
Abstract
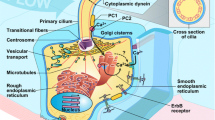
Inherited renal cystic diseases constitute an important set of single-gene disorders that frequently progress to end stage renal disease (ESRD). Transmitted as autosomal dominant, autosomal recessive, or X-linked traits, renal cystic diseases are phenotypically diverse with respect to age at onset, rate of disease progression, and associated extra-renal manifestations. These disorders involve defects in a set of gene products commonly referred to as cystoproteins that, while functionally distinct, appear to co-localize, at least in part, with the cilia/centrosome complex. Therefore, investigations are increasingly focused on the role of this complex in the pathogenesis of renal cystic disease. Sorting out the functional relationship between these cystoproteins and the cilia/centrosome complex will undoubtedly provide a better understanding of renal cystic disease pathogenesis and, potentially, identify new targets for therapeutic intervention.



Similar content being viewed by others


References
Fick G, Gabow P (1994) Hereditary and acquired cystic disease of the kidney. Kidney Int 46:951–964
D’Agata I, Jonas M, Perez-Atayde A, Guay-Woodford L (1994) Combined cystic disease of the liver and kidney. Semin Liver Dis 14:215–228
Guay-Woodford L (2003) Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 285:F1034–F1049
Wilson P (2004) Polycystic kidney disease. N Engl J Med 350:151–164
Gabow P, Grantham J (1997) Polycystic kidney disease. In Schrier R, Gottschalk C (eds) Diseases of the kidney. Little, Brown, Boston, pp 521–560
Chapman A, Gabow P (1997) Hypertension in autosomal dominant polycystic kidney disease. Kidney Int 61 [Suppl]:S71–S73
Hateboer N, v Dijk MV, Bogdanova N, Coto E, Saggar-Malik AK, San Millan JL, Torra R, Breuning M, Ravine D (1999) Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet 353:103–107
Torres VE (2005) Vasopressin antagonists in polycystic kidney disease. Kidney Int 68:2405–2418
Torres V, Harris P (2006) Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol 2:40–55
Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33:129–137
Ong A, Harris P (2005) Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int 67:1234–1247
Zerres K, Muecher G, Becker J, Steinkamm C, Rudnik-Schoneborn S, Heikkila P, Rapola J, Salonen R, Germino G, Onuchic L, Somlo S, Avner E, Harman L, Stockwin J, Guay-Woodford L (1998) Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): molecular genetics, clinical experience, and fetal morphology. Am J Med Genet 76:137–144
Guay-Woodford L, Desmond R (2003) Autosomal recessive polycystic kidney disease (ARPKD): the clinical experience in North America. Pediatrics 111:1072–1080
Ward C, Hogan M, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham J, Bacallao R, Ishibashi M, Milliner D, Torres V, Harris P (2002) The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30:259–269
Onuchic L, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, Rudnik-Schöneborn S, Mrug M, Sweeney W, Avner E, Zerres K, Guay-Woodford L, Somlo S, Germino G (2002) PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple IPT domains and PbH1 repeats. Am J Hum Genet 70:1305–1317
Xiong H, Chen Y, Yi Y, Tsuchiya K, Moeckel G, Cheung J, Liang D, Tham K, Xu X, Chen X, Pei Y, Zhao Z, Wu G (2002) A novel gene encoding a TIG multiple domain protein is a positional candidate for autosomal recessive polycystic kidney disease. Genomics 80:96–104
Ward C, Yuan D, Masyuk T, Wang X, Punyashthiti R, Whelan S, Bacallao R, Torra R, LaRusso N, Torres V, Harris P (2003) Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet 12:2703–2710
Menezes F, Cai Y, Nagasawa Y, Silva A, Watkins M, Silva A, Somlo S, Guay-Woodford L, Germino G, Onuchic L (2004) Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium and cytoplasm. Kidney Int 66:1345–1355
Zhang M, Mai W, Li C, Cho S, Hao C, Moeckel G, Zhao R, Kim I, Wang J, Xiong H, Wang H, Sato Y, Wu Y, Nakanuma Y, Lilova M, Pei Y, Harris R, Li S, Coffey R, Sun L, Wu D, Chen X, Breyer M, Zhao Z, McKanna J, Wu G (2004) PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci USA 101:2311–2316
Salomon R, Gubler M, Antignac C (2005) Nephronophthisis. In: Davidson A, Cameron J, Grunfeld J, Ponticelli C, Ritz E, Winearb C, Van Ypersele C (eds) Oxford text book of clinical nephrology. Oxford University Press, Oxford, pp 2325–2334
Saunier S, Salomon R, Antignac C (2005) Nephronophthisis. Curr Opin Genet Dev 15:324–331
Hildebrandt F, Otto E (2005) Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet 6:928–940
Parisi M, Bennett C, Eckert M, Dobyns W, Gleeson J, Shaw D, McDonald R, Eddy A, Chance P, Glass I (2004) The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet 75:82–91
Dixon-Salazar T, Silhavy J, Marsh S, Louie C, Scott L, Gururaj A, Al-Gazali L, Al-Tawari A, Kayserili H, Sztriha L, Gleeson J (2004) Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 75:979–987
Ferland R, Eyaid W, Collura R, Tully L, Hill R, Al-Nouri D, Al-Rumayyan A, Topcu M, Gascon G, Bodell A, Shugart Y, Ruvolo M, Walsh C (2004) Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 36:1008–1013
Utsch B, Sayer J, Attanasio M, Pereira R, Eccles M, Hennies H, Otto E, Hildebrandt F (2006) Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol 21:32–35
Parisi M, Doherty D, Eckert M, Shaw D, Ozyurek H, Aysun S, Giray O, Al Swaid A, Al Shahwan S, Dohayan N, Bakhsh E, Indridason O, Dobyns W, Bennett C, Chance P, Glass I (2006) AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet 43:334–339
Valente E, Brancati F, Silhavy J, Castori M, Marsh S, Barrano G, Bertini E, Boltshauser E, Zaki M, Abdel-Aleem A, Abdel-Salam G, Bellacchio E, Battini R, Cruse R, Dobyns W, Krishnamoorthy K, Lagier-Tourenne C, Magee A, Pascual-Castroviejo I, Salpietro C, Sarco D, Dallapiccola B, Gleeson J (2006) AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neurol 59:527–534
Beales P (2005) Lifting the lid on Pandora’s box: the Bardet–Biedl syndrome. Curr Opin Genet Dev 15:315–323
Parfrey PS, Davidson WS, Green JS (2002) Clinical and genetic epidemiology of inherited renal disease in Newfoundland. Kidney Int 61:1925–1934
Katsanis N (2004) The oligogenic properties of Bardet–Biedl syndrome. Hum Mol Genet 13 Spec No 1:R65–R71
Moore S, Green J, Fan Y, Bhogal A, Dicks E, Fernandez B, Stefanelli M, Murphy C, Cramer B, Dean J, Beales P, Katsanis N, Bassett A, Davidson W, Parfrey P (2005) Clinical and genetic epidemiology of Bardet–Biedl syndrome in Newfoundland: a 22-year prospective, population-based, cohort study. Am J Med Genet 132:352–360
Nishimura D, Swiderski R, Searby C, Berg E, Ferguson A, Hennekam R, Merin S, Weleber R, Biesecker L, Stone E, Sheffield V (2005) Comparative genomics and gene expression analysis identifies BBS9, a new Bardet–Biedl syndrome gene. Am J Hum Genet 77:1021–1033
Badano J, Leitch C, Ansley S, May-Simera H, Lawson S, Lewis R, Beales P, Dietz H, Fisher S, Katsanis N (2006) Dissection of epistasis in oligogenic Bardet–Biedl syndrome. Nature 439:326–330
Badano J, Teslovich T, Katsanis N (2005) The centrosome in human genetic disease. Nat Rev Genet 6:194–205
Salonen R, Paavola P (1998) Meckel syndrome. J Med Genet 35:497–501
Simpson J, Mills J, Rhoads G, Cunningham G, Conley M, Hoffman H (1991) Genetic heterogeneity in neural tube defects. Ann Genet 34:279–286
Smith U, Consugar M, Tee L, McKee B, Maina E, Whelan S, Morgan N, Goranson E, Gissen P, Lilliquist S, Aligianis I, Ward C, Pasha S, Punyashthiti R, Malik Sharif S, Batman P, Bennett C, Woods C, McKeown C, Bucourt M, Miller C, Cox P, Algazali L, Trembath R, Torres V, Attie-Bitach T, Kelly D, Maher E, Gattone V, Harris P, Johnson C (2006) The transmembrane protein meckelin (MKS3) is mutated in Meckel–Gruber syndrome and the wpk rat. Nat Genet 38:191–196
Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M (2006) MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet 38:155–157
Feather S, Woolf A, Donnai D, Malcolm S, Winter R (1997) The oral-facial-digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2-Xp22.3. Hum Mol Genet 6:1163–1167
Ferrante M, Giorgio G, Feather S, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf A, Sylvie O, Bernard L, Malcolm S, Winter R, Ballabio A (2001) Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet 68:569–576
Romio L, Fry A, Winyard P, Malcolm S, Woolf A, Feather S (2004) OFD1 is a centrosomal/basal body protein expressed during mesenchymal–epithelial transition in human nephrogenesis. J Am Soc Nephrol 15:2556–2568
Ferrante M, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, Dolle P, Franco B (2006) Oral-facial-digital type I protein is required for primary cilia formation and left–right axis specification. Nat Genet 38:112–117
Pazour GJ (2004) Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol 15:2528–2536
Davenport J, Yoder B (2005) An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol 289:F1159–F1169
Pazour G, Rosenbaum J (2002) Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol 12:551–555
Praetorius H, Spring K (2005) A physiological view of the primary cilium. Annu Rev Physiol 67:515–529
Yamaguchi T, Wallace DP, Magenheimer BS, Hempson SJ, Grantham JJ, Calvet JP (2004) Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J Biol Chem 279:40419–40430
Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello O, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G (2005) Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37:537–543
Germino G (2005) Linking cilia to Wnts. Nat Genet 37:455–457
Chauvet V, Tian X, Husson H, Grimm D, Wang T, Hieseberger T, Igarashi P, Bennett A, Ibraghimov-Beskrovnaya O, Somlo S, Caplan M (2004) Mechanical stimuli induce the cleavage and nuclear translocation of the polycystin-1 C-terminus. J Clin Invest 114:1433–1443. For corrected article see J Clin Invest 2005;115:788
Acknowledgment
Supported in part by a Clinical Scientist Award in Translational Research from the Burroughs-Wellcome Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Guay-Woodford, L.M. Renal cystic diseases: diverse phenotypes converge on the cilium/centrosome complex. Pediatr Nephrol 21, 1369–1376 (2006). https://doi.org/10.1007/s00467-006-0164-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-006-0164-9