaran N, Moore AT, Weleber RG, Michaelides M (2017) Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol 101:1147–1154
Chacon-Camacho OF, Zenteno JC (2015) Review and update on the molecular basis of Leber congenital amaurosis. World J Clin Cases 3:112–124
Warburg M (1993) Classification of microphthalmos and coloboma. J Med Genet 30:664–669
Nakamura KM, Diehl NN, Mohney BG (2011) Incidence, ocular findings, and systemic associations of ocular coloboma: a population-based study. Arch Ophthalmol 129:69–74
Stoll C, Alembik Y, Dott B, Roth MP (1992) Epidemiology of congenital eye malformations in 131,760 consecutive births. Ophthalmic Paediatr Genet 13:179–186
Schubert HD (2005) Structural organization of choroidal colobomas of young and adult patients and mechanism of retinal detachment. Trans Am Ophthalmol Soc 103:457–472
Olsen TW, Summers CG, Knobloch WH (1996) Predicting visual acuity in children with colobomas involving the optic nerve. J Pediatr Ophthalmol Strabismus 33:47–51
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E (2000) Ocular colobomata. Surv Ophthalmol 45:175–194
Bavbek T, Ogüt MS, Kazokoglu H (1993) Congenital lens coloboma and associated pathologies. Doc Ophthalmol 83:313–322
Lee BJ, Traboulsi EI (2008) Update on the morning glory disc anomaly. Ophthalmic Genet 29:47–52
Beyer WB, Quencer RM, Osher RH (1982) Morning glory syndrome. A functional analysis including fluorescein angiography, ultrasonography, and computerized tomography. Ophthalmology 89:1362–1367
Wang SF, Kowal TJ, Ning K, Koo EB, Wu AY, Mahajan VB, Sun Y (2018) Review of Ocular Manifestations of Joubert Syndrome. Genes (Basel) 9:605
Harris CM, Shawkat F, Russell-Eggitt I, Wilson J, Taylor D (1996) Intermittent horizontal saccade failure (‘ocular motor apraxia’) in children. Br J Ophthalmol 80:151–158
Saunier S, Morin G, Calado J, Benessy F, Silbermann F, Antignac C (1997) Large deletions of the NPH1 region in Cogan syndrome (CS) associated with familial juvenile nephronophthisis (NPH). Am J Hum Genet 61:A346–A346
Weiss AH, Doherty D, Parisi M, Shaw D, Glass I, Phillips JO (2009) Eye movement abnormalities in Joubert syndrome. Invest Ophthalmol Vis Sci 50:4669–4677
Cornec-Le Gall E, Alam A, Perrone RD (2019) Autosomal dominant polycystic kidney disease. Lancet 393:919–935
Imhoff O, Marion V, Stoetzel C, Durand M, Holder M, Sigaudy S, Sarda P, Hamel CP, Brandt C, Dollfus H, Moulin B (2011) Bardet-Biedl syndrome: a study of the renal and cardiovascular phenotypes in a French cohort. Clin J Am Soc Nephrol 6:22–29
Devuyst O, Olinger E, Weber S, Eckardt KU, Kmoch S, Rampoldi L, Bleyer AJ (2019) Autosomal dominant tubulointerstitial kidney disease. Nat Rev Dis Primers 5:60
Schimmenti LA (2011) Renal coloboma syndrome. Eur J Hum Genet 19:1207–1212
Chittiboina P, Lonser RR (2015) Von Hippel-Lindau disease. Handb Clin Neurol 132:139–156
Randle SC (2017) Tuberous sclerosis complex: a review. Pediatr Ann 46:e166–e171
Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, Marro B, Desmettre T, Cohen SY, Roullet E, Dracon M, Fardeau M, Van Agtmael T, Kerjaschki D, Antignac C, Ronco P (2007) COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 357:2687–2695
Habib R, Gubler MC, Hinglais N, Noel LH, Droz D, Levy M, Mahieu P, Foidart JM, Perrin D, Bois E, Grunfeld JP (1982) Alport’s syndrome: experience at Hopital Necker. Kidney Int Suppl 11:S20-28
Gibson J, Fieldhouse R, Chan MMY, Sadeghi-Alavijeh O, Burnett L, Izzi V, Persikov AV, Gale DP, Storey H, Savige J, Genomics England Research Consortium (2021) Prevalence estimates of predicted pathogenic COL4A3-COL4A5 variants in a population sequencing database and their implications for Alport syndrome. J Am Soc Nephrol 32:2273–2290
Hysi PG, Choquet H, Khawaja AP, Wojciechowski R, Tedja MS, Yin J, Simcoe MJ, Patasova K, Mahroo OA, Thai KK, Cumberland PM, Melles RB, Verhoeven VJM, Vitart V, Segre A, Stone RA, Wareham N, Hewitt AW, Mackey DA, Klaver CCW, MacGregor S, Consortium for Refractive Error and Myopia, Khaw PT, Foster PJ, UK Eye and Vision Consortium, Guggenheim JA, 23andMe Inc., Rahi JS, Jorgenson E, Hammond CJ (2020) Meta-analysis of 542,934 subjects of European ancestry identifies new genes and mechanisms predisposing to refractive error and myopia. Nat Genet 52:401–407
Khawaja AP, Cooke Bailey JN, Wareham NJ, Scott RA, Simcoe M, Igo RP Jr, Song YE, Wojciechowski R, Cheng CY, Khaw PT, Pasquale LR, Haines JL, Foster PJ, Wiggs JL, Hammond CJ, Hysi PG, UK Biobank Eye and Vision Consortium, NEIGHBORHOOD Consortium (2018) Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat Genet 50:778–782
Raile K, Klopocki E, Holder M, Wessel T, Galler A, Deiss D, Muller D, Riebel T, Horn D, Maringa M, Weber J, Ullmann R, Gruters A (2009) Expanded clinical spectrum in hepatocyte nuclear factor 1b-maturity-onset diabetes of the young. J Clin Endocrinol Metab 94:2658–2664
Bower M, Salomon R, Allanson J, Antignac C, Benedicenti F, Benetti E, Binenbaum G, Jensen UB, Cochat P, DeCramer S, Dixon J, Drouin R, Falk MJ, Feret H, Gise R, Hunter A, Johnson K, Kumar R, Lavocat MP, Martin L, Morinière V, Mowat D, Murer L, Nguyen HT, Peretz-Amit G, Pierce E, Place E, Rodig N, Salerno A, Sastry S, Sato T, Sayer JA, Schaafsma GC, Shoemaker L, Stockton DW, Tan WH, Tenconi R, Vanhille P, Vats A, Wang X, Warman B, Weleber RG, White SM, Wilson-Brackett C, Zand DJ, Eccles M, Schimmenti LA, Heidet L (2012) Update of PAX2 mutations in renal coloboma syndrome and establishment of a locus-specific database. Hum Mutat 33:457–466
Sanyanusin P, Schimmenti LA, McNoe LA, Ward TA, Pierpont ME, Sullivan MJ, Dobyns WB, Eccles MR (1995) Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 9:358–364
Amiel J, Audollent S, Joly D, Dureau P, Salomon R, Tellier AL, Augé J, Bouissou F, Antignac C, Gubler MC, Eccles MR, Munnich A, Vekemans M, Lyonnet S, Attié-Bitach T (2000) PAX2 mutations in renal-coloboma syndrome: mutational hotspot and germline mosaicism. Eur J Hum Genet 8:820–826
Schimmenti LA, Manligas GS, Sieving PA (2003) Optic nerve dysplasia and renal insufficiency in a family with a novel PAX2 mutation, Arg115X: further ophthalmologic delineation of the renal-coloboma syndrome. Ophthalmic Genet 24:191–202
Bower MA, Schimmenti LA, Eccles MR (1993) PAX2-Related Disorder. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews. University of Washington, Seattle
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH (2003) von Hippel-Lindau disease. Lancet 361:2059–2067
Singh AD, Shields CL, Shields JA (2001) von Hippel-Lindau disease. Surv Ophthalmol 46:117–142
Varshney N, Kebede AA, Owusu-Dapaah H, Lather J, Kaushik M, Bhullar JS (2017) A review of Von Hippel-Lindau Syndrome. J Kidney Cancer VHL 4:20–29
Binderup MLM, Stendell AS, Galanakis M, Moller HU, Kiilgaard JF, Bisgaard ML (2018) Retinal hemangioblastoma: prevalence, incidence and frequency of underlying von Hippel-Lindau disease. Br J Ophthalmol 102:942–947
Agarwal A, Kumari N, Singh R (2016) Intravitreal bevacizumab and feeder vessel laser treatment for a posteriorly located retinal capillary hemangioma. Int Ophthalmol 36:747–750
Slim E, Antoun J, Kourie HR, Schakkal A, Cherfan G (2014) Intravitreal bevacizumab for retinal capillary hemangioblastoma: a case series and literature review. Can J Ophthalmol 49:450–457
Zubair T, Callaway NF, Ludwig CA, Tang PH, Shields RA, Ji MH, Vail D, Powers MA, Moshfeghi DM (2020) Von Hippel-Lindau syndrome phenotype with prominent vitreoretinal neovascularization treated with early PPV: a case series and literature review. Ophthalmic Surg Lasers Imaging Retina 51:109–115
Karimi S, Arabi A, Shahraki T, Safi S (2020) Von Hippel-Lindau disease and the eye. J Ophthalmic Vis Res 15:78–94
Robertson DM (1991) Ophthalmic manifestations of tuberous sclerosis. Ann N Y Acad Sci 615:17–25
Rowley SA, O’Callaghan FJ, Osborne JP (2001) Ophthalmic manifestations of tuberous sclerosis: a population based study. Br J Ophthalmol 85:420–423
Hodgson N, Kinori M, Goldbaum MH, Robbins SL (2017) Ophthalmic manifestations of tuberous sclerosis: a review. Clin Exp Ophthalmol 45:81–86
Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M (2009) Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 16:691–696
Lucchese NJ, Goldberg MF (1981) Iris and fundus pigmentary changes in tuberous sclerosis. J Pediatr Ophthalmol Strabismus 18:45–46
Geffrey AL, Geenen KR, Abati E, Greenstein SH, VanderVeen DK, Levy RL, Davidson SL, McGarrey MP, Thiele EA, Aronow ME (2020) Juvenile cataract in association with tuberous sclerosis complex. Ophthalmic Genet 41:345–349
Savige J, Harraka P (2021) Pathogenic variants in the genes affected in Alport Syndrome (COL4A3-COL4A5) and their association with other kidney conditions: A review. Am J Kidney Dis 78:857–864
Savige J, Sheth S, Leys A, Nicholson A, Mack HG, Colville D (2015) Ocular features in Alport syndrome: pathogenesis and clinical significance. Clin J Am Soc Nephrol 10:703–709
Tan R, Colville D, Wang YY, Rigby L, Savige J (2010) Alport retinopathy results from “severe” COL4A5 mutations and predicts early renal failure. Clin J Am Soc Nephrol 5:34–38
Colville D, Savige J, Branley P, Wilson D (1997) Ocular abnormalities in thin basement membrane disease. Br J Ophthalmol 81:373–377
Savige J, Wang Y, Crawford A, Smith J, Symons A, Mack H, Nicholls K, Wilson D, Colville D (2017) Bull’s eye and pigment maculopathy are further retinal manifestations of an abnormal Bruch’s membrane in Alport syndrome. Ophthalmic Genet 38:238–244
Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY (2003) Thin basement membrane nephropathy. Kidney Int 64:1169–1178
Chen Z, Migeon T, Verpont MC, Zaidan M, Sado Y, Kerjaschki D, Ronco P, Plaisier E (2016) HANAC syndrome Col4a1 mutation causes neonate glomerular hyperpermeability and adult glomerulocystic kidney disease. J Am Soc Nephrol 27:1042–1054
Alamowitch S, Plaisier E, Favrole P, Prost C, Chen Z, Van Agtmael T, Marro B, Ronco P (2009) Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology 73:1873–1882
Wiley HE, Krivosic V, Gaudric A, Gorin MB, Shields C, Shields J, Aronow ME, Chew EY (2019) Management of retinal hemangioblastoma in von Hippel-Lindau disease. Retina 39:2254–2263
McGarrey MP, Patel S, Aronow ME, Thiele EA (2020) Ophthalmic and genetic findings in a large cohort of patients with tuberous sclerosis complex. Invest Ophthal Vis Sci 61:5176–5176
Garafalo AV, Cideciyan AV, Heon E, Sheplock R, Pearson A, WeiYang YuC, Sumaroka A, Aguirre GD, Jacobson SG (2020) Progress in treating inherited retinal diseases: early subretinal gene therapy clinical trials and candidates for future initiatives. Prog Retin Eye Res 77:100827
Acknowledgements
We would like particularly to acknowledge the assistance of the Genomics England PanelApp, and the OMIM, Human Protein Atlas, and Mouse Genome Informatics websites.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
OS was a medical student who undertook this research project as part of his degree. None of the authors has a financial or non-financial Conflict of Interest to declare.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salehi, O., Mack, H., Colville, D. et al. Ocular manifestations of renal ciliopathies. Pediatr Nephrol 39, 1327–1346 (2024). https://doi.org/10.1007/s00467-023-06096-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-023-06096-5