
Abstract
We investigated 52 cases of de novo unbalanced translocations, consisting in a terminally deleted or inverted-duplicated deleted (inv-dup del) 46th chromosome to which the distal portion of another chromosome or its opposite end was transposed. Array CGH, whole-genome sequencing, qPCR, FISH, and trio genotyping were applied. A biparental origin of the deletion and duplication was detected in 6 cases, whereas in 46, both imbalances have the same parental origin. Moreover, the duplicated region was of maternal origin in more than half of the cases, with 25% of them showing two maternal and one paternal haplotype. In all these cases, maternal age was increased. These findings indicate that the primary driver for the occurrence of the de novo unbalanced translocations is a maternal meiotic non-disjunction, followed by partial trisomy rescue of the supernumerary chromosome present in the trisomic zygote. In contrast, asymmetric breakage of a dicentric chromosome, originated either at the meiosis or postzygotically, in which the two resulting chromosomes, one being deleted and the other one inv-dup del, are repaired by telomere capture, appears at the basis of all inv-dup del translocations. Notably, this mechanism also fits with the origin of some simple translocations in which the duplicated region was of paternal origin. In all cases, the signature at the translocation junctions was that of non-homologous end joining (NHEJ) rather than non-allelic homologous recombination (NAHR). Our data imply that there is no risk of recurrence in the following pregnancies for any of the de novo unbalanced translocations we discuss here.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Unbalanced translocations leading to monosomy and trisomy for the distal parts of two different chromosomes account for about 0,1% of chromosomal imbalances detected at amniocentesis in cases mainly ascertained for advanced maternal age or ultrasound abnormalities (Chang et al. 2013), and for about 1% in subjects with developmental delay and intellectual disability (Robberecht et al. 2013; Weckselblatt et al. 2015). Of these, 13/18 in the prenatal sample (Chang et al. 2013) and about 1/3 in the postnatal one (Robberecht et al. 2013) resulted to be de novo.
Sometimes, the de novo unbalanced chromosome mimicks a recombinant from a parental pericentric inversion (Rivera et al. 2013) in which the deleted arm ends with the distal portion of the opposite arm. The frequency of this type of translocation, to which we will refer as “de novo unbalanced inversion”, is unknown. A more complex category of de novo unbalanced translocations is characterized by three breakpoints, two associated with the formation of an inverted-duplicated deleted chromosome and the third with the capture of a stabilizing telomere from another chromosome. The inv-dup del chromosome derives from a mirror dicentric chromosome after its asymmetric breakage generating a simple deleted and an inverted-duplicated deleted product (Zuffardi et al. 2009). In any case, the products of the dicentric’s breakage need to acquire a new telomere by telomerase-mediated telomere healing or by telomere capture from another chromosome or the opposite arm of the same chromosome (Kostiner et al. 2002; Pham et al. 2014), generating either a complex translocation or an inversion. Again, the frequency of these rearrangements is unknown.
The occurrence of de novo unbalanced translocations has been the source of tantalizing discussions regarding their origin and any possible risk of recurrence in the following pregnancies that would suggest the opportunity for invasive prenatal diagnosis. The recent literature on the unexpected high frequency of cryptic mosaics (Campbell et al. 2014; Rios and Delgado 2015) does not permit to exclude a priori that the derivative chromosome was inherited from a healthy parent who carried an undetected mosaic rearrangement. On the other hand, the finding in some patients of unbalanced translocations in mosaic with a normal cell line (see Gijsbers et al. 2011 for a review) or with the deleted and the duplicated region of the derivative chromosome having different parental origin (cases 8 and 12 in Robberrecht et al. 2013; Eggermann et al. 1997; Sarri et al. 1997; Giorda et al. 2008; Sakazume et al. 2012) strongly suggests a postzygotic event. To clarify the mechanism of origin of de novo unbalanced translocations (Robberrecht et al. 2013) and (Weckselblatt et al. 2015) analyzed the breakpoint junctions in a total of 16 cases. The two studies reported equivocal results, indicating non-allelic homologous recombination between retrotransposable elements as the main mechanism in one study and non-homologous end joining or microhomology-mediated break-induced replication in the second one.
To find a more definitive answer, we analyzed 52 de novo unbalanced rearrangements, consisting of 37 simple translocations, 6 translocations mimicking an inversion, and 9 complex translocations, using a combination of array comparative genomic hybridization (aCGH), fluorescence in situ hybridization (FISH), and mate-pair whole-genome sequencing (WG-MPS). Moreover, parent of origin investigations of the unbalanced chromosomes/segments was possible in 46 of the 52 trios.
Materials and methods
Subjects
The study was approved by the Ethical Committee at the “Eugenio Medea” Scientific Institute.
We collected 52 unique de novo unbalanced chromosomal rearrangements ascertained by G- or Q-banding karyotype (28 cases, 54%), subtelomeric FISH (13 cases, 25%) or aCGH (11 cases, 21%), in subjects with developmental delay, prenatal ultrasound abnormalities (cases 12, 13, 45, and 46), or couple infertility (case 24). In all cases, the rearrangements were characterized by the simultaneous presence of at least two terminal imbalances: a deletion and a duplication, as detected by aCGH and confirmed by FISH with subtelomeric probes (Online Resource 1: Table S1). The genomic imbalances involved the distal portions of two different chromosomes in 45 cases (unbalanced translocations) or the opposite arm of the same chromosome in 7 cases (unbalanced inversions).
The translocations/inversions were present in all cells, except for cases 24, 25, and 37, where conventional karyotyping revealed that the imbalance was present in mosaic with a normal cell line, and in case 24 harboring a third cell line trisomic for the same chromosome that is translocated in the unbalanced cell line. In case 45, two different abnormal cell lines were detected. In non-mosaic cases, FISH with probes from both unbalanced regions was performed on the parental metaphases, demonstrating the de novo origin of the rearrangements, while parental karyotype analysis in subjects with an unbalanced inversion excluded the presence of a pericentric inversion.
Cytogenetics, microarray, and FISH investigations
Microarray analysis with Agilent kits (G4411B, G4449A, G4890A, G4447A) was performed in all cases following standard manufacturer protocols. Scanning was done by the Feature Extraction V.9.1 software and data analysis performed by CGH Analytics V.3.4.27 or Cytogenomics software V.2.15.8.1 (Agilent Technologies). DNA from cases 1–9, 12, 13, 17, 19, 22, 26, 27, 29, 30, 34, and 36 and cases 51–52 was also studied by SNP array with Human Omni Express Exome ILLUMINA v1.2 constituted by 964,193 SNP probes and analyzed using the Illumina Genome Studio v.2011.1 software as well as Illumina CNV Partition (ver 2.3.4) and PennCNV software (version June 2011). DNA from cases 25, 37, and 45 was analyzed by CGH–SNP array (Agilent kit G4890A). The pan-telomeric peptide nucleic acid (PNA) probe (PNA FISH kit/Cy3, Dako, Denmark), which recognizes the consensus sequence (TTAGGG)n of human pan-telomeres, was hybridized to metaphases from case 25 (fibroblasts) and 37 (lymphocytes), according to manufacturer’s instructions. The whole-chromosome painting and locus-specific probes (Vysis, Abbott Park Illinois USA; Kreatech, Amsterdam NL) were hybridized to metaphases from case 24, following manufactures protocols. To better characterize the complex translocation in case 38, double-color FISH on nuclei was also performed with BAC clones, as described in Bonaglia et al. (2009).
Parent of origin determination
We genotyped family trios in 46 subjects (Fig. 1, Online Resource 1: Table S2) either by amplification with primers labeled with fluorescent probes (ABI 5-Fam, Hex and Tet) followed by analysis on a ABI 310 Genetic Analyzer (Applied Biosystems, Monza, Italy) or SNP array (Human Omni Express Exome ILLUMINA v1.2). In the remaining subjects (cases 23, 24, 33, 38, 43, and 47), parental DNA was not available.

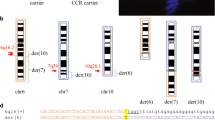
Schematic representation of the events leading to the origin of de novo unbalanced translocations. First box: Parental origin of the deletion and duplication leading to the four classes of de novo translocations (34 of class A, 6 of class B, 5 of class C, and 1 of class D) in which STS/SNPs could be investigated. (*) reported cases re-analyzed in this study; (§) amniotic fluid; (°) chorionic villi and (✓) mosaic cases; del, dup: parental origin of the deletion and duplication (paternal blue, maternal pink; two pink squares, light and bright, indicate two different maternal haplotypes within the duplication region; ❖: UPD excluded by SNP genotyping; ⁄ : not informative. Second box: (A) Trisomy rescue Mat-MI or Mat-MII non-disjunction. The segregation of the supernumerary chromosome within the micronucleus, where shattering occurs, followed by the retrieval of its telomeric portion by a different chromosome. The final translocation may arise by postzygotic NHEJ/alt-NHEJ joining the left telomeric portion of the supernumerary chromosome to a recipient chromosome (in grey) that loses its terminal portion. Although the parental origin of the recipient chromosome might be random (maternal or paternal: M/P in the figure), in most cases, it is maternal and only a few rearrangements have a biparental origin of the deletion and the duplication. Third box: (B) Deletion-induced repair. A chromosome deletion (upper) may lead to the creation of an unstable intermediate dicentric (III) whose asymmetric breakage results into an inv-dup del and a simply deleted chromosome. Both of them may be stabilized by the telomere capture from the opposite end of the broken chromosome (1) or from another chromosome whose parental origin is random (2, 3). Notably, the inv-dup del(8p) cases (42, 43, and 44) originate at Mat-MI by NAHR and not by a previous deletion. In any case, although the parental origin of the telomere-donor chromosome should be random, in most cases it has the same origin of the recipient chromosome and only a few rearrangements have a biparental origin of the deletion and the duplication
Sequencing translocation/inversion junctions
Breakpoint junctions sequencing could be done in the 26 subjects (Online Resource 1: Table S1 Online Resource 2: Figure S1), where DNA was available for quantitative PCR (qPCR), long-range PCR and mate-pair whole genome sequencing.
On the basis of the coordinates from the aCGH experiments, primer pairs for qPCR were selected within non-repeated portions of the chromosome using the Primer Express software (Applied Biosystems, Foster City, CA, USA) and performed on an ABI PRISM 7900HT sequence detection system (Applied Biosystems).
Long-range PCR primers were performed with JumpStart Red ACCUTaq LA DNA polymerase (Sigma) and the following protocol: 30 s at 96 °C, 35 cycles of 15 s at 94 °C/20 s at 58 °C/15 min at 68 °C, 15 min final elongation time. Sequencing reactions were performed with a Big Dye Terminator Cycle Sequencing kit 3.1 (Life Technologies) and run on an ABI Prism 3500AV Genetic Analyzer.
Mate-Pair Whole Genome Sequencing
Genome-wide mate-pair sequencing was used to narrow the breakpoint regions in cases 2–5, 9, 10, 12, 22, 43, 49, 50, and 52 (Online Resource 1: Table S1). Mate-pair libraries were constructed using 1 µg of DNA following the instruction for a gel-free preparation of 2 kb effective insert size library (Mate-Pair Library v2, Illumina). Final libraries were quantified using Pico Green (Quant-iT, Invitrogen). Ten different indexed libraries were pooled together in a single flowcell and sequenced on a NextSeq (Illumina, San Diego, CA, USA) (2 × 75 bp). Raw sequence reads were trimmed with cutadapt (Martin 2011) to remove the adaptors. The remaining sequences passing Illumina Chastity filtering (> 0.6) were mapped to the human reference genome (GRCh37/hg19) using Burrows Wheeler Aligner (BWA). Reads not aligning uniquely were removed and discordant paired reads with unexpected orientation or alignment to different chromosomes were extracted using SVDetect (http://svdetect.sourceforge.net/) and Delly (http://www.korbel.embl.de/software.html). Usually, the predicted SVs are compared with > 200 in-house mate-pair data sets to identify sample-specific SVs and exclude non-unique rearrangements, but for unbalanced translocations/inversions the estimated region detected by aCGH was also used to find the approximate breakpoint regions. By uploading the BAM files (containing all the reads, both concordant and discordant) into Integrative Genomics Viewer (IGV) (Broad Institute, Cambridge, MA, USA), we could visualize the genomic imbalances related to the unbalanced translocation/inversion using the depth of coverage of the aligned mate-pair, together with the cluster of reads that indicated the breakpoint regions. All cases were compared with at least two controls to identify potential deletions or duplications. Depending on the insert size and, in a few cases, split reads, mate-pair sequencing narrowed the breakpoint regions to 3 kb–1 bp. Mate-pair sequencing also identified genes that were truncated by the breakpoints. Sanger sequencing confirmed all breakpoints except for case 49, where further DNA was unavailable.
Fusion gene prediction
For breakpoints that interrupted coding genes oriented in the same direction, we predicted the reading frame of possible fusion genes using the ExPASy’s Translation Tool (http://web.expasy.org/translate/) and EMBOSS Transeq (https://www.ebi.ac.uk/Tools/st/emboss_transeq/) for comparison. We obtained the main gene transcript sequence in the Ensembl 88 database to predict whether the reading frame was preserved following the rearrangement. We predicted the generation of novel fusion protein motifs using ScanProsite (http://prosite.expasy.org/scanprosite/), as previously reported (Newman et al. 2015; Weckselblatt et al. 2015). We evaluated any possible fusion transcripts already collected from various public resources by ChimerDB 2.0 (Kim et al. 2010).
Results
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Cytogenetic investigations
Array CGH was used to size the imbalances and define the boundaries of each deletion and duplication segment. From this, we classified the group of unbalanced translocations/inversions into four structurally distinct subclasses (Online Resource 1: Table S1):
Class A: Simple unbalanced translocations (n = 37) with monosomy and trisomy of distal segments of two different chromosomes.
Class B: Inv-dup del translocations (n = 8) characterized by three breakpoints, two associated with the formation of the inv-dup del recipient chromosome and the third one with the telomeric portion of another donor chromosome (Online Resource 2: Figures S2, S3, S4, S5).
Class C: Simple unbalanced inversions (n = 6) presenting deletion and duplication of the distal parts of opposite arms of the same chromosome (Online Resource 2: Figure S6).
Class D: Inv-dup del unbalanced inversion (n = 1) showing an inv-dup del chromosome ending with the distal portion of the opposite arm of the same chromosome (Online Resource 2: Figure S7).
A normal copy region interposed between the distal deletion and the proximal duplication characterized all the inv-dup del recipient chromosomes (classes B and D), as clearly demonstrated by aCGH or by breakpoint sequencing analysis. Dual-color FISH analysis of case 38 (Online Resource 2: Figure S2) and breakpoint cloning of cases 39, 40, and 52 (Online Resource 2: Figures S3, S4, S7) demonstrated that the duplicated region of inv-dup del was in an inverted orientation.
Chromosome mosaicism
In four subjects, three of class A (cases 24–25 and 37), and one of class B (case 45), the translocation occurred as a mosaic (Online Resource e 1: Table S1).
Case 24 (class A, Online Resource 2: Figure S8). In this infertile man, conventional karyotype in 100 blood metaphases and FISH analysis with whole-chromosome painting and locus-specific probes revealed the presence of three cell lines: a minor one with trisomy 9 (2%), a second with an unbalanced translocation t(9;14)(q11;p11) (20%) and the prevalent one with a normal karyotype (78%).
Case 25 (class A, Online Resource 2: Figure S9). The karyotype of this female child (case 8 in Vetro et al. 2018), affected by severe neurodevelopmental delay, showed a simple unbalanced translocation der(2)t(2;14)(q37.2;q24.3) in 80% of peripheral blood cells and an apparently normal karyotype in the remaining ones. Array CGH and subtelomeric FISH analysis performed in both blood and fibroblasts revealed a terminal 2q deletion in 100% of the cells, while the 14q duplication was detected in about 70% of blood cells and 30% of fibroblasts. In addition, pan-telomeric sequences were present on both the der(2) and the 2q deleted chromosome in 100% of fibroblast nuclei (Online Resource 2: Figure S10).
Case 37 (class A, Online Resource 2: Figure S11). The blood karyotype of this female child, with severe neurodevelopmental delay, hypotonia, and thin corpus callosum, showed a mosaic with an unbalanced translocation der(11)t(7;11)(q33;q25) in 50% of the metaphases, confirmed by FISH investigation with subtelomeric probes. In addition, pan-telomeric sequences were present on both the normal and the der(11). Array CGH showed that the duplicated 7q region had a log2 ratio of + 0.4 for 19.5 Mb at 7q34–36.3 and of + 0.6 for 3.5 Mb at 7q33-q34. SNP array along the 7q duplicated region consistently showed that the duplication was of maternal origin, while a biparental origin was evident along the non-duplicated portion of chromosome 7 (Online Resource 1: Table S2).
Case 45 (class B, Online Resource 2: Figure S12). In the chorionic villi from the 12 weeks’ gestation fetus with increased nuchal translucency and severe IUGR, a 5p- was detected in all of the 30 analyzed metaphases from the direct slide culture, whereas a 5p + was found in the cultured cell line. Array CGH demonstrated a 17.7 Mb distal 5p deletion and a 5.6 Mb 5p duplication in direct villi, characterized by two different log2 ratios of + 0.3 and + 0.6, respectively (see figure S12), whereas a homogeneous log2 ratio of 0.6 was found in the cultured cell line. The deletion and duplication were separated by a normal copy region of 750 kb. A 61.3 Mb distal duplication of chromosome 3q was revealed in both direct and cultured villi, with an average log2 ratio of + 0.3 and + 0.6, respectively. Array CGH + SNP performed in the trio showed a paternal origin of the 5p rearrangement and a maternal origin of the 3q duplication, further confirmed by microsatellite analysis (Fig. 1, Online Resource 1: Table S2).
Breakpoint junctions sequencing
Improvements in resolution of translocation breakpoint intervals were obtained by qPCR and/or mate-pair whole genome sequencing in the 26 individuals with available DNA (Online Resource 1: Table S1). The translocation junctions were successfully cloned in 22 subjects, including 15 cases of class A (cases 2–3, 5–9, 12, 18, 22, 26–27, and 29–31), 5 cases of class B (cases 38–40, 43, and 44), 1 case of class C (case 50), and 1 case of class D (case 52) (Table 1, Online Resource 2: Figure S1). In four cases (15%), we have only been able to narrow the breakpoint regions to less than 3 kb because of the presence of GC-rich sequences, large repeats or cryptic complexity (cases 4, 10, and 28 of class A and case 51 of class C, Online Resource 1: Table S1).
Eight junctions (4 cases of class A, 3 cases of class B, and 1 case of class C) had 2–6 base pairs (bp) of microhomology (36%, 8/22) and four (3 cases of class A and 1 case of class B) had blunt ends (18%, 4/22) (Table 1). Insertions of 1–36 bp were present in 36% (8/22), of which 23% (5/22) originated from local sequences (templated insertions) (cases 3, 9, 18, 30, and 52, Online Resource 2: Figure S1). Notably, three templated local insertions were in direct orientation (cases 3, 9, and 30 Online Resource 2: Figure S1) and two were in inverted orientation with respect to the reference genome (case 18 and 52 Online Resource 2: Figure S1). The 36 bp templated insertion in case 8 (Online Resource 2: Figure S1) was identical to two mitochondrial piRNA sequences (piR-34804, piR-31490), similarly to what has been reported in balanced translocations (Willett-Brozick et al. 2001), and to two sequences from chromosome 1.
Finally, homologies of 885 bp and 1.7 kb with 93–94% sequence identity at both sides of the junction were present in 9% (cases 2 and 38: 2/22) (Online Resource 2: Figure S13). In case 2, the formation of the der(13)t(3;13) occurs at the edge of a 885 bp homology with 95% identity, spanning an L1HS retrotransposon on chromosome 13 and LIPA3 on chromosome 3. The L1HS contained an AluYg6 element, which is distributed along different chromosomes, including chromosome 13 (Styles and Brookfield 2007). In case 38, the translocation junction lies within LIPA2 on chromosome X and LIPA4 on chromosome 5, sharing a 94% identity for 1.7 kb (Online Resource 2: Figure S13). Alu–Alu recombination was predicted for 2 (9%) of the cases: in case 18 (class A), the breakpoints occurred between AluSx3 and AluY repeats sharing 82% identity, showing 5 bp templated inverted insertion; in case 40 (class B), the breakpoints occurred between two AluSx sharing 83% identity with a 4 bp michromology (Online Resource 1: Table S1).
Disrupted and fused genes at translocation junctions
Disrupted genes were present in about 58% of the rearrangements in every of the four classes, as detected by breakpoint cloning (19 cases) or aCGH breakpoint intervals (11 cases) (Online Resource 1: Table S1). In 22 cases (42%), no gene was disrupted at either chromosome breakpoints, while in 6 cases (11%), long non-coding RNAs (lncRNA) were disrupted: at both breakpoints in case 47, at the deleted chromosome in cases 11 and 43, and at the duplicated one in cases 10, 30, and 40 (Online Resource 1: Table S1).
Twenty-two cases carried a gene disruption at one breakpoint only, including three independent (cases 7, 9, and 26) truncations of MYO16 (MIM: 615479) at 13q13.33, although scattered along the entire gene (Online Resource 1: Table S1). In two out of the eight cases, where the gene was disrupted at both breakpoints, an out-of-frame fusion transcript was created (cases 22 and 27, Online Resource 1: Table S1; Online Resource 2: Figures S13, S14). In case 22, the translocation between chromosomes 8 and 9 joins CSMD1 exon 1 (MIM: 608397) and GLIS3 exons 5–10 (MIM: 610192). The resulting CSMD1-GLIS3 fusion protein apparently does not contain any predicted functional domain, as assessed by ExPASy’s Translation Tool, EMBOSS Transeq, ScanProsite and ChimerDB 2.0 (Online Resource 2: Figure S14) (Kim et al. 2010).
In case 27, the translocation between chromosomes 1 and 3 joins AKT3 exons 12–13 (MIM: 611223) to ATP13A3 exon 31 (MIM: 610232). The resulting AKT3–ATP13A3 fusion protein maintains the ATP13A3-specific E1–E2 ATPases phosphorylation site in position 498–504, while it has lost the AGC-kinase C-terminal domain of AKT3. On the other hand, consecutive phosphothreonine and N-myristoylation sites are juxtaposed at position 1171–1173 and 1174–1179, respectively, with possible gain-of-function effects (Online Resource 2: Figure S15). However, RNA for this case was not available, so we could not confirm the presence of the fusion transcript.
Parental origin analysis
We investigated the parental origin of both deletions and duplications in 46 subjects and determined the origin of both imbalances in 35 of them (74%) and of one imbalance only (deletion or duplication) in 11 (26%) (Fig. 1; Online Resource 1: Table S2).
Among the 35 fully informative cases, 19 (54%) were of maternal origin, 10 (29%) of paternal origin, and 6 (17%) showed a biparental origin (paternal deletion/maternal duplication or vice versa). The 11 partly informative cases included 5 cases (11%) of maternal and 7 (15%) of paternal origin: in 4 cases only the deleted segment was informative (maternal in cases 7, 14, and 35 and paternal in case 41), while in 8, only the duplicated segment was informative (paternal in cases 16, 20, 21, and 36 and maternal in cases 32, 51, and 37). (Fig. 1).
Among simple unbalanced translocations (class A), the origin of the duplicated segment was maternal in 20 out 37 cases (54%) (cases 1–2, 4–6, 9–13, 15, 17, 19, 22, 26, 28, 30, 32, 34, and 37). In eight of these, the duplication region showed consistently three alleles, with two maternal and one paternal allele (cases 1, 2, 9, 11, 15, 19, 28, and 32). No cases with two different paternal alleles within the duplication region were detected.
The origin of the deleted segments was maternal in 20 cases (1, 2, 4–7, 9, 11, 13–15, 17, 19, 22, 26–28, 31, 34, and 35) and paternal in eight (3, 8, 10, 12, 18, 25, 29, and 30). In five cases (cases 10, 12, 27, 30, and 31), the duplication and the deletion were of biparental origin (Fig. 1, Online Resource 1: Table S2).
Four out of the five simple unbalanced inversions (class C), we could analyze presented a paternal origin of both their deleted and duplicated segments (Fig. 1, Online Resource 1: Table S2). In the six cases of inv-dup del translocations (class B), the rearrangement was of maternal origin in two cases (cases 42 and 44), paternal in three cases (cases 39–41), and of biparental origin in case 45, where the inv-dup del was of paternal origin and the translocated segment of maternal origin (Fig. 1, Online Resource 1: Table S2). The only case of inv-dup del unbalanced inversion (class D) showed a paternal origin of all imbalances.
Discussion
Simple unbalanced translocations and inversions (classes A and C): mechanisms of origin
The straightforward result of this study is that in 20 out of 37 of the de novo unbalanced translocations, the duplication has a maternal origin (Fig. 1, Online Resource 1: Table S2). Since the investigation of the parental origin was not informative or not possible in six cases (cases 7, 14, 23, 24, 33, and 35), the de novo unbalanced translocations where the duplicated region is of maternal origin represent 64,5% (20/31) of the fully informative cases. Combining these data with the finding that in eight of these cases (cases 1, 2, 9, 11, 15, 19, 28, and 32) three alleles (Fig. 1, Online Resource 1: Table S2), two maternal and one paternal, were present at least in part of the duplication region, the emerging scenario is that in no less than 25% (8 out 31), the primary driver for the occurrence of de novo unbalanced translocations is a maternal meiotic non-disjunction, followed by a partial trisomy rescue of the supernumerary maternal chromosome (Fig. 1a). The most likely explanation for the presence of three alleles within the duplicated region, two maternal and one paternal, would be non-disjunction at the maternal meiosis I (mat-MI) (Fig. 1A.I). Alternatively, a meiotic recombination proximal to or at the translocation breakpoint must have preceded a non-disjunction in maternal meiosis II (mat-MII). In contrast, in the 12 cases (4–6, 10, 12, 13, 17, 22, 26, 30, 34, and 37) with the same maternal duplicated haplotype other mechanisms cannot be excluded (Fig. 1b), although they could also result either from mat-MI non-disjunction (Fig. 1A.I), after a telomeric crossing-over, according to the preferential occurrence of telomeric crossovers demonstrated for some non-disjoined chromosome (Oliver et al. 2014) or a maternal MII (mat-MII) non-disjunction (Fig. 1A.II), as reported in a number of cases of trisomy rescue (reviewed in Chantot-Bastaraud et al. 2017).
It is well known that anaphase lagging of the supernumerary chromosome followed by its trapping within a micronucleus is at the basis of many chromothripsis events (Zhang et al. 2015; Ly et al. 2017; Zhu et al. 2018). As a consequence, in one daughter cell, the supernumerary chromosome is eliminated, while in the other one, the shattering of the micronuclear genomic material, followed by the re-embedding of all or some fragments inside the main nucleus may result in a massive reorganization of the original chromosome that either maintains most of its material although reorganized in a new order, or loses some of its portions (Fig. 1a), which portions of the original supernumerary chromosome are lost and which are preserved could depend on stochastic events, on the proneness of the broken ends to integrate with each other or with other parts of the genome, and/or on the subsequent selection of cells that can survive and multiply in the presence of minimal segmental imbalances. In theory, centric fragments may be preserved as supernumerary marker chromosomes by their circularization with formation of ring chromosomes, thus overcoming the absence of telomeric sequence at both ends. The preservation of supernumerary interstitial acentric fragments requires the formation of a neocentromere as well or, alternatively, the capture of the fragment by another chromosome with the formation of a de novo insertional translocation. Indeed, Kato et al. (Kato et al. 2017) recently reported an instance of de novo interstitial translocation derived by chromothripsis of a supernumerary chromosome present in a trisomic zygote. Finally, the preservation of supernumerary fragments equipped with a telomere at one end requires either a telomere capture, e.g., a translocated supernumerary marker chromosome as reported in Vetro et al. (2012) (cases 2 and 3) or its capture by another chromosome, the recipient one, that loses its distal portion thus forming a 46-chromosome cell with an unbalanced de novo translocation, as described here. Indeed, case 6 with the duplication of the same maternal haplotype (Fig. 1, Online Resource 1: Table S2) illustrates the occurrence of a chromothripsis event (Fig. 1a), as shown by the presence of two non-contiguous duplicated regions separated by ~ 1.3 Mb of which the interstitial one has an inverted orientation (Online Resource 2: Figure S15). Chromothripsis events have been observed in a number of cases of de novo unbalanced translocations (Weckselblatt et al. 2015), and most of them have been reported to be of paternal origin and to involve more than two chromosomes (Marcozzi et al. 2018). In contrast, the unbalanced translocation in our case 6 was of maternal origin and indeed de novo, in agreement with FISH investigations of parental metaphases. Moreover, at least as judged by the high-resolution (1M) aCGH and FISH investigations, it involved just two chromosomes, while a more complex rearrangement was excluded. Therefore, it is tempting to speculate that the zygote or the early embryo was trisomic for chromosome 8 because of either a meiotic or postzygotic maternal non-disjunction. The shattering of the supernumerary chromosome 8 was followed by the recovery of only two non-contiguous portions, including the telomeric one which, when already stitched back together, were acquired by the recipient 18q (Fig. 1a).
The model of formation of the de novo unbalanced translocations, starting from a trisomic zygote, also fits with case 24, although we could not perform any supporting molecular investigation. However, the presence of a residual cell line trisomic for chromosome 9, together with a main one with a normal karyotype and a third one with the unbalanced t(14;9) translocation suggests how, starting from a trisomic zygote, various cell lines may be formed, some with the total loss of the supernumerary chromosome and others with the loss of a part of it (9p) and its residual portion (the entire 9q) captured by another chromosome. This type of three cell lines mosaic have rarely been documented (Phillips et al. 1997), but in prenatal diagnosis mosaics with one normal cell line and a second one with a de novo unbalanced translocation are relatively common and an increased maternal age can be detected in most of them (Kovaleva and Cotter 2017; Van Opstal et al. 2018).
An independent support for the hypothesis that maternal non-disjunction is a main trigger for de novo unbalanced translocations, is an increased maternal age. Indeed, in our eight cases (cases 1, 2, 9, 11, 15, 19, 28, and 32), where the duplication was without any doubt linked to a maternal meiotic non-disjunction (two different maternal alleles within the duplication region), an increase in the average maternal age was documented (34.75 years, Online Resource 1: Table S1), taking into account that the mean maternal age in Italy in 2016 was of 31.8 years (ISTAT, https://www.istat.it/). An increase (33.5 years) was also recognized in the 12 cases (cases 4, 5, 6, 10, 12, 13, 17, 22, 26, 30, 34, and 37) with duplication of the same maternal allele, compatible with a maternal non-disjunction as an initiating event. Even in the cases, where the duplication was paternal, an increase, although much more limited, was shown in the mean maternal age (32.6 years), possibly indicating that mechanisms other than maternal non-disjunction may play a role in the formation of these unbalanced translocations. Indeed, in the four cases (3, 8, 29, and 36) analyzed by SNP array no maternal hetero-isodisomy was present in the non-duplicated portion of the homologous chromosomes (Online Resource 1: Table S2), as one would have expected for a trisomic zygote in which the supernumerary paternal chromosome had undergone chromothripsis with the rescue of only the telomeric portion. Thus, taking also into consideration the negligible frequency of trisomy originating from missegregation in paternal meiosis (Nagaoka et al. 2012), the occurrence of unbalanced translocations ignited by partial trisomic rescue of a paternal-of-origin trisomic zygote appears highly unlikely. Instead other mechanisms, such as an inherited or postzygotic double-strand paternal breakage requiring to be repaired by telomere capture, seem more plausible (Fig. 1b). Indeed, case 25 which is mosaic for two cell lines, present in blood and in fibroblasts, with a terminal 2q deletion and a derivative chromosome der(2)t(2q;14q) of paternal origin (Online Resource 2:Figure S9), fits with this hypothesis. Thus, in the presence of a terminal deletion, different repair mechanisms may occur at different times in different cells of the early embryo, eventually leading to a mosaic condition. In this model, the deleted chromosome initiates the translocation event, acting as the recipient one, while the duplicated is the donor, operating as injury repairing (Fig. 1b). However, it is not possible to discern whether the deletion is actually the first event leading to the translocation or is instead secondary to the formation of a dicentric chromosome (Fig. 1BIII and IV) which, as a result of its asymmetric rupture, generates an inv-dup del chromosome and a simply deleted one, the latter being then repaired by telomere capture (Fig. 1b). It is possible that the persistence of the dicentric beyond the first embryonic divisions results in breakage of different size in the different cells, again repaired either by telomerase or telomere capture.
Remarkably, whatever the mechanism, a high prevalence of cases, where both imbalances have the same parental origin was detected (Fig. 1, Online Resource 1: Table S2), despite a biparental origin of deletion and duplication would be expected in half of the cases, according to a postzygotic final event. The demonstration of a distinct compartmentalization of the two sets of parental chromosomes in mice embryos until the 8-cell stage (Du et al. 2017) may provide an explanation. Since this period coincides with very high chromosomal instability, (McCoy et al. 2017), it is tempting to speculate that a mechanism such as telomere capture, necessary for the stabilization of an earlier structural abnormality, occurs on the same parental chromosomal set of the original anomaly. In contrast, further later readjustments would be expected to occur randomly, involving chromosomes of both parental origin, which would lead to an unbalanced translocated chromosome of biparental origin as indeed we found in five cases (cases 10, 12, 27, 30, and 31; Fig. 1, Online Resource 1: Table S2).
As for class C rearrangements, we did not find any peculiarity to the junction breakpoints that could clarify if their stabilization by telomere capture of the chromosomal portion opposite to the deleted one is due to a different mechanism compared to the cases in which the capture of the telomere is dependent on another chromosome. However, this analysis could be done in two cases only. The finding that in 4 out of the 5 cases both deletion and duplication were of paternal origin suggests that they derive either by an original deletion or the breakage of a dicentric chromosome.
Complex unbalanced translocations and inversions (classes B and D)
There is general agreement that these rearrangements result by an intermediate dicentric, after its asymmetric breakage leading to an inv-dup del and a simply deleted chromosome (Fig. 1B.III). The acquirement of a stabilizing telomeric sequence may occur by a range of modalities: telomerase-mediated addition of de novo TTAGGG sequences, telomere capture of the distal portion of another chromosome (Yu and Graf 2010) or of the opposite portion of the same chromosome (Buysse et al. 2009; Fan and Siu et al. 2001), or even formation of a ring chromosome (Rossi et al. 2008). The two translocated inv-dup del(8p) cases we could genotype (cases 42 and 44) originated, as expected (Giglio et al. 2001), by NAHR at mat-MI, as demonstrated by the presence of two maternal and one paternal alleles in the 8p duplication region (Online Resource 1: Table S2). This recurrent abnormal recombination is mediated by highly identical segmental duplications located at 8p23 within a normal and an inverted chromosome 8 (Giglio et al. 2001). In the zygote, the resulting dicentric chromosome will likely undergo different breakage events in different cells, sometime leading to a mosaic condition with a del(8p) cell line and a second one with the inv-dup del(8p) (Hand et al. 2010), or in exceptional cases even a third one with the inv-dup del(8p) ending with the distal region of another chromosome (Pramparo 2004). These mosaics are more frequently detected in prenatal diagnosis, whereas in postnatal life a single cell line with the inv-dup del(8p), either translocated or not, is the most frequent finding at least in blood. In our three translocated cases (cases 42, 43, and 44), telomeres were donated by 6q, 17p, and Xq, respectively, all of maternal origin, and with the same duplication haplotype of the maternal chromosomes 6, 17, and X, as expected for a stabilization event that occurred postzygotically. In the remaining three cases (cases 39, 40, and 45), which, as all the inv-dup del rearrangements not-involving 8p, are non-recurrent, the translocated inv-dup was of paternal origin with identical paternal alleles in the duplication region, whereas telomere capture was provided by a paternal-derived chromosome in two cases (cases 39 and 49, Online Resource 2: Figures S3, S4) and a maternal one in the third case (case 45), a fetus in which the translocated chromosome t(inv-dup5p;3q) was in mosaic with a cell line containing a non-translocated inv-dup del(5p) (Online Resource 2: Figure S12). Notably, the 5p duplication is of different size in the two cell lines (Online Resource 1: Table S1), indeed showing that the original dicentric chromosome presumably present in the zygote or in the very early embryo (Fig. 1b), underwent different breakages in the different cells, beyond the cleavage-stage embryo.
Identical duplicated alleles have been reported bona fide in all the inv-dup del rearrangements not-involving 8p, thus indicating an intrachromosomal origin of these rearrangements (Hermetz et al. 2014). Accordingly, these inv-dup translocated rearrangements appear to be the final result of an initial mitotic event, possibly in the early embryo (Voet 2011), such as a double-strand break followed by intrastrand pairing at relatively close sites of inverted homologous sequences, leading to the generation of the dicentric chromosome with an interposed normal copy region (Hermetz et al. 2014; Rowe et al. 2009). As already mentioned, the reciprocal product of the inv-dup del, namely, a deleted chromosome, may be repaired and stabilized by the capture of the distal portion of a non-homologous chromosome, or the opposite arm of the same chromosome, resulting in a simple unbalanced translocation or inversion, respectively, as we propose for some of the rearrangements listed in classes A and C (Fig. 1b).
Breakpoint junctions
The sequencing of breakpoint translocation junction could be done in 26 cases (Online Resource 1: Table S1, Online Resource 2: Figure S1) although in 4 cases cryptic complexity (cases 4, 10, 28, and 51) impaired a fine sequencing. Alternative non-homologous end joining (alt-NHEJ) or microhomology-mediated break-induced replication (MMBIR) features such as microhomologies and templated or non-templated small insertions were detected in 16 cases (cases 3, 5, 6, 7, 8, 9, 12, 18, 22, 27, 30, 39, 40, 44, 50, and 52) (Online Resource 2: Figure S1). Remarkably, although in cases 18 and 40 the rearrangement occurred within Alu sequences, the presence of 5 bp short templated insertion and 4 bp microhomology, respectively, showed that the rearrangement did not accomplish a NAHR mediated mechanism but rather an alt-NHEJ or MMBIR. Canonical NHEJ (c-NHEJ, blunt ends junctions) occurred in four cases (cases 26, 29, 31, and 43). In cases 2 and 38, the breakpoint junctions, were at the edge of homologous retrotransposon (Online Resource 2: Figure S13). This finding suggests that in a minority of cases, repair mechanisms such as telomere capture are made easier by these sequences. Although Robberecht et al. found that most of their de novo unbalanced translocations were mediated by NAHR between LINEs, HERVs, or segmental duplications (Robberecht et al. 2013), we can exclude a NAHR event at least in case 2, where the presence of two duplicated maternal haplotypes, one translocated to the recipient chromosome, clearly indicates a mat-MI non-disjunction (Fig. 1, Online Resource 1: Table S2), thus making it impossible to attribute the translocation to a meiotic event. Indeed, LINE-1-mediated retrotransposition events have been shown to occur in somatic cells of the early human embryo, in human embryonic stem cells, and at least in mice mainly in embryogenesis (for a review Kazazian and Moran 2017), supporting that in our cases, the formation of the translocation was a postzygotic event as well.
Combined, these features indicate that repair-based mechanisms (c-NHEJ and alt-NHEJ) and replicative repair mechanisms (MMBIR) are responsible for the junction of the donor chromosome with the recipient one, or the opposite portion of the same chromosome (Fig. 1).
Genotype–phenotype relationship
Although genotype–phenotype correlations in large unbalanced rearrangements, such as those reported in our study, are in general rather coarse, the data we report add some further details also in this context.
The strong evidence that some unbalanced translocations originate from a zygote with a supernumerary chromosome that undergoes a chromothriptic event, suggests that the duplication region, although apparently composed only of the distal region of the chromothriptic chromosome, may contain fragments from other portions, as it is the case of our case 6 and as shown by Weckselblatt et al. (2015). In these cases, if the size of the shattered pieces is below the one detectable by array CGH, the risk is to attribute the abnormal phenotype only to the genes that appear duplicated or deleted, whereas further morbidity may be due to abnormal gene expression generated by TAD disruption (Fukami et al. 2017).
Furthermore, in unbalanced translocations originating from partial trisomy rescue, if the chromotripsis would occur on the supernumerary chromosome of paternal origin, the following maternal hetero/isodisomy for the remaining two chromosomes might generate further pathogenicity (Niida et al. 2018) either by reducing to homozygous state disease-causing variants present in the mother at heterozygous state, or due to the presence of imprinted genes that are expressed by the paternal allele only.
As a final consideration, it is tempting to speculate that the choice of which telomere is captured for the stabilization of the broken chromosome, may not be random but influenced by the deregulation of the original TADs (Topologically Associated Domains), in turn creating a favourable substrate for specific inter-chromosomal contacts and spatial 3D chromatin compartmentalization (Dekker and Mirny et al. 2016).
Conclusions
Our findings show that de novo unbalanced rearrangements has a complex history. In some of them (simple translocations) the first event is meiotic non-disjunction followed by postzygotic chromothripsis of the supernumerary chromosome with preservation of at least its distal portion. The latter is rescued by transposition to a distal region of an acceptor chromosome with simultaneous deletion of its distal part of the acceptor chromosome. Remarkably, this sequence of events parallels the one demonstrated in complex insertions (Gu et al. 2016), indicating that the formation of an insertion instead of an unbalanced translocation depends on the absence or the presence of a telomeric region in the translocated donor segment.
The finding that in the four simple translocations in which the duplicated segment was of paternal origin (cases 3, 8, 18, and 25) with absence of maternal hetero/isodisomy (Online Resource 1: Table S2), indicates that other mechanisms beside meiotic non-disjunction and partial trisomic rescue adds to the complex history of de novo unbalanced translocations. A probable hypothesis is that in these cases the first start is a mirror dicentric chromosome whose asymmetric breakage generates an inv-dup del and a simple deleted chromosome, triggering for both of them the usual mechanism of telomere capture by either repair or replicative repair. In these cases, the formation of the dicentric, with the exception of the mirror chromosome 8p23, is likely to occur in the early embryo characterized by high genome instability. We cannot even exclude that the original event was a simple deletion either repaired by telomere capture or triggering a bridge fusion breakage cycle.
Our data combined with those reported by Weckselblatt et al. (2015), impute repair-based mechanisms as those mainly responsible for de novo non-recurrent unbalanced translocations. In contrast, those mediated by NAHR between (retro)transposable elements and especially long interspersed elements (Robberecht et al. 2013), would play a minor role. Although replicative-based mechanisms, characterized by microhomologies and templated insertions, are detected in a number of breakpoint junctions of both complex chromosomal insertions (Gu et al. 2016) and non-recurrent balanced translocations (Nilsson et al. 2017), further investigation is still necessary to clarify any possible role of retrotransposable elements.
Whatever the mechanism, the puzzling observation is that in most cases of unbalanced translocations, both the deletion and the duplication have the same parental origin, possibly indicating that the genomic damage occurred and was repaired in the very early embryonic stages when the maternal and paternal genomes are compartmentalized as observed in mice preimplantation embryos (Du et al. 2017) and partly in humans (Wu et al. 2018). If this hypothesis holds true, the cases with imbalances of biparental origin should reflect events of breakage–fusion–bridge cycles, where the selection of the final cell line(s) occurs later.
The final and important implication of our data is that there in no risk of recurrence in the following pregnancies for any of the de novo unbalanced translocations discussed here.
References
Bonaglia MC, Giorda R, Massagli A, Galluzzi R, Ciccone R, Zuffardi O (2009) A familial inverted duplication/deletion of 2p25.1-25.3 provides new clues on the genesis of inverted duplications. Eur J Hum Genet 17:179–186. https://doi.org/10.1038/ejhg.2008.160
Buysse K, Antonacci F, Callewaert B, Loeys B, Fränkel U, Siu V, Mortier G, Speleman F, Menten B (2009) Unusual 8p inverted duplication deletion with telomere capture from 8q. Eur J Med Genet 52:31–36. https://doi.org/10.1016/j.ejmg.2008.10.007
Campbell IM, Shaw CA, Stankiewicz P, Lupski JR (2015) Somatic mosaicism: implications for disease and transmission genetics. Trends Genet 31:382–392. https://doi.org/10.1016/j.tig.2015.03.013
Chang YW, Wang PH, Li WH, Chen LC, Chang CM, Sung PL, Yang MJ, Cheng LY, Lai YL, Cheng YY, Yeh CC, Chang WH, Wang SY, Chen SR, Yen MS, Chao KC (2013) Balanced and unbalanced reciprocal translocation: an overview of a 30-year experience in a single tertiary medical center in Taiwan. J Chin Med Assoc 76:153–157. https://doi.org/10.1016/j.jcma.2012.11.002
Chantot-Bastaraud S, Stratmann S, Brioude F, Begemann M, Elbracht M, Graul-Neumann L, Harbison M, Netchine I, Eggermann T (2017) Formation of upd(7)mat by trisomic rescue: SNP array typing provides new insights in chromosomal nondisjunction. Mol Cytogenet 25:28. https://doi.org/10.1186/s13039-017-0329-1
Dekker J, Mirny L (2016) The 3D genome as moderator of chromosomal communication. Cell 10:1110–1121. https://doi.org/10.1016/j.cell.2016.02.007
Du Z, Zheng H, Huang B, Ma R, Wu J, Zhang X, He J, Xiang Y, Wang Q, Li Y, Ma J, Zhang X, Zhang K, Wang Y, Zhang MQ, Gao J, Dixon JR, Wang X, Zeng J, Xie W (2017) Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 12(547):232–235. https://doi.org/10.1038/nature23263
Eggermann T, Engels H, Heidrich-Kaul C, Moderau I, Schwanitz G (1997) Molecular investigation of the parental origin of a de novo unbalanced translocation 13/18. Hum Genet 99:521–522
Fan YS, Siu VM (2001) Molecular cytogenetic characterization of a derivative chromosome 8 with an inverted duplication of 8p21.3–>p23.3 and a rearranged duplication of 8q24.13–>qter. Am J Med Genet 15:102:266–271
Fukami M, Shima H, Suzuki E, Ogata T, Matsubara K, Kamimaki T (2017) Catastrophic cellular events leading to complex chromosomal rearrangements in the germline. Clin Genet 91:653–660. https://doi.org/10.1111/cge.12928
Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O (2001) Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 68(4):874–883. https://doi.org/10.1086/319506
Gijsbers AC, Dauwerse JG, Bosch CA, Boon EM, van den Ende W, Kant SG, Hansson KM, Breuning MH, Bakker E, Ruivenkamp CA (2011) Three new cases with a mosaicism involving a normal cell line and a cryptic unbalanced autosomal reciprocal translocation. Eur J Med Genet 54:e409–e412. https://doi.org/10.1016/j.ejmg.2011.05.002
Giorda R, Bonaglia MC, Milani G, Baroncini A, Spada F, Beri S, Menozzi G, Rusconi M, Zuffardi O (2008) Molecular and cytogenetic analysis of the spreading of X inactivation in a girl with microcephaly, mild dysmorphic features and t(X;5)(q22.1;q31.1). Eur J Hum Genet 16(8):897–905. https://doi.org/10.1038/ejhg.2008.28
Gu S, Szafranski P, Akdemir ZC, Yuan B, Cooper ML, Magriñá MA, Bacino CA, Lalani SR, Breman AM, Smith JL, Patel A, Song RH, Bi W, Cheung SW, Carvalho CM, Stankiewicz P, Lupski JR (2016) Mechanisms for complex chromosomal insertions. PLoS Genet 12:e1006446. https://doi.org/10.1371/journal.pgen.1006446
Hand M, Gray C, Glew G, Tsuchiya KD (2010) Mild phenotype in a patient with mosaic del(8p)/inv dup del(8p). Am J Med Genet A 152A:2827–2831. https://doi.org/10.1002/ajmg.a.33669
Hermetz KE, Newman S, Conneely KN, Martin CL, Ballif BC, Shaffer LG, Cody JD, Rudd MK (2014) Large inverted duplications in the human genome form via a fold-back mechanism. PLoS Genet 30:10:e1004139. https://doi.org/10.1371/journal.pgen.1004139
Kato T, Ouchi Y, Inagaki H, Makita Y, Mizuno S, Kajita M, Ikeda T, Takeuchi K, Kurahashi H (2017) Genomic characterization of chromosomal insertions: insights into the mechanisms underlying chromothripsis. Cytogenet Genome Res 153:1–9. https://doi.org/10.1159/000481586
Kazazian HH Jr, Moran JV (2017) Mobile DNA in health and disease. N Engl J Med 27(377):361–370. https://doi.org/10.1056/NEJMra1510092
Kim P, Yoon S, Kim N, Lee S, Ko M, Lee H, Kang H, Kim J, Lee S (2010) ChimerDB 2.0–a knowledgebase for fusion genes updated. Nucleic Acids Res 38(Database issue):D81–D85. https://doi.org/10.1093/nar/gkp982
Kostiner DR, Nguyen H, Cox VA, Cotter PD (2002) Stabilization of a terminal inversion duplication of 8p by telomere capture from 18q. Cytogenet Genome Res 98:9–12. https://doi.org/10.1159/000068536
Kovaleva NV, Cotter PD (2017) Mosaicism for structural non-centromeric autosomal rearrangement in prenatal diagnoses: evidence for sex-specific selection against chromosomal abnormalities. Mol Cytogenet 10:45. https://doi.org/10.1186/s13039-017-0346-0
Ly P, Teitz LS, Kim DH, Shoshani O, Skaletsky H, Fachinetti D, Page DC, Cleveland DW (2017) Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19:68–75. https://doi.org/10.1038/ncb3450
Marcozzi A, Pellestor F, Kloosterman WP (2018) The genomic characteristics and origin of chromothripsis. Methods Mol Biol 1769:3–19. https://doi.org/10.1007/978-1-4939-7780-2_1
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17:10–12 https://doi.org/10.14806/ej.17.1.200. http://journal.embnet.org/index.php/embnetjournal/article/view/200
McCoy RC (2017) Mosaicism in preimplantation human embryos: when chromosomal abnormalities are the norm. Trends Genet 33:448–463. https://doi.org/10.1016/j.tig.2017.04.001
Nagaoka SI, Hassold TJ, Hunt PA (2012) Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet 13(7):493–504. https://doi.org/10.1038/nrg3245
Newman S, Hermetz KE, Weckselblatt B, Rudd MK (2015) Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet 96:208–220. https://doi.org/10.1016/j.ajhg.2014.12.017
Niida Y, Ozaki M, Shimizu M, Ueno K, Tanaka T (2018) Classification of uniparental isodisomy patterns that cause autosomal recessive disorders: proposed mechanisms of different proportions and parental origin in each pattern. Cytogenet Genome Res 154:137–146. https://doi.org/10.1159/000488572
Nilsson D, Pettersson M, Gustavsson P, Förster A, Hofmeister W, Wincent J, Zachariadis V, Anderlid BM, Nordgren A, Mäkitie O, Wirta V, Käller M, Vezzi F, Lupski JR, Nordenskjöld M, Lundberg ES, Carvalho CMB, Lindstrand A (2017) Whole-genome sequencing of cytogenetically balanced chromosome translocations identifies potentially pathological gene disruptions and highlights the importance of microhomology in the mechanism of formation. Hum Mutat 38:180–192. https://doi.org/10.1002/humu.23146
Oliver TR, Middlebrooks CD, Tinker SW, Allen EG, Bean LJ, Begum F, Feingold E, Chowdhury R, Cheung V, Sherman SL (2014) An examination of the relationship between hotspots and recombination associated with chromosome 21 nondisjunction. PLoS One 9:e99560. https://doi.org/10.1371/journal.pone.0099560
Pham J, Shaw C, Pursley A, Hixson P, Sampath S, Roney E, Gambin T, Kang SH, Bi W, Lalani S, Bacino C, Lupski JR, Stankiewicz P, Patel A, Cheung SW (2014) Somatic mosaicism detected by exon-targeted, high-resolution aCGH in 10,362 consecutive cases. Eur J Hum Genet 22:969–978. https://doi.org/10.1038/ejhg.2013.285
Phillips OP, Velagaleti GV, Tharapel AT, Shulman LP (1997) Discordant direct and culture results following chorionic villus sampling and the diagnosis of a third cell line in the fetus. Prenat Diagn 17:170–172
Pramparo T, Giglio S, Gregato G, de Gregori M, Patricelli MG, Ciccone R, Scappaticci S, Mannino G, Lombardi C, Pirola B, Giorda R, Rocchi M, Zuffardi O (2004) Inverted duplications: how many of them are mosaic? Eur J Hum Genet 12:713–717
Rios JJ, Delgado MR (2015) Using whole-exome sequencing to identify variants inherited from mosaic parents. Eur J Hum Genet 23:547–550. https://doi.org/10.1038/ejhg.2014.125
Rivera H, Dominguez MG, Vasquez-Velasquez AI, Lurie IW (2013) De novo dup p/del q or dup q/del p rearranged chromosomes: review of 104 cases of a distinct chromosomal mutation. Cytogenet Genome Res 141:58–63. https://doi.org/10.1159/000351184
Robberrecht C, Voet T, Zamani Esteki M, Nowakowska BA, Vermeesch JR (2013) Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res 23:411–418. https://doi.org/10.1101/gr.145631.112
Rossi E, Riegel M, Messa J, Gimelli S, Maraschio P, Ciccone R, Stroppi M, Riva P, Perrotta CS, Mattina T, Memo L, Baumer A, Kucinskas V, Castellan C, Schinzel A, Zuffardi O (2008) Duplications in addition to terminal deletions are present in a proportion of ring chromosomes: clues to the mechanisms of formation. J Med Genet 45:147–154
Rowe LR, Lee JY, Rector L, Kaminsky EB, Brothman AR, Martin CL, South ST (2009) U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J Med Genet 46:694–702. https://doi.org/10.1136/jmg.2008.065052
Sakazume S, Ohashi H, Sasaki Y, Harada N, Nakanishi K, Sato H, Emi M, Endoh K, Sohma R, Kido Y, Nagai T, Kubota T (2012) Spread of X-chromosome inactivation into chromosome 15 is associated with Prader-Willi syndrome phenotype in a boy with a t(X;15)(p21.1;q11.2) translocation. Hum Genet 131:121–130. https://doi.org/10.1007/s00439-011-1051-4
Sarri C, Gyftodimou J, Avramopoulos D, Grigoriadou M, Pedersen W, Pandelia E, Pangalos C, Abazis D, Kitsos G, Vassilopoulos D, Brøndum-Nielsen K, Petersen MB (1997) Partial trisomy 17q22-qter and partial monosomy Xq27-qter in a girl with a de novo unbalanced translocation due to a postzygotic error: case report and review of the literature on partial trisomy 17qter. Am J Med Genet 1997 70:87–94
Styles P, Brookfield JF (2007) Analysis of the features and source gene composition of the AluYg6 subfamily of human retrotransposons. BMC Evol Biol 7:102. https://doi.org/10.1186/1471-2148-7-102
Van Opstal D, van Maarle MC, Lichtenbelt K, Weiss MM, Schuring-Blom H, Bhola SL, Hoffer MJV, Huijsdens-van Amsterdam K, Macville MV, Kooper AJA, Faas BHW, Govaerts L, Tan-Sindhunata GM, den Hollander N, Feenstra I, Galjaard RH, Oepkes D, Ghesquiere S, Brouwer RWW, Beulen L, Bollen S, Elferink MG, Straver R, Henneman L, Page-Christiaens GC, Sistermans EA (2018) Origin and clinical relevance of chromosomal aberrations other than the common trisomies detected by genome-wideNIPS: results of the TRIDENT study. Genet Med 20:480–485. https://doi.org/10.1038/gim.2017.132
Vetro A, Manolakos E, Petersen MB, Thomaidis L, Liehr T, Croci G, Franchi F, Marinelli M, Meneghelli E, Dal Bello B, Cesari S, Iasci A, Arrigo G, Zuffardi O (2012) Unexpected results in the constitution of small supernumerary marker chromosomes. Eur J Med Genet 55:185–190. https://doi.org/10.1016/j.ejmg.2012.01.010
Vetro A, Goidin D, Lesende I, Limongelli I, Ranzani GN, Novara F, Bonaglia MC, Rinaldi B, Franchi F, Manolakos E, Lonardo F, Scarano F, Scarano G, Costantino L, Tedeschi S, Giglio S, Zuffardi O (2018) Diagnostic application of a capture based NGS test for the concurrent detection of variants in sequence and copy number as well as LOH. Clin Genet 93:545–556. https://doi.org/10.1111/cge.13060
Voet T, Vanneste E, Van der Aa N, Melotte C, Jackmaert S, Vandendael T, Declercq M, Debrock S, Fryns JP, Moreau Y, D’Hooghe T, Vermeesch JR (2011) Breakage-fusion-bridge cycles leading to inv dup del occur in human cleavage stage embryos. Hum Mutat 32:783–793. https://doi.org/10.1002/humu.21502
Weckselblatt B, Hermetz KE, Rudd MK (2015) Unbalanced translocations arise from diverse mutational mechanisms including chromothripsis. Genome Res 25:937–947. https://doi.org/10.1101/gr.191247.115
Willett-Brozick JE, Savul SA, Richey LE, Baysal BE (2001) Germ line insertion of mtDNA at the breakpoint junction of a reciprocal constitutional translocation. Hum Genet 109:216–223
Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S, Zhang N, Duan F, Ming J, Zhang X, Niu W, Song W, Jin H, Guo Y, Dai S, Hu L, Fang L, Wang Q, Li Y, Li W, Na J, Xie W, Sun Y (2018) Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557:256–260. https://doi.org/10.1038/s41586-018-0080-8
Yu S, Graf WD (2010) Telomere capture as a frequent mechanism for stabilization of the terminal chromosomal deletion associated with inverted duplication. Cytogenet Genome Res 129:265–274. https://doi.org/10.1159/000315887
Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, Pellman D (2015) Chromothripsis from DNA damage in micronuclei. Nature 522:179–184. https://doi.org/10.1038/nature14493
Zhu J, Tsai HJ, Gordon MR, Li R (2018) Cellular stress associated with aneuploidy. Dev Cell 44(4):420–431. https://doi.org/10.1016/j.devcel.2018.02.002
Zuffardi O, Bonaglia M, Ciccone R, Giorda R (2009) Inverted duplications deletions: underdiagnosed rearrangements?? Clin Genet 75:505–513. https://doi.org/10.1111/j.1399-0004.2009.01187.x
Acknowledgements
The authors wish to thank the patients and their families for contributing to this research study. They also wish to acknowledge the Ring14 International Society and Dr Chiara Baldo from the Galliera Genetic Bank, Laboratory of Human Genetics, EO Ospedale Galliera, Genova, Italy for providing material from case 25.
Funding
MCB was supported by grants of the Italian Ministry of Health (Ricerca Corrente 2018). NT was supported by the Lundbeck Foundation (2013–14290), the UCPH Programme for Interdisciplinary Research (Global Genes, Local Concerns) and The Danish Council for Independent Research - Medical Sciences (4183-00482B). OZ was supported by Telethon Italian Foundation (Grant GGP13060).
Author information
Authors and Affiliations
Contributions
MCB and OZ conceived and designed the study. MCB, EE, SB, SS, and MMM performed the experiments. MCB, NEK, EE, MMM, NT, and OZ analyzed and validated the data. Contribution of reagents/materials/analysis tools: MCB, EE, SB, SS, MMM, AP, DV, VP, FN, PR, MCD, GD, RG, MAD, CBS, APG, DNA, SG, IR, FF, PP, CS, MAM, JN, NT, and OZ. MCB and OZ were the major contributors in writing the paper, where NEK, EE, and NT critically discussed the final editing of the manuscript. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval
The study has E. Medea Scientific Institute Research Ethics Committee approval (Prot. no. 007/15-CE).
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bonaglia, M.C., Kurtas, N.E., Errichiello, E. et al. De novo unbalanced translocations have a complex history/aetiology. Hum Genet 137, 817–829 (2018). https://doi.org/10.1007/s00439-018-1941-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-018-1941-9