
Abstract
Background
There is currently a limited number of studies on transglutaminase type 1 (TGM1) in tumors. The objective of this study is to perform a comprehensive analysis across various types of cancer to determine the prognostic significance of TGM1 in tumors and investigate its role in the immune environment.
Method
Pan-cancer and mutational data were retrieved from the TCGA database and analyzed using R (version 3.6.4) and its associated software package. The expression difference and prognosis of TGM1 were examined, along with its correlation with tumor heterogeneity, stemness, mutation landscape, and RNA modification. Additionally, the relationship between TGM1 expression and tumor immunity was investigated using the TIMER method.
Results
TGM1 is expressed differently in various tumors and normal samples and is associated with the overall survival and progression-free time of KIRC, ACC, SKCM, LIHC, and STES. In LICH, we found a negative correlation between TGM1 expression and 6 indicators of tumor stemness. The mutation frequencies of BLCA, LIHC, and KIRC were 1.7%, 0.3%, and 0.3% respectively. In BLCA and BRCA, there was a significant correlation between TGM1 expression and the infiltration of CD4 + T cells, CD8 + T cells, neutrophils, and dendritic cells.
Conclusion
TGM1 has the potential to serve as both a prognostic marker and a drug target.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
With the increase in population aging, the burden of disease on health is expected to become increasingly heavy (Zhang et al. 2023). Statistics indicate that the likelihood of cancer in individuals aged 65 and above is ten times higher compared to those under 65, and the aging process is closely linked to tumorigenesis (Wang et al. 2022). Advanced age is associated with several cancer risk factors, including prostate and bladder cancer (Feng et al. 2022a; Jin et al. 2023). Consequently, as the global population continues to age, malignant tumors are poised to become increasingly significant threats to human life and well-being. This scenario presents a formidable challenge to global healthcare (Siegel et al. 2023; Yan et al. 2022). While current treatment methods have undoubtedly improved the prognosis for cancer patients, there remains a subset of patients who exhibit poor response to treatment, along with instances of metastasis and recurrence, ultimately leading to low survival rates (Klein 2020; Phillips 2022). Therefore, it is imperative to quest new markers for the diagnosis and treatment of tumors.
Previous studies have primarily focused on transglutaminase type 2 (TGM2) within the TGM family, which has been associated with tumor proliferation, invasion, and drug resistance (Lee et al. 2018; Tempest, et al. 2021; Eckert et al. 2015).The enzyme TGM2 encodes the catalysis of cross-linking between glutamine and lysine residues, which plays a crucial role in stabilizing the extracellular matrix, cytoskeletal function, adhesion, and signaling (Bianchi et al. 2018). The conformation of TGM2 is regulated by cellular stimuli, such as changes in calcium levels, resulting in different biological effects (Lai and Greenberg 2013). Several studies have shown that TGM2 exhibits strong expression and activity in the stromal tissue surrounding tumors, indicating its involvement in tumor aggressiveness (Malkomes et al. 2023; Zhang et al. 2023). Due to its diverse functions and implications in disease, TGM2 has emerged as a potential therapeutic target. However, there has been limited research on transglutaminase type 1 (TGM1) in relation to tumors (Zhong et al. 2019). TGM1 plays a crucial role in the formation of the cornified cell envelope and is essential for maintaining skin barrier function (Boeshans et al. 2007). The potential of TGM1 in tumors remains largely unexplored, and there is a lack of comprehensive studies investigating the correlation between TGM1 expression and various types of tumors. To address this gap, we conducted a pan-cancer analysis using data from The Cancer Genome Atlas (TCGA). Our findings demonstrate that TGM1 exhibits prognostic value across a range of tumors and holds promise as a potential biomarker.
Methods
Date acquisition and differential and analysis
Consistent with our previous study (Feng et al. 2022b; Shi et al. 2023), we obtained the TCGA pan-cancer dataset from the USCS database ( (Goldman et al. 2020). In addition, we integrated the TCGA prognostic dataset from previous study and extracted the expression data of TGM1 in each sample (Liu et al. 2018). Samples were screened for an expression level of 0, starting from Solid Tissue Normal, Primary tumor and Primary Blood Derived Cancer-Peripheral Blood. Subsequently, a log2 (x + 0.001) transformation was performed on each expression quality, and cancer types with a sample size of less than 10 were eliminated, and the unpaired Wilconxon rank sum test and sign test were used for significant difference analysis.
Pan-cancer survival analysis and relationship with clinical features
We screened metastatic samples from Primary Blood Derived Cancer-Peripheral Blood (TCGA-LAML), Primary Tumor, and TCGA-SKCM databases. After excluding samples with an expression level of 0 or a follow-up time of less than 30 days, we obtained expression data, overall survival (OS), and progression-free interval (PFI) data for 39 cancer types. The data were analyzed using a Cox proportional hazards regression model and the log-rank test was used to obtain prognostic significance (Andersen and Gill 1982). Furthermore, the unpaired Wilcoxon rank sum test, sign test, and Kruskal test were utilized to evaluate the correlation between TGM1 expression and clinical stage, gender, and other clinical characteristics. In addition, we investigated the correlation between TGM1 mRNA expression and the age of the patients. We utilized proportional hazards hypothesis testing and Cox regression analysis to investigate various urological tumors. Subsequently, we conducted nomogram analysis and visualization.
Analysis of tumor heterogeneity, stemness and gene mutation
We calculated tumor stemness indicators by analyzing tumor methylation and mRNA expression signatures. This included six indexes namely DNA methylation based (DNAss), differentially methylated probes-based (DMPss), enhancer elements/DNA methylation-based (ENHss), RNA expression-based (RNAss), epigenetically regulated DNA methylation-based (EREG-METHss) and epigenetically regulated RNA methylation-based (EREG-METHss). Additionally, we conducted Spearman analysis to determine the correlation between tumor stemness characteristics and TGM1 expression. Tumor mutation burden (TMB), mutant-allele tumor heterogeneity (MATH), tumor ploidy, tumor purity, loss of heterozygosity (LOH), neoantigen (NEO), and homologous recombination deficiency (HRD) are indicators that reflect tumor heterogeneity (Ozga et al. 2021). These indicators were obtained from GDC (https://protal.gdc.cancer.gov/) and processed using Mutcet2 software and the R package 'maftools’ (Beroukhim et al. 2010), and microsatellite instability (MSI) was analyzed to examine the relationship between TMG1 expression and tumor heterogeneity, using the Spearman rank correlation coefficient (Bonneville et al. 2017). The Mutect2 software was utilized to process a simple nucleotide variation dataset for the analysis of gene mutations. Following the integration of the data, gene expression and mutations were detected in ACC, BLCA, KIRC, and LIHC. To evaluate the difference in mutation frequency among each group of samples, the chi-square test was employed.
Analysis of RNA modifications, tumor immune microenvironment and drug sensitivity
We conducted an analysis to examine the correlation between TGM1 mRNA expression levels and a total of 36 stimulatory and 24 heterogeneous checkpoints (Ozga et al. 2021), as well as 150 immune regulatory genes (receptor, MHC, chemokine, immunoinhibitory, immunostimulator (Shen et al. 2011)). The correlation of TGM1 expression with 44 genes in three RNA modification types was analyzed using data matrix, including 10 m1A genes, 13 m5C genes and 21 m6A genes. The tumor microenvironment was assessed using Timer (Li et al. 2017), a tool available in the R package 'IOBR' (Ru et al. 2019). Furthermore, we investigated the Genomics of Drug Sensitivity in Cancer (GDSC) and the Cancer Therapeutics Response Portal (CTRP) drug sensitivity in pan-cancer via GSCALite (Liu et al. 2018). In addition, we mapped the gene expression profiles of tumors to GeneSymbol. To assess the stromal scores of each patient in each tumor, we utilized the R software package ESTIMATE and calculated them based on gene expression (Yoshihara et al. 2013).
Statistical analysis
All the analysis were performed with the use of R software (version 3.6.3) and its suitable packages. A p-value below 0.05 was considered statistical significance. The Wilcoxon rank sum test and sign test were utilized to analyze paired differences, while the Kruskal test was employed to test multiple groups of samples. Correlation analysis between two variables was conducted using the Spearman test.
Results
Differential expression and clinical characteristics of TGM1
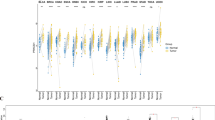
Compared to normal samples, we found that the TGM1 mRNA expression was significantly upregulated in Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), Colon adenocarcinoma (COAD), Rectum adenocarcinoma (READ), Esophageal carcinoma (ESCA), Stomach and Esophageal carcinoma (STES), Thyroid carcinoma (THCA), Bladder Urothelial Carcinoma (BLCA) and Cholangiocarcinoma (CHOL) while downregulated in Lung adenocarcinoma (LUAD), Breast invasive carcinoma (BRCA), Kidney renal clear cell carcinoma (KIKP),Kidney renal papillary cell carcinoma (KIPAN),Prostate adenocarcinoma (PRAD),Head and Neck squamous cell carcinoma (NHSC) and Kidney Chromophobe (KICH) patients (Fig. 1A). In terms of OS, we observed a significant association between high expression of TGM1 and poor prognosis in several cancer types, including KIPAN, KIRC, Adrenocortical carcinoma (ACC), Skin Cutaneous Melanoma (SKCM), LIHC, and Pheochromocytoma and Paraganglioma (PCPG) (Fig. 1B). in terms of PFI, we observed a significant correlation between high expression of TGM1 and poor prognosis in patients with KIPAN, KIRC, ACC, STES, LIHC, Stomach adenocarcinoma (STAD), and BCLA and low expression of TGM1 was associated with poor prognosis in patients with GBMLGG and Thymoma (THYM) (Fig. 1C). The expression level of TGM1 in LUAD was found to be associated with the T stage, N stage, and M stage (Additional file 1: Fig.S1A–C). There are significant differences in TGM1 expression in different clinical stages and grades of Head and Neck squamous cell carcinoma (NHSC) (Additional file 1: Fig.S1D-F). Additionally, TGM1 expression levels in Lymphoid Neoplasm Diffuse Large B-cell Lymphoma (DLBC) and THYM are correlated with age (Additional file 1: Fig.S1G). In this study, we developed a prognostic nomogram that incorporates clinicopathological characteristics and TGM1 expression in patients with ACC, BLCA, and KIRC. The nomogram aims to predict the prognosis of these cancers based on these factors (Additional file 2: Fig.S2A-C).

Differential expression and prognosis analysis of TGM1. A pan-cancer analysis of TGM1 for differential expression between tumor and normal tissues; B pan-cancer analysis of TGM1 for OS; C pan-cancer analysis of TGM1 for PFI; OS: overall survival; PFI: progression-free interval
In summary, we discovered several common cancer types and prognosis-related tumors that showed differential expression between tumor tissues and normal samples. The cancer types identified were ACC, KIKPAN, KIRC, and LIHC. Furthermore, our analysis revealed an age-related expression pattern of TGM1, with a positive correlation in THYM and a negative correlation in DLBC and GBMLGG.
Relationship of TGM1 with tumor heterogeneity, stemness and gene mutation
We further investigated the correlation between the expression level of TGM1 and tumor heterogeneity and stemness. Our study revealed significant correlations between TGM1 expression levels and HRD status in 14 tumors (Fig. 2A). Additionally, we observed a negative correlation between TGM1 expression and LOH in 9 tumors (Fig. 2B). In terms of MATH, we found a positive correlation between TGM1 mRNA expression and 4 tumors (Fig. 2C). MSI and NEO are indicators of tumor response to immunotherapy. Our findings demonstrated a significant correlation between TGM1 expression and MSI in 15 tumors, including HNSC, BRCA, and KICH. However, NEO was only correlated with TGM1 expression in 7 tumors (Fig. 2D, E). In our analysis of tumor stemness, we observed a negative correlation between the expression level of TGM1 in LIHC and all six tumor stemness (Fig. 3A–F).

The pan-cancer Spearman analysis of tumor heterogeneity and TGM1 expression. A the correlation between HRD and TGM1 level; B the correlation between LOH and TGM1 level; C the correlation between MATH and TGM1 level; D the correlation between MSI and TGM1 level; E the correlation between NEO and TGM1 level; (F) the correlation between tumor ploidy and TGM1 level; G the correlation between tumor purity and TGM1 level; H the correlation between TMB and TGM1 level. HRD: homologous recombination deficiency; LOH loss of heterozygosity, MATH: mutant-allele tumor heterogeneity, MSI microsatellite instability, NEO neoantigen, TMB tumor mutation burden

The pan-cancer Spearman analysis of tumor stemness and TGM1 expression. A The correlation between tumor stemness and TGM1 level using DMPss; B the correlation between tumor stemness and TGM1 level using DNAss; C the correlation between tumor stemness and TGM1 level using ENHss; D the correlation between tumor stemness and TGM1 level using EREG.EXPss; E the correlation between tumor stemness and TGM1 level using EREG-METHss; F the correlation between tumor stemness and TGM1 level using RNAss. DNAss DNA methylation based, DMPss differentially methylated probes-based, EHNss enhancer elements/DNA methylation-based, RNAss RNA expression-based, EREG-METHss epigenetically regulated DNA methylation-based, EREG-METHss epigenetically regulated RNA methylation-based
Some targeted therapeutic responses do not depend on tumor type, but rather on mutations. Tumor gene mutations are closely associated with their biological behavior. The study revealed mutation frequencies of 1.3% for GMB, 0.3% for KIRC, and 1.4% for LUAD (Fig. 4A). TGM1 was divided into high expression and low expression groups based on its expression level. The possible mechanism of TGM1 in tumorigenesis and development was explored through its mutated gene. No statistically significant gene mutations were found in ACC (Fig. 4B). In BLCA, significant gene mutations including TP53, RB1, PCLO, CDKN1A, and VPS13D were observed between the high expression group and low expression group (Fig. 4C). Likewise, KIRC showed significant gene mutations, such as PTEN, SPEN, ADAMTS12, RYR2, and CHD6, between the two groups (Fig. 4D). In LIHC, CTNNB1, BAP1, KEAP1, KNDC1, and MEGF8 were found to have significant differences in gene expression between the low and high expression groups (Fig. 4E).

Mutation landscape of TGM1. A mutation landscapes of TGM1 for pan-cancer; B the top 5 mutation genes between high and low-expression of TGM1 in ACC patients; C the top 5 mutation genes between high and low-expression of TGM1 in BLCA patients; D the top 5 mutation genes between high and low-expression of TGM1 in KIRC patients; E the top 5 mutation genes between high and low-expression of TGM1 in LIHC patients
Relationship between TGM1 expression with immune regulation, checkpoints, RNA modification and drug sensitivity
Our findings indicated that the expression levels of TGM1 in various cancer types were associated with multiple immune regulatory genes and immune checkpoint (IC) genes (Fig. 5A, B). Figure 5 demonstrates a correlation between the expression of TGM1 and various immune checkpoint genes and immunomodulatory genes in BLCA, LIHC, ACC, and even PRAD. However, it is worth noting that previous studies have indicated a limited effectiveness of immunotherapy in PRAD. In terms of RNA modification, our study revealed a negative correlation between TGM1 expression in ESCA and the expression of YTHDF1, LRPPRC, RBM15, ZC3H13, NSUN7, NSUN6, and TRMT61A (Fig. 6A). In the case of BLCA, we observed a positive correlation between TGM1 expression and multiple m1A, m5C, and m6A modifications (Fig. 6A). The results from Timer analysis demonstrated a positive correlation between TGM1 expression and the infiltration of several immune cell types in the tumor microenvironment (TME) of PRAD, LIHC, LUAD, and BRCA (Fig. 6B). For instance, in LIHC, TGM1 expression was positively correlated with the infiltration of B cells, CD4 + T cells, neutrophils, macrophages, and dendritic cells (Fig. 6B). We also observed an inverse correlation between TGM1 expression and macrophage infiltration in ACC (Fig. 6B). The correlation between TGM1 expression and drug sensitivity was analyzed in the pan-cancer analysis of GDSC and CTRP, as depicted in Fig. 6C and Fig. 6D, respectively. Among the tested drugs, Crizotinib, ML320, KW-2449, Dabrafenib, Tubastatin A, and CAY10603 demonstrated relatively favorable outcomes. We calculated Spearman's correlation coefficient between genes and immune infiltration scores in each tumor to determine the immune infiltration scores that showed significant correlation. Our analysis revealed that gene expression was significantly correlated with immune infiltration in 18 different cancer types. Among these, 7 cancer types showed a significant positive correlation, while 11 cancer types showed a significant negative correlation (Fig. 7).

The Spearman analysis of TGM1 expression and regulatory genes and immune checkpoints. A the correlation of TGM1 expression with immune regulatory genes; B the correlation of TGM1 expression with immune checkpoint genes

The Spearman analysis of TGM1 expression and RNA modification; Tumor immune environment and its correlation with TGM1 expression and drug sensitivity analysis. A the correlation of TGM1 expression with genes of RNA modification; B the correlation of TGM1 expression with immune infiltrating cells using TIMER; C the correlation between gene expression and the sensitivity of GDSC drugs (top 10) in pan-cancer; D the correlation between gene expression and the sensitivity of CTRP drugs (top 10) in pan-cancer

The correlation of TGM1 expression with stromal score
Discussion
Understanding the biological behavior of tumors and their treatment methods from multiple dimensions has always been a significant challenge due to the complex mechanism of tumor formation and development. The high mortality rate and treatment cost impose a significant burden on society (Carrera et al. 2018). However, with the advancements in high-throughput sequencing technology, we now have the ability to explore the genetic characteristics of tumors and identify new therapeutic targets (Song et al. 2023). Notably, tumor cells exhibit unlimited proliferation when cellular homeostasis is disrupted (Shen et al. 2022), making the gene TGM1, which is associated with cell proliferation and keratinization, a potential breakthrough in tumor therapy. TGM1 gene is composed of 15 exons and is located on chromosome 14q11.2 (Polakowska et al. 1991). It is a protein-coding gene that encodes the TGase-1 enzyme, which has a molecular weight of 89 kDa and consists of 817 amino acids (Hu et al. 2019). Its primary function is to catalyze the cross-linking of proteins and the combination of polyamines and proteins, playing a role in cell proliferation (Zhang et al. 2016). Previous studies have linked TGM1 to autosomal recessive lamellar ichthyosis and non-bullous congenital ichthyosis erythroderma (Farasat et al. 2009; Kawashima et al. 2005; Landegren et al. 2021). While there is limited research on TGM1 in tumors, Ding et al. (2021) have shown that TGM1 is upregulated in ascites-derived ovarian cancer cells, potentially contributing to metastasis and spread. In vitro experiments have also demonstrated a relationship between TGM1 and cell proliferation in gastric cancer cells (Huang et al. 2017).
In this study, we conducted a bioinformatic analysis of patient data from each dataset to assess the expression of TGM1 in tissues. Our findings revealed that TGM1 mRNA levels were significantly upregulated in 12 tumors, including bladder cancer, when compared to their corresponding normal tissues. These results are consistent with previous studies conducted by Huang et al. (2017), which also reported an upregulation of TGM1 in gastric cancer cells compared to normal tissues. Furthermore, the study suggested that knockdown of TGM1 could potentially enhance apoptosis. This has also been confirmed in a study on Alzheimer's disease. Tripthy et al. (2020) observed an increase in the expression of TGM1 in the hippocampus and primary cortical neurons of a mouse model during the late stage of the disease, leading to cell death. Based on these findings, it is hypothesized that the progression of TGM1 in tumors might be associated with apoptosis. However, further experiments are required to validate this hypothesis. We also investigated the prognostic value of TGM1 by analyzing tumor OS and PFI. However, the exact role of TGM1 in predicting prognosis remains unclear. In our study, we observed that TGM1 was associated with poor prognosis in some tumors, but interestingly, it acted as a protective factor in tumors like GBMLGG. Our findings showed that the expression level of TGM1 was correlated with the poor prognosis of SKCM patients, a malignancy that mainly occurs in the elderly (Long et al. 2022). This aligns with a study by Li et al. (2021), where they discovered that TGM1 was significantly down-regulated in metastatic SKCM tissues compared to primary SKCM tissues, suggesting potential involvement of TGM1 in the occurrence and spread of SKCM tumors. We hypothesize that the differences in TGM1 expression levels and prognosis could be attributed to epigenetic regulation. We investigated the correlation between TGM1 expression and clinical features, specifically TNM stages. Our findings revealed that TGM1 expression levels varied across different T stages, N stages, and M stages of LUAD. This suggests that the expression of TGM1 may be influenced by epigenetic regulation triggered by heterogeneity during tumor development or metastasis. Furthermore, our results indicate that the role of TGM1 may differ at different stages of various tumors, making it a potential predictor for clinical staging and guiding therapy. Our study also demonstrates a correlation between age and TGM1 expression. Aging is a well-known risk factor for various tumors, as it results in reduced cell proliferation, homing, and differentiation processes (Feng et al. 2023; Hu et al. 2022; Feng et al. 2020). These abnormalities in the human body contribute to the promotion and development of tumors (Ruiz et al. 2023). Hence, it is crucial to identify the genetic intersection between aging and tumors and subsequently intervene in this process.
Tumors consist of a diverse population of cells with high heterogeneity, which can be categorized into intra-tumor heterogeneity and inter-tumor heterogeneity (Dagogo-Jack and Shaw 2018). This heterogeneity arises from variations in genetics, epigenetics, and the tumor microenvironment (Chu et al. 2022; Liu et al. 2023a). Understanding tumor heterogeneity is crucial for studying tumor growth, metastasis, apoptosis and drug resistance, and it holds significant promise for identifying potential therapeutic approaches. We conducted an analysis of 8 indicators that reflect tumor heterogeneity, including TMB and MSI. TMB measures the number of genomic mutations, while the number of neoantigens expressed in tumor cells increases in proportion to the mutational burden (Jardim et al. 2021). This increase in neoantigens enhances the likelihood of positive responses to checkpoint blockade immunotherapy, making it a potential biomarker for anti-PD-1/PD-L1 therapy (Goodman et al. 2017; Joshi and Durden 2019). Previous studies have demonstrated that melanoma patients with a high mutational burden experienced improved survival following ipilimumab treatment (Gupta et al. 2015). Additionally, TMB has been found to predict the survival rate of non-small cell lung cancer patients undergoing immunotherapy (Hellmann et al. 2022). Our study discovered a significant association between the expression of TGM1 and TMB in 14 tumors. This finding indicates that the expression level of TGM1 has an impact on tumor heterogeneity, resulting in alterations in TMB. Consequently, these changes may influence the response of patients to immunotherapy. Two major forms of genomic instability commonly found in tumors are chromosomal instability and MSI (Ogino and Goel 2008). MSI-high tumors exhibit defects in the mismatch repair system, resulting in phenotypic hypermutation and the generation of immunogenic neoantigens (Grasso et al. 2018). This frequently leads to a significant infiltration of lymphocytes in these tumors, which is associated with favorable clinical outcomes (Ge et al. 2023; Pei et al. 2023). Our study demonstrated a positive correlation between the expression level of TGM1 and MSI in seven tumors, including COAD and BLCA. Previous studies have also indicated the prognostic value of high MSI in these two tumor types (Awadalla et al. 2022; Lochhead et al. 2013). Therefore, our results may serve as a valuable reference for the selection of immunotherapy approaches.
Through genome sequencing of tumor samples from cancer patients, the relative abundance of gene mutations has been determined for various forms of cancer (Xu et al. 1986). Identifying commonly mutated genes is crucial for drug development, as some oncoproteins can be targeted by drugs (Bollag et al. 2010; Loriot et al. 2019). This knowledge can help allocate public resources to benefit a larger number of patients. In this study, we aim to investigate the role of TGM1 in tumors by analyzing gene mutations based on its expression levels. Our findings reveal that PTEN mutation is the most common in KICH, and it exerts an anti-tumor effect by inhibiting the activation of PI3K/AKT (Peglion et al. 2022). Voss et al. (2019) conducted a study on patients with advanced KICH who were treated with everolimus and sunitinib. The study found that patients with positive PTEN protein expression had a better prognosis when treated with everolimus. However, no significant difference in prognosis was observed in patients treated with sunitinib. These findings could potentially assist clinicians in making informed decisions regarding treatment options for patients with advanced KICH. Moreover, PTEN is expressed in prostate cancer cell lines PC3 and DU145, which are androgen-independent and represent advanced stages of the disease (Brussel et al. 1999; Huang et al. 2001; Calastretti et al. 2014). PTEN mutation may can serve as a marker to predict treatment response and the developmental stage of cancer cells. Previous studies have demonstrated that TP53 mutations can result in the inactivation of wild-type p53 and contribute to tumor progression (Kotler, et al. 2018; Hu et al. 2021). In addition to TP53 mutations, our findings reveal a higher mutation frequency in RB1 in BLCA. Importantly, a study conducted by Liu et al. (2023b) observed that BLCA tissues with co-mutations in TP53 and RB1 exhibited a greater presence of immune effectors, which showed a significant correlation with the response to immune checkpoint inhibitors (ICIs). Mutated KEAP1 has a higher mutation frequency in the LIHC group with low TGM1 expression, and KEAP1 mutation is associated with poor prognosis in many tumors (Romero et al. 2020). Our findings revealed a positive correlation between the expression level of TGM1 and several RNA modification genes, including m1A, m5C, and m6A. Notably, this correlation was particularly evident in LIHC, KIRC, BRCA, CHOL, and ACC. Previous studies have partially explored the prognostic significance of m6A modification in BRCA and CHOL (Feng et al. 2022c; Wei et al. 2023). Furthermore, m5C can serve as a potential biomarker for predicting the efficacy of LIHC immunotherapy (Liu et al. 2022).
Our findings indicate that the expression of TGM1 plays a crucial role in tumor immunity within the TME. TME encompasses various components, including immune cells, cytokines, and oxidative stress-related products, which collectively influence tumor growth and survival (Xiao and Yu 2021; Xiong et al. 2023a). To investigate the relationship between TGM1 expression and tumor-infiltrating immune cells in the TME, we utilized the TIMER method to evaluate the infiltration score of different cell types. Our results revealed a positive correlation between TGM1 expression and CD4 + T cells as well as neutrophils in most tumors. CD4 + T cells play a vital role in enhancing the intratumoral cytotoxic T lymphocyte response through signal transduction, thereby promoting the effectiveness of immunotherapy (Borst et al. 2018). Additionally, neutrophils exhibit tumor-killing capabilities by releasing catalytically active elastase (Cui, et al. 2021). Furthermore, TGM1 expression showed a positive correlation with multiple immune regulatory genes, suggesting that its high expression level may indicate increased sensitivity to immunotherapy. In addition, immune checkpoint genes have the ability to influence immune cell function. IC signaling is activated as tumors progress, enabling immune evasion (Feng et al. 2019). Therefore, examining the relationship between TGM1 expression and immune checkpoint markers can offer fresh opportunities for the development of innovative immunosuppressants. We conducted an analysis on 60 immune checkpoint genes and found that TGM1 expression exhibited a positive correlation with immune checkpoints in the majority of tumors (Feng et al. 2023b). This suggests the potential of TGM1 as a viable drug target. Medicines are undoubtedly the most important resource in the treatment of cancer. Our analysis revealed a correlation between the sensitivity of multiple drugs and the expression of TGM1. Crizotinib, a multi-target protease inhibitor, is commonly used in tumor patients with abnormal AKL, ROS, and MET kinase activities (Blackhall and Cappuzzo 2016). It has demonstrated promising therapeutic effects in tumors (Shaw et al. 2020; Pal et al. 2021). Based on our study, we propose that TGM1 could serve as a potential target gene of crizotinib for clinical application.
Our study reveals that the expression of TGM1 in BLCA is associated with a poor prognosis. However, the mechanism of TGM1 in BLCA remains unexplored. Our previous study have indicated that TGM1 may utilize exogenous metabolic pathways, such as cytochrome P450 and steroid hormone biosynthesis, to contribute to the development and progression of tumors (Wang et al. 2023). Additionally, Huang et al. discovered that TGM1 activates the Wnt signaling pathway, promoting the development of gastric cancer (Huang et al. 2017). The Wnt/β-catenin signaling pathway regulates mesenchymal changes by controlling the levels of cell adhesion molecules. It is possible that this pathway also plays a role in tumor epithelial-mesenchymal transition, but its involvement in bladder cancer cells is yet to be determined. Further investigations are required to verify this hypothesis.
Like BLRC, KIRC is a prevalent malignant tumor in urinary system. Among the different pathological types of renal cancer, KIRC is the most observed. Our study revealed that the expression of TGM1 in normal tissues of KIRC is higher compared to tumor tissues. Moreover, a high expression of TGM1 is associated with a poor prognosis. These findings suggest that TGM1 may not act as a tumor suppressor in KIRC. However, its expression shows a strong correlation with immune checkpoints and has the potential to be a therapeutic target. Further research is still required to validate these findings.
Our study demonstrates that the role of TGM1 in tumors is not limited to a single tumor type. We examined the correlation between TGM1 expression and prognosis, clinical characteristics, tumor heterogeneity, gene mutation, and immune infiltration in pan-cancer patients. This investigation sheds light on the role of TGM1 in tumorigenesis and development, and comprehensively evaluates its potential as a prognostic marker and drug target from various perspectives. However, it is important to acknowledge the limitations of our work. Firstly, the heterogeneity of different databases may slightly impact the reliability of our analysis results. Moreover, our findings are based on public database analysis and encompass multiple tumor types, necessitating further experiments to elucidate the mechanism of TGM1. Nevertheless, our pan-cancer analysis of TGM1 provides a solid foundation and novel insights for future research.
Conclusion
TGM1 has the potential to serve as both a prognostic marker and a drug target.
Availability of data and materials
The data sets presented in this study are available in online repositories. The name and join number of the repository can be found in the article/supplement.
Abbreviations
- ACC:
-
Adrenocortical carcinoma
- BLCA:
-
Bladder Urothelial Carcinoma
- BRCA:
-
Breast invasive carcinoma
- CESC:
-
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- CHOL:
-
Cholangiocarcinoma
- COAD:
-
Colon adenocarcinoma
- READ:
-
Rectum adenocarcinoma Esophageal carcinoma
- DLBC:
-
Lymphoid Neoplasm Diffuse Large B-cell Lymphoma
- ESCA:
-
Esophagus carcinoma
- GBM:
-
Glioblastoma multiforme
- GBMLGG:
-
Glioma
- HNSC:
-
Head and Neck squamous cell carcinoma
- KICH:
-
Kidney Chromophobe
- KIPAN:
-
Pan-kidney cohort (KICH + KIRC + KIRP)
- KIRC:
-
Kidney renal clear cell carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- LAML:
-
Acute Myeloid Leukemia
- LGG:
-
Brain Lower Grade Glioma
- LIHC:
-
Liver hepatocellular carcinoma
- LUAD:
-
Lung adenocarcinoma
- LUSC:
-
Lung squamous cell carcinoma
- MESO:
-
Mesothelioma
- OV:
-
Ovarian serous cystadenocarcinoma
- PAAD:
-
Pancreatic adenocarcinoma
- PCPG:
-
Pheochromocytoma and Paraganglioma
- PRAD:
-
Prostate adenocarcinoma
- READ:
-
Rectum adenocarcinoma
- SARC:
-
Sarcoma
- STAD:
-
Stomach adenocarcinoma
- SKCM:
-
Skin Cutaneous Melanoma
- STES:
-
Stomach and Esophageal carcinoma
- TGCT:
-
Testicular Germ Cell Tumors
- THCA:
-
Thyroid carcinoma
- THYM:
-
Thymoma
- UCEC:
-
Uterine Corpus Endometrial Carcinoma
- UCS:
-
Uterine Carcinosarcoma
- UVM:
-
Uveal Melanoma
- TARGET-OS:
-
Osteosarcoma
- TARGET-ALL:
-
Acute Lymphoblastic Leukemia
- TARGET-NB:
-
Neuroblastoma
- TARGET-WT:
-
High-Risk Wilms Tumor
- TGM2:
-
Transglutaminase type 2
- TGM1:
-
Transglutaminase type 1
- TCGA:
-
The Cancer Genome Atlas
- OS:
-
Overall survival
- PFI:
-
Progression-free interval
- DNAss:
-
DNA methylation based
- DMPss:
-
Differentially methylated probes-based
- EHNss:
-
Enhancer elements/DNA methylation-based
- RNAss:
-
RNA expression-based
- EREG-METHss:
-
Epigenetically regulated DNA methylation-based
- EREG-METHss:
-
Epigenetically regulated RNA methylation-based
- TMB:
-
Tumor mutation burden
- MATH:
-
Mutant-allele tumor heterogeneity
- LOH:
-
Loss of heterozygosity
- NEO:
-
Neoantigen
- HRD:
-
Homologous recombination deficiency
- MSI:
-
Microsatellite instability
- IC:
-
Immune checkpoint
- TME:
-
Tumor microenvironment
- GDSC:
-
Genomics of Drug Sensitivity in Cancer
- CTRP:
-
Cancer Therapeutics Response Portal
References
Andersen PK, Gill RD (1982) Cox’s regression model for counting processes: a large sample study. Ann Stat. https://doi.org/10.1214/aos/1176345976
Awadalla A et al (2022) Prognostic influence of microsatellite alterations of muscle-invasive bladder cancer treated with radical cystectomy. Urol Oncol 40(2):64.e9-64.e15
Beroukhim R et al (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463(7283):899–905
Bianchi N, Beninati S, Bergamini CM (2018) Spotlight on the transglutaminase 2 gene: a focus on genomic and transcriptional aspects. Biochem J 475(9):1643–1667
Blackhall F, Cappuzzo F, Crizotinib: from discovery to accelerated development to front-line treatment. Ann Oncol, 2016. 27 Suppl 3: p. iii35-iii41.
Boeshans KM, Mueser TC, Ahvazi B (2007) A three-dimensional model of the human transglutaminase 1: insights into the understanding of lamellar ichthyosis. J Mol Model 13(1):233–246
Bollag G et al (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467(7315):596–599
Bonneville R et al (2017) Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol. https://doi.org/10.1200/PO.17.00073
Borst J et al (2018) CD4 (+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol 18(10):635–647
Calastretti A et al (2014) Down-modulation of Bcl-2 sensitizes PTEN-mutated prostate cancer cells to starvation and taxanes. Prostate 74(14):1411–1422
Carrera PM, Kantarjian HM, Blinder VS (2018) The financial burden and distress of patients with cancer: Understanding and stepping-up action on the financial toxicity of cancer treatment. CA Cancer J Clin 68(2):153–165
Chu X, Zhang Y, Cheng S (2022) Heterogeneity of tumor-infiltrating myeloid cells in era of single-cell genomics. Chin J Cancer Res 34(6):543–553
Cui C et al (2021) Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell 184(12):3163-3177.e21
Dagogo-Jack I, Shaw AT (2018) Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 15(2):81–94
Ding Y et al (2021) Molecular characteristics and tumorigenicity of ascites-derived tumor cells: mitochondrial oxidative phosphorylation as a novel therapy target in ovarian cancer. Mol Oncol 15(12):3578–3595
Eckert RL et al (2015) Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol Carcinog 54(10):947–958
Farasat S et al (2009) Novel transglutaminase-1 mutations and genotype-phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J Med Genet 46(2):103–111
Feng M et al (2019) Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 19(10):568–586
Feng D et al. (2023) Senescence-associated lncRNAs indicate distinct molecular subtypes associated with prognosis and androgen response in patients with prostate cancer. Acta Materia Medica 2(3)
Feng D et al (2022a) Developing an immune-related gene prognostic index associated with progression and providing new insights into the tumor immune microenvironment of prostate cancer. Immunology 166(2):197–209
Feng D et al (2022b) A pan-cancer analysis of the oncogenic role of leucine zipper protein 2 in human cancer. Exp Hematol Oncol 11(1):55
Feng ZH et al (2022c) m6A-immune-related lncRNA prognostic signature for predicting immune landscape and prognosis of bladder cancer. J Transl Med 20(1):492
Feng D, et al. Scientific advancements in drug development and trials for urothelial carcinoma: insights from the 2023 ASCOGU cancers symposium. Aging Dis, 2023.
Feng DC et al (2023c) Identification of senescence-related molecular subtypes and key genes for prostate cancer. Asian J Androl 25(2):223–229
Ge W et al (2023) Review and prospect of immune checkpoint blockade therapy represented by PD-1/PD-L1 in the treatment of clear cell renal cell carcinoma. Oncol Res 31(3):255–270
Goldman MJ et al (2020) Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol 38(6):675–678
Goodman AM et al (2017) Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 16(11):2598–2608
Grasso CS et al (2018) Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 8(6):730–749
Gupta S et al (2015) Gender disparity and mutation burden in metastatic melanoma. J Natl Cancer Inst. https://doi.org/10.1093/jnci/djv221
Hellmann MD et al. (2018) Genomic features of response to combination immunotherapy in patients with advanced non-small-cell lung cancer. Cancer Cell 33(5)
Hu Q et al (2019) Analysis of clinical phenotype and TGM1 gene mutation in a child with neonatal congenital ichthyosis. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 36(4):357–359
Hu J et al (2021) Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol 14(1):157
Hu C et al (2022) Cellular senescence in cardiovascular diseases: a systematic review. Aging Dis 13(1):103–128
Huang H et al (2001) PTEN induces chemosensitivity in PTEN-mutated prostate cancer cells by suppression of Bcl-2 expression. J Biol Chem 276(42):38830–38836
Huang H, Chen Z, Ni X (2017) Tissue transglutaminase-1 promotes stemness and chemoresistance in gastric cancer cells by regulating Wnt/beta-catenin signaling. Exp Biol Med (maywood) 242(2):194–202
Jin Y.-H et al. (2022) Treatment and surveillance for non-muscle-invasive bladder cancer: a clinical practice guideline (2021 edition). Mil Med Res 9(1):44
Jardim DL et al (2021) The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell 39(2):154–173
Joshi S, Durden DL (2019) Combinatorial approach to improve cancer immunotherapy: rational drug design strategy to simultaneously hit multiple targets to kill tumor cells and to activate the immune system. J Oncol 2019:5245034
Kawashima J et al (2005) Structural, enzymatic and molecular studies in a series of nonbullous congenital ichthyosiform erythroderma patients. Clin Exp Dermatol 30(4):429–431
Klein CA (2020) Cancer progression and the invisible phase of metastatic colonization. Nat Rev Cancer 20(11):681–694
Kotler E et al (2018) A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell 71(1):178-190.e8
Lai TS, Greenberg CS (2013) TGM2 and implications for human disease: role of alternative splicing. Front Biosci (landmark Ed) 18(2):504–519
Landegren N et al (2021) A gene-centric approach to biomarker discovery identifies transglutaminase 1 as an epidermal autoantigen. Proc Natl Acad Sci U S A. https://doi.org/10.1073/pnas.2100687118
Long GV et al. (2023) Cutaneous melanoma. Lancet 402(10400):485–502
Lee HT et al (2018) Transglutaminase 2 promotes migration and invasion of lung cancer cells. Oncol Res 26(8):1175–1182
Li T et al (2017) TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res 77(21):e108–e110
Li K et al (2021) Identification of keratinocyte differentiation-involved genes for metastatic melanoma by gene expression profiles. Comput Math Methods Med 2021:9652768
Liu CJ et al (2018) GSCALite: a web server for gene set cancer analysis. Bioinformatics 34(21):3771–3772
Liu J et al (2018) An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173(2):400-416.e11
Liu P et al (2022) Prognostic stratification based on m (5)C regulators acts as a novel biomarker for immunotherapy in hepatocellular carcinoma. Front Immunol 13:951529
Liu W, Qu C, Wang X (2023a) Comprehensive analysis of the role of immune-related PANoptosis lncRNA model in renal clear cell carcinoma based on RNA transcriptome and single-cell sequencing. Oncol Res 31(4):543–567
Liu N et al (2023b) Identification and validation of RB1 as an immune-related prognostic signature based on tumor mutation burdens in bladder cancer. Anticancer Drugs 34(2):269–280
Lochhead P et al (2013) Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst 105(15):1151–1156
Loriot Y et al (2019) Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med 381(4):338–348
Malkomes P et al (2023) Transglutaminase 2 is associated with adverse colorectal cancer survival and represents a therapeutic target. Cancer Gene Ther 30(10):1346–1354
Ogino S, Goel A (2008) Molecular classification and correlates in colorectal cancer. J Mol Diagn 10(1):13–27
Ozga AJ, Chow MT, Luster AD (2021) Chemokines and the immune response to cancer. Immunity 54(5):859–874
Pal SK et al (2021) A comparison of sunitinib with cabozantinib, crizotinib, and savolitinib for treatment of advanced papillary renal cell carcinoma: a randomised, open-label, phase 2 trial. Lancet 397(10275):695–703
Peglion F et al (2022) PTEN inhibits AMPK to control collective migration. Nat Commun 13(1):4528
Pei B et al (2023) Effect of microbiome group on immune checkpoint inhibitors in treatment of gastrointestinal tumors. Chin J Cancer Res 35(3):252–265
Polakowska RR et al (1991) Epidermal type I transglutaminase (TGM1) is assigned to human chromosome 14. Cytogenet Cell Genet 56(2):105–107
Romero R et al (2020) Keap1 mutation renders lung adenocarcinomas dependent on Slc33a1. Nat Cancer 1(6):589–602
Ru B et al (2019) TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics 35(20):4200–4202
Ruiz E et al (2023) An integrative multi-omics analysis of the molecular links between aging and aggressiveness in thyroid cancers. Aging Dis 14(3):992–1012
Shen W et al. (2022) Sangerbox: a comprehensive, interaction‐friendly clinical bioinformatics analysis platform. iMeta 1(3)
Shaw AT et al (2020) First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N Engl J Med 383(21):2018–2029
Shi X et al (2023) A pan-cancer analysis of the oncogenic and immunological roles of apolipoprotein F (APOF) in human cancer. Eur J Med Res 28(1):190
Siegel RL et al (2023) Cancer statistics, 2023. CA Cancer J Clin 73(1):17–48
Song Z et al (2023) Overexpression of RACGAP1 by E2F1 promotes neuroendocrine differentiation of prostate cancer by stabilizing EZH2 expression. Aging Dis. https://doi.org/10.14336/AD.2023.0202
Tempest R et al (2021) The biological and biomechanical role of transglutaminase-2 in the tumour microenvironment. Cancers (basel). https://doi.org/10.3390/cancers13112788
Tripathy D et al (2020) Increased transcription of transglutaminase 1 mediates neuronal death in in vitro models of neuronal stress and Abeta1-42-mediated toxicity. Neurobiol Dis 140:104849
van Brussel JP et al (1999) Chemosensitivity of prostate cancer cell lines and expression of multidrug resistance-related proteins. Eur J Cancer 35(4):664–671
Voss MH et al (2019) PTEN expression, not mutation status in TSC1, TSC2, or mTOR, correlates with the outcome on everolimus in patients with renal cell carcinoma treated on the randomized RECORD-3 trial. Clin Cancer Res 25(2):506–514
Wang J et al (2023) TGM1 could predict overall survival for patients with urinary bladder cancer. Asian J Surg 46(11):5373–5375
Wang J, Wei J, Inuzuka H (2023) Aging and cancer hallmarks as therapeutic targets. Acta Materia Medica 2(3)
Wei F et al (2023) Expression of m6A RNA methylation regulators and their clinical predictive value in intrahepatic cholangiocarcinoma. Front Biosci (landmark Ed) 28(6):120
Xu Y et al. (2023) The Largest Chinese Cohort Study Indicates Homologous Recombination Pathway Gene Mutations as Another Major Genetic Risk Factor for Colorectal Cancer with Heterogeneous Clinical Phenotypes. Research (Washington, D.C.), vol 6, p. 0249
Xiong D, Zhang L, Sun ZJ (2023) Targeting the epigenome to reinvigorate T cells for cancer immunotherapy. Mil Med Res 10(1):59
Xiao Y, Yu D (2021) Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther 221:107753
Yan X et al.(2023) Stomach cancer burden in China: Epidemiology and prevention. Chin J Cancer Res 35(2):81–91
Yin Y et al. (2023) Novel Combination Therapy for Triple-Negative Breast Cancer based on an Intelligent Hollow Carbon Sphere. Research (Wash D C), vol 6: p. 0098
Yoshihara K et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4:2612
Zhang SQ et al (2016) Identification and functional characterization of a novel transglutaminase 1 gene mutation associated with autosomal recessive congenital ichthyosis. Int J Dermatol 55(2):201–207
Zhang S et al (2023b) Transglutaminases are oncogenic biomarkers in human cancers and therapeutic targeting of TGM2 blocks chemoresistance and macrophage infiltration in pancreatic cancer. Cell Oncol (Dordr) 46(5):1473–1492
Zhong X et al (2019) Identification of crucial miRNAs and genes in esophageal squamous cell carcinoma by miRNA-mRNA integrated analysis. Medicine (baltimore) 98(27):e16269
Acknowledgements
We appreciated the Figdraw (www.figdraw.com) and Chengdu Basebiotech Co,Ltd for their assistance in drawing and data process.
Funding
This research was funded by Chinese Scholarship Council (grant no. 202206240086) .
Author information
Authors and Affiliations
Contributions
RCW and DXL proposed the project, conducted data analysis, interpreted the data, and wrote the manuscript; JW, ZTT, SXZ, KC, AM, KHY, WRW and CZ conducted data analysis, interpreted the data; DCF and PH supervised the project, and interpreted the data. All authors reviewed and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors have no conflicts of interest to declare.
Ethics approval and consent to participate
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Consent for publication
Not available.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
432_2024_5640_MOESM1_ESM.tif
Supplementary file1: Fig. S1. The correlation of TGM1 expression with Clinicopathological features A The correlation of TGM1 expression with gender; B The correlation of TGM1 expression with grade; C The correlation of TGM1 expression with clinical stages; D The correlation of TGM1 expression with T stages; E the correlation of TGM1 expression with N stage; F The correlation of TGM1 expression with M stages; G The correlation of TGM1 expression with age(TIF 2139 KB)
432_2024_5640_MOESM2_ESM.tif
Supplementary file2: Fig. S2. Nomogram of TGM1 expression and tumor pathological characteristics. A Nomogram of ACC; B Nomogram of BLCA; C Nomogram of KICH. ACC Adrenocortical carcinoma, BLCA Bladder Urothelial Carcinoma, KICH Kidney Chromophobe(TIF 2301 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, R., Li, D., Zhang, S. et al. A pan-cancer analysis of the oncogenic and immunological roles of transglutaminase 1 (TGM1) in human cancer. J Cancer Res Clin Oncol 150, 123 (2024). https://doi.org/10.1007/s00432-024-05640-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00432-024-05640-6