
Abstract
Background
Using the Clinical Audit Research and Evaluation of Motor Neuron Disease (CARE-MND) database and the Scottish Regenerative Neurology Tissue Bank, we aimed to outline the genetic epidemiology and phenotypes of an incident cohort of people with MND (pwMND) to gain a realistic impression of the genetic landscape and genotype–phenotype associations.
Methods
Phenotypic markers were identified from the CARE-MND platform. Sequence analysis of 48 genes was undertaken. Variants were classified using a structured evidence-based approach. Samples were also tested for C9orf72 hexanucleotide expansions using repeat-prime PCR methodology.
Results
339 pwMND donated a DNA sample: 44 (13.0%) fulfilled criteria for having a pathogenic variant/repeat expansion, 53.5% of those with a family history of MND and 9.3% of those without. The majority (30 (8.8%)) had a pathogenic C9orf72 repeat expansion, including two with intermediate expansions. Having a C9orf72 expansion was associated with a significantly lower Edinburgh Cognitive and Behavioural ALS Screen ALS-Specific score (p = 0.0005). The known pathogenic SOD1 variant p.(Ile114Thr), frequently observed in the Scottish population, was detected in 9 (2.7%) of total cases but in 17.9% of familial cases. Rare variants were detected in FUS and NEK1. One individual carried both a C9orf72 expansion and SOD1 variant.
Conclusions
Our results provide an accurate summary of MND demographics and genetic epidemiology. We recommend early genetic testing of people with cognitive impairment to ensure that C9orf72 carriers are given the best opportunity for informed treatment planning. Scotland is enriched for the SOD1 p.(Ile114Thr) variant and this has significant implications with regards to future genetically-targeted treatments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The genetics of motor neuron disease (MND) is an evolving landscape. People with MND (pwMND) are becoming increasingly aware of, and interested in, pursuing genetic testing. For those who proceed with genetic testing, interpretation of variant implications brings another dimension to their complex disease.
Classification of variant pathogenicity is problematic for many genetic diseases but becomes particularly difficult within the scope of a rare disease with multiple genetic links such as MND [1]. Barriers to firm classification have been acknowledged, including the relative paucity of functional studies and large pedigrees for assessment of co-segregation [2]. Variants of Uncertain Clinical Significance (VUS) are inevitable and bring their own diagnostic difficulties. There remains no consensus classification system for assessment of MND variant causality [3]. Latterly, the American College of Medical Genetics and Association for Molecular Pathology (ACMG-AMP) framework has been adopted [4,5,6,7,8].
The implications for MND are clear when we consider the imminent advent of genetically stratified therapies, which have the potential to involve prolonged commitment to invasive treatments. The recently Food and Drug Administration (FDA) approved drug, Tofersen, for MND mediated by the SOD1 gene is currently under review by UK and European drug regulators [9]. Accurate description of the genetic epidemiology of MND in Scotland will be important for planning potential delivery of these treatments. On a more immediate level there is the concern of burdening patients and their relatives with the anxiety of uncertain future risk [10].
In a previous study of the genetic epidemiology of MND in Scotland, 17% of pwMND had a potential genetic cause of their disease using a limited gene panel [5]. Key genetic mutations in this population include C9orf72 expansions and a Scottish founder variant in the SOD1 gene (p.(Ile114Thr)) [5, 7, 11]. In 2014, a neurodegenerative gene panel comprising 11 genes was incorporated into clinical practice in Scotland. However, with the emergence of new discoveries regarding genetic associations in MND, this quickly became outdated. The burden of MND-associated rare genes in this population is unknown. This information is required to inform clinical and diagnostic testing, outline priorities for future disease modelling studies and identify families for whom genetically-targeted treatments may be an option.
Scotland benefits from a longstanding national register for pwMND, now hosted by the CARE-MND Platform (Clinical Audit Research and Evaluation for MND) [12]. A broad selection of variables are available to allow us to appreciate the phenotypes of pwMND.
We aimed to study the genetic epidemiology and phenotypes of a well-characterised incident population of pwMND in Scotland diagnosed between 2015 and 2017. This cohort was carefully studied at the inception of the CARE-MND platform and coincided with a boost in MND nursing care in Scotland [12, 13]. Using an inclusive and contemporary research gene panel and adopting stringent variant classification methods, we aimed to obtain a realistic representation of the clinical impact of genetics in the Scottish MND population and identify any genotype–phenotype associations. Findings would inform clinical gene-panel testing pathways.
Methods
Gene-panel selection
A review was undertaken in 2015–2016 to update the existing 11-gene neurodegenerative disease gene panel. Existing UK-based MND-related gene panels and resources were examined [14,15,16,17,18,19]. The final panel consisted of 49 MND-associated genes for research study: ALS2, ANG, ANXA11, APP, ATL1, BSCL2, CCNF, CHCHD10, CHMP2B, CSF1R, DAO, DCTN1, ERBB4, FIG4, FUS, GRN, hnRNPA1, hnRNPA2/BA, HTRA1, ITM2B, MAPT, MATR3, NEFH, NEK1, NIPA1, NOTCH3, OPTN, PFN1, PLP1, PRNP, PRPH, PSEN1, PSEN2, REEP1, SETX, SIGMAR1, SOD1, SPAST/SPG4, SPG11, SPG20, SQSTM1, TAF15, TARDBP, TBK1, TUBA4A, UBQLN2, VAPB, VCP and the C9orf72 repeat expansion (previously published, https://doi.org/10.1007/s00415-022-11505-0) [7].
Recruitment and ethical approvals
All people diagnosed with MND in Scotland are invited to participate in the Scottish MND Register via the CARE-MND platform (ethical approvals MREC/98/0/56 1989–2010, 10/MRE00/78 2011–2015, and the Scotland A Research Ethics Committee 15/SS/0126 2015 onwards). DNA samples were donated to the Scottish MND DNA Bank and the Scottish Regenerative Neurology Tissue Bank (MREC/98/0/56 1989–2010, 10/MRE00/77 2011 to 2013, 13/ES/0126 2013–2015, 15/ES/0094 2015-present). The Lothian Birth Cohorts (LBC) – a research population of Scottish adults born in 1921 and 1936—were used as ancestry-matched genetic controls [20]. Ethical permission for the LBC1936 study protocol was obtained from the Multi-Centre Research Ethics Committee for Scotland (Wave 1: MREC/01/0/56), the Lothian Research Ethics Committee (Wave 1: LREC/2003/2/29), and the Scotland A Research Ethics Committee (Waves 2, 3,4 and 5: 07/MRE00/58). Ethical permission for the LBC1921 study protocol was obtained from the Lothian Research Ethics Committee (Wave 1: LREC/1998/4/183; Wave 2: LREC/2003/7/23; Wave 3: LREC1702/98/4/183), the Scotland A Research Ethics Committee (Waves 4 and 5: 10/MRE00/87).
Genotyping
Samples were genotyped using QiaSeq Amplicon Sequencing. Sequence analysis of a panel of 48 genes causally associated with neurodegeneration was carried out using a custom-designed QIAseq assay for library construction as per manufacturer’s instructions (QIAGEN). In brief, 80 ng of DNA was fragmented followed by adaptor ligation. Target enrichment was carried out by single primer extension, followed by sample indexing and amplification. Equal volumes of libraries were combined, and quantified using a Quantus™ Fluorometer as per manufacturer’s instructions. Paired-end sequencing of the resulting DNA library (at a concentration of 10 pM) was performed using an Illumina MiSeq instrument. Alignment and variant calling was performed using the QIAGEN CLC Genomics Workbench as per in-house standard operating procedure. Sequence read coverage was assessed against a browser extensible data (BED) file containing the genomic regions of interest.
All samples were also screened for C9orf72 hexanucleotide expansions using repeat-prime PCR methods [21], taking the total gene count to 49. Expansions > 30 repeats were considered pathogenic.
Variant classification
Each variant was systematically reviewed using the ACMG-AMP 28-point system and adhering to the Association for Clinical Genomic Science (ACGS) UK 2020 guidelines [6, 22]. A modified Delphi approach[23, 24] was taken to outline consensus criteria for the major MND-associated genes. Classification criteria have been used by our group previously[7] and are detailed in Supplementary Material 1.
Phenotyping and genotype–phenotype associations
Data were available for individuals who had provided written informed consent to data-sharing via the Scottish MND Register. A wide breadth of premorbid demographical (sex, ethnicity), environmental (smoking, heavy metal or pesticide exposure) and health-related variables (exercise, history of head injury, history of autoimmune disease, cardiovascular disease, malignancy, psychiatric illness), family history (of MND, dementia, early-onset dementia, other neurological disease, psychiatric disease) as well as markers of disease (age of onset, time to diagnosis, site of onset, classification of MND, riluzole use, feeding tube insertion, non-invasive ventilaton (NIV) use) were extracted and pre-processed from the CARE-MND database. MND-specific tools were used, including the validated and globally recognised Edinburgh Cognitive and Behavioural Amyotrophic Lateral Sclerosis Screen (ECAS)[25] and the revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R), a measure of limb, bulbar and respiratory function in daily living. Rate of ALSFRS-R decline was calculated using the concept of the ‘preslope’ or ALSFRS-R-based linear estimate of rare of disease progression, which is a recognised measurement in MND observational studies and clinical trials [26].
The following group were studied for genotype–phenotype associations: (i) pwMND with C9orf72 expansions, (ii) pwMND with SOD1 mutations, (iii) pwMND with SOD1 p.(Ile114Thr) variants. Descriptive statistics for phenotypic variables by group were summarised. In view of the large number of variables included relative to the number of individuals studied, univariate statistics were used to determine significance.
Statistical analyses
Data were formatted and analysed using R statistical programming [27]. Krippendorff’s alpha (k-alpha) statistic was used to assess formally inter-rater reliability; k-alpha score ranges from 0 (no concordance) to 1 (complete concordance) with good agreement considered ≥ 0.80 [28]. Univariate statistics (Fisher’s exact tests, t tests and Wilcoxon rank-sum tests) were used for association testing with correction for multiple testing using the Bonferroni method.
Results
Of 619 people with MND diagnosed in Scotland in 2015–2017, 437 (70.6%) consented to share their medical record data via the MND Register. The number of DNA samples donated by incident pwMND 2015–2017 was 339; this is representative of 54.8% of the incident MND cohort 2015–17 [13].
Variant classification
After three rounds of honing classification approaches using the modified Delphi method, classification concordance between raters was 84%, with an error rate of 3.0% and a mean k-alpha of 0.91 (95% CI 0.87, 0.95). In a review of use of the ACMG-AMP criteria amongst nine laboratories, average intra-laboratory k-alpha was 0.91 [29]. As our third-round k-alpha was ≥ 0.80 and compatible with clinical sequencing laboratory agreements, the consensus methods were considered to be appropriate for use.
Genotyping: C9orf72
Repeat-prime PCR for the C9orf72 expansion identified 29/339 (8.6%) individuals with > 30 GGGGCC repeats. Of these, one patient had an unusual intermediate-length expansion (70 repeats). One further patient had 28 repeats. Meta-analysis suggests that intermediate expansions 24–30 repeats in length are associated with MND [30]. In view of this evidence, our intermediate-length samples were both considered significant giving a final population frequency of 30/339 (8.8%).
Genotyping: panel sequencing
On gene panel sequencing, depth of coverage (≥ 20X) was, on average, 98% across the regions of interest. After VarSeq variant filtering, 503 variants were identified in 339 samples. Variants (including benign variants and VUS) were identified in 278/339 (82.0%) of samples. Fifteen (15/339, 4.4%) had a variant meeting criteria for pathogenicity (Table 2). The number of pwMND with a VUS in an MND-associated gene was 88 (88/339, 25.9%). Of these, 38 individuals (38/339, 1.1%) had a VUS which met some pathogenic ACMG-AMP criteria (‘hot’ VUS). These are summarised in Supplementary Material 2. One patient had both a pathogenic missense variant and a pathogenic C9orf72 expansion. A further six individuals had two variants of interest (including VUS meeting some criteria for pathogenicity) and these are summarised in Supplementary Material 3.
After the C9orf72 expansion, the most common variant in this MND cohort was the SOD1 p.(Ile114Thr) variant (n = 9), previously described as a founder mutation in the Scottish population [5, 7, 11]. Three further SOD1 variants were identified. One of these, p.(Ala146Asp), had previously been seen in a different Scottish individual with MND[5] and was absent from the control population and gnomAD. Two other SOD1 missense variants were observed: p.(Gln23His) and p.(Gly73Cys). These variants have not previously been identified in the Scottish population. The frequency of SOD1 mutations in this cohort was therefore 12/339, 3.5%.
A loss of function (LoF) variant (p.Tyr479Metfs*50) was observed in the FUS gene. This is a novel variant in a genomic location near to previously described MND-associated frameshift mutations.
Two individuals were found to have a LoF variant in exon 21 of NEK1, which was absent from gnomAD and classified as pathogenic (p.(Glu634Lysfs*11)). As far as we can determine from patient histories, the patients were unrelated. A further LoF NEK1 splice donor variant (NM_012224.2:c.868 + 1G > C) was identified in a different individual; this is predicted to abolish the canonical splice donor site and initiate nonsense-mediated decay. However, the variant is present in gnomAD and so it did not meet criteria for pathogenicity (see Supplementary Material 2).
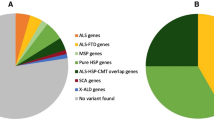
In summary, a total of 44/339 (13.0%) of individuals (including those with C9orf72 expansions) had a potential genetic explanation for their disease (Fig. 1) (see Table 1).

Summary of genetic epidemiology of incident MND cohort 2015–2017, VUS variant of uncertain clinical significance
Genotype–phenotype associations
Phenotypic characteristics of the genotyped cohort (n = 339) as well as those of individuals with i) C9orf72 pathogenic expansions, (ii) SOD1 pathogenic variants and (iii) SOD1 p(.Ile114Thr) variants are summarised in Table 2.
Univariate statistics with Bonferroni correction revealed that a family history of MND and lower ECAS: ALS-Specific Score were associated with having a C9orf72 expansion (Table 2). There were two individuals with intermediate-length repeat expansions. Both individuals were diagnosed in their early sixties with upper limb-onset disease and had diagnoses of ALS with cognitive impairment identified via ECAS measurements (ECAS total score 73 in patient with 28 repeats, 79 in patient with 70 repeats). Neither had a family history of MND nor past medical or family history of psychiatric conditions. The individual with 28 repeats died 2.6 years after symptom onset whereas the individual with 70 repeats was alive 1.6 years after onset.
There was a significant association between SOD1 carriers and having a family history of MND (p = 0.0001) (Table 2). SOD1 p(.Ile114Thr) carriers were studied separately and they were also significantly associated with having a family history of MND (p = 0.0003) (Table 2). Family-history details of p(.Ile114Thr) carriers are described in Table 3. The individual with both the C9orf72 expansion and SOD1 p(.Ile114Thr) variant was a male who had lower limb-onset ALS age 68 and who developed cognitive impairment as assessed by ECAS. Interestingly, he had no family history of MND or other neurological conditions. He died 65 months (5.4 years) after symptom onset.
The patient with the FUS LoF mutation had young-onset ALS with short survival (20 months from onset) and a family history of MND. The individuals with NEK1 LoF mutations did not have family histories of MND.
Of those with a family history of MND (28/339, 8.3%), 9/28 (32.1%) had pathogenic C9orf72 expansions and 6/28 (21.4%) had SOD1 mutations. Including all pathogenic variants and expansions, there was an overall mutation rate of 16/28, 57.1%. However, even in those without a clear family history of MND the frequency of C9orf72 was 21/311 (6.8%) and SOD1 6/311 (1.9%) with an overall pathogenic mutation rate of 28/311, 9.0%.
Discussion
Genetic epidemiology of MND in Scotland
Thirteen per cent of pwMND met criteria for having a pathogenic mutation/expansion. A further 11.2% of the cohort had potential ‘hot’ VUS but there was insufficient evidence to classify these variants as causative and they would not have been reportable clinically. As such, we report a realistic impression of the MND genetic landscape in Scotland. Almost two thirds of those with family history of MND and almost a tenth of those without a family history of MND had a potentially pathogenic mutation. The rates amongst apparently sporadic cases are very similar to those reported in a recent large study of ALS genomes, using a 90-gene panel and ACMG-AMP led classification [8].
The most important regions of interest in this incident Scottish cohort are: the C9orf72 expansion and the SOD1 and NEK1 genes. The proportion of C9orf72 expansion carriers (8.8%) was lower than in a previous Scottish study (10.2%) [5]. However, the 2015–17 cohort is more unselected than our historical cohort and so more representative. The C9orf72 expansion is the commonest cause of familial MND affecting 32.1% of cases and 6.8% of apparently sporadic cases. Similar rates have been reported in people of European, USA and Australian origin [8, 31, 32]. SOD1 mutations were identified in 3.5% of cases overall, but 21.4% of familial cases. This is again lower than in our selected 1989–2014 cohort (5% of all cases, 29% of familial cases) though figures are higher than global population estimates (1–2% sporadic and 12% familial cases [32, 33]). As before, the p.(Ile114Thr) variant is the biggest contributor to this observation, implicated in 17.9% (5/28) of familial cases. The relative ethnic homogeneity of the Scottish MND population is likely a factor in the persistence of this variant [13]. This has significant implications as antisense oligonucleotide (ASO) gene-modifying treatments for SOD1 carriers appear promising, with the potential to treat up to 4% of the Scottish MND population [9].
We also discovered potentially pathogenic LoF variants in the NEK1 gene. This gene is as yet poorly characterised in MND populations but loss of function is considered mechanistic [34]. Of note, the NEK1 missense mutation p.(Arg261His) was also identified in this study (Supplementary Material 2). First identified in an isolated community in the Netherlands, it was thought to be a risk variant for ALS using meta-analysed data (p = 4.8 × 10–5, OR 2.4 in cases versus controls) [35]. In the Scottish 1989–2014 cohort, five cases and two controls had this variant and it was considered Likely Pathogenic. In the 2015–17 cohort, it was present in nine cases and seven controls. Other clinical samples reported to ClinVar suggest that the variant may be a VUS, Likely Benign or Benign. In the absence of functional studies, we would now classify this variant as likely benign, present in a total of 1.8% of cases and 0.7% of controls in Scottish MND cohorts (1989–2017).
Genotype–phenotype associations
Univariate association testing of C9orf72 expansion carriers showed that they have significantly poorer ALS-Specific ECAS scores; this finding parallels other studies and supports a link between C9orf72 and MND-FTD (Motor Neuron Disease with Frontotemporal Dementia) spectrum disorders [36,37,38,39]. This highlights the utility and importance of early cognitive assessment using the ECAS assessment tool following diagnosis of MND. The penetrance for the C9orf72 expansion is incomplete but is thought to be higher in MND than in pure FTD; it is also unaffected by prior family history of disease and increases with age [40]. Although previous trials of ASO therapies for C9orf72 expansions were terminated due to lack of efficacy, exploration of other genetically-targeted drug treatments for C9orf72 expansions are ongoing and an urgent priority for the MND/ALS community. Early identification of cognitive impairment will therefore be crucial to guide appropriate genetic testing and potential drug trial participation before people with C9orf72 expansions lose capacity. Whilst a family history of MND was significantly associated with having a C9orf72 expansion, having a family history of young-onset dementia, psychiatric disease or other neurological disease did not reach correct significance (p = 0.1, p = 0.2, and p = 0.03 respectively). This is perhaps surprising [41] but likely reflects the low patient numbers and potential under-reporting of family histories.
Whilst it did not meet Bonferroni-corrected threshold for significance, fewer C9orf72 expansion carriers in our population were initiated on NIV (p = 0.003). C9orf72 expansion carriers are thought to have fast respiratory decline [42]. We might infer that this population might not have had opportunity to commence on NIV due to inability to consent and comply with treatment (because of cognitive impairment) and because of rapidly progressive disease. Inclusion of Forced Vital Capacity respiratory measures in a longitudinal survival study might help to validate these findings. In the meantime, early assessment of C9orf72 status and ECAS cognitive assessment in a clinical setting could guide intervention strategies and help to maximise patient access to available intervention. The male-to-female ratio in C9orf72 expansion carriers was 50:50, ie., more females that would be expected in a typical MND cohort. In a Scottish study, we found that significantly fewer females than males were commenced on NIV (p < 0.0001) [13]. This was unexplained but, on reflection, C9orf72 status might be a contributor.
We identified two patients with intermediate-length repeat expansions who both had classical ALS phenotypes and cognitive impairment. Intermediate repeats are more common in those with neuropsychiatric disease (including FTD) and our results provide further evidence of this phenotype [30].
In contrast to C9orf72, more SOD1 mutation carriers started NIV, though this did not reach corrected significance (p = 0.007). This may be due to their having more predictable limb-onset ALS disease. ECAS scores of SOD1 carriers were reflective of the population as a whole; patients with SOD1 mutations tend not to have significant cognitive impairment and so this was anticipated [43]. Individuals who had the p.(Ile114Thr) mutation all had limb-onset ALS suggesting that the variant may result in a ‘typical’ SOD1 phenotype, as has been described in recent meta-analyses [44]. Indeed, none required gastrostomy insertion by the time of censorship (p = 0.03), suggesting that bulbar disease was not a prominent feature. Family histories of individuals with the p.(Ile114Thr) variant revealed histories of limb-onset MND with similar ages to the proband, although details about disease site and onset are missing for some. Family histories of multiple sclerosis were also apparent, perhaps implying either a shared genetic aetiology or a historical misdiagnosis of phenotype.
The individual with MND with the FUS LoF variant has young-onset rapid progressive disease, meeting the expected phenotypic profile for this gene [45]. The NEK1 gene is relatively newly described and genotype–phenotype observations are limited but there is a suggestion in the literature that carriers are more likely to have the flail arm/bibrachial phenotype [46]. Our two NEK1-variant carriers, however, had ALS (bulbar onset and limb onset, respectively). Interestingly, we identified one patient with both a C9orf72 expansion and the SOD1 p.(Ile114Thr) variant. This individual had a typical age of onset and relatively slow progression, perhaps in-keeping with a SOD1 phenotype, but had cognitive impairment in line with C9orf72 phenotype.
Strengths and limitations
Due to the well-established and robust nature of the CARE-MND platform and the Scottish MND register we have been able to gain a wealth of information about our MND population. The 2015–2017 cohort was the first to be studied within the CARE-MND platform and is comprehensive, with ascertainment of 99% of pwMND in Scotland, using capture-recapture methodology [13]. These data were also unaffected by the coronavirus pandemic. We would therefore anticipate that patients diagnosed subsequently would have similar characteristics and future and ongoing analysis of CARE-MND Register data should support this. However, just over half of the incident MND population (54.8%) contributed to this genetic study. Reasons for not achieving higher ascertainment might include patient choice, limited discussions regarding research options prior to the CARE-MND initiative and rapidly deteriorating disease, meaning that patients were less willing to devote time and efforts to research. Currently, genetic research in Scotland does not offer feedback of results and does not lead to treatment modification and so benefits to patients at an individual level are limited. Allowing for these factors, we consider our recruitment figures to be appropriate and reflective of the generosity of the Scottish MND patient community.
We acknowledge that that we have not confirmed variants identified in the incident cohort using Sanger sequencing. Whilst concordance between next-generation sequencing (NGS) and Sanger techniques is now excellent, the risk of false positives with NGS may be 1.3% [47]. One reason that this was not pursued was that sequencing was performed as part of a research study only, with results not being fed back to patients. In Scotland, all patients with MND are encouraged to store DNA in an NHS clinical-approved laboratory and if a drug were to become available for an MND-associated gene, the patient or their family members could activate confirmatory testing. We have also adopted a panel-sequencing approach for this study to make the results more generalisable to clinical practice. Advances in gene sequencing technology, including long-read sequencing, may allow the detection of even more rare genetic variation in future studies [48].
The number of patients with mutations also is small to make firm conclusions, especially with regard to genotype–phenotype associations. Bonferroni correction of statistical testing gives us conservative estimates which are hypothesis-generating, though near-significant results do confirm clinical observations in practice.
The majority of pwMND in Scotland are of White Scottish origin[13] (98.8% in this study). The SOD1 p.(Ile114Thr) variant has been detected in European, North American, and Australasian populations but our results may not be generalisable to other ethnic groups or populations.
Conclusions
Our results show that the CARE-MND database provides a wealth of information about people diagnosed with MND which can be used to inform pwMND and stakeholders. By employing structured variant classification and using an extensive gene-panel approach we have provided a realistic estimate of the frequency of rare variants in the Scottish MND population for the first time. This information has subsequently informed patient information sources. We have confirmed the frequency of a specific SOD1 variant (SOD1 p.(Ile114Thr)) in the population and have detailed associated phenotypic characteristics; awareness of such key local mutations is essential for the delivery of future genetically-targeted drug trials and drug approvals. NEK1 LoF variant carriers (0.6%) make up a small but important subset of patients. As a consequence of this study, the Scottish Neurodegenerative Disease Gene Panel has been updated to include NEK1 (https://www.nhsggc.org.uk/media/271442/germlinetestdirectory_v10.pdf). As NEK1 carriers were all apparently sporadic cases, future work into functional and pathological correlates is merited. We have also demonstrated that the SOD1 p.(Ile114Thr) variant and the C9orf72 expansion can co-exist and should be tested simultaneously—to our knowledge this is not widely described. However, more systematic gene testing of pwMND would likely reveal further cases and help to determine which gene phenotype is more strongly manifested. From a practical perspective, we suggest that early clinical gene testing may guide management, either by prompting consideration of NIV prior to cognitive decline (C9orf72) or by preparing patients early for the likelihood of NIV and/or reduced pressure to consider urgent gastrostomy insertion (SOD1).
Data Availability
Data supporting the genetic findings of this study are available within the article and supplementary material. Raw CARE-MND data are not available due to their containing information that could compromise the privacy of research participants. Further information about the CARE-MND database can be found at: https://www.care-mnd.org.uk/.
References
MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, et al (2014) Guidelines for investigating causality of sequence variants in human disease. Nature [Internet] 508(7497):469–76. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4180223&tool=pmcentrez&rendertype=abstract. Accessed 24 Apr 2014
Marangi G, Traynor BJ (2015) Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res [Internet] 1607:75–93. https://www.sciencedirect.com/science/article/pii/S0006899314013614?via%3Dihub. Accessed 14 May 2015
Dekker AM, Seelen M, van Doormaal PTC, van Rheenen W, Bothof RJP, van Riessen T, et al (2016) Large-scale screening in sporadic amyotrophic lateral sclerosis identifies genetic modifiers in C9orf72 repeat carriers. Neurobiol Aging [Internet] 39:220.e9–220.e15. http://www.sciencedirect.com/science/article/pii/S0197458015006168?via%3Dihub. Accessed 1 Mar 2016
Keogh MJ, Wei W, Wilson I, Coxhead J, Ryan S, Rollinson S, et al (2017) Genetic compendium of 1511 human brains available through the UK Medical Research Council Brain Banks Network Resource. Genome Res [Internet] 27(1):165–73. http://www.ncbi.nlm.nih.gov/pubmed/28003435. Accessed Jan 2017
Black HA, Leighton DJ, Cleary EM, Rose E, Stephenson L, Colville S, et al (2017) Genetic epidemiology of motor neuron disease-associated variants in the Scottish population. Neurobiol Aging [Internet] 51:178.e11–178.e20. http://linkinghub.elsevier.com/retrieve/pii/S0197458016303190. Accessed Mar 2017
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med [Internet] 17(5):405–24. http://www.ncbi.nlm.nih.gov/pubmed/25741868. Accessed May 2015
Leighton DJ, Ansari M, Newton J, Parry D, Cleary E, Colville S et al (2023) Genotype–phenotype characterisation of long survivors with motor neuron disease in Scotland. J Neurol [Internet]. 270(3):1702–1712. https://doi.org/10.1007/s00415-022-11505-0. Accessed 14 Mar 2023
Van Daele SH, Moisse M, van Vugt JJFA, Zwamborn RAJ, van der Spek R, van Rheenen W, et al (2023) Genetic variability in sporadic amyotrophic lateral sclerosis. Brain [Internet]. 146(9):3760–9. http://www.ncbi.nlm.nih.gov/pubmed/37043475. Accessed 1 Sep 2023
Miller TM, Cudkowicz ME, Genge A, Shaw PJ, Sobue G, Bucelli RC et al (2022) Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N Engl J Med [Internet]. 387(12):1099–1110. https://doi.org/10.1056/NEJMoa2204705. Accessed 22 Sep 2022
Roggenbuck J, Quick A, Kolb SJ (2017) Genetic testing and genetic counseling for amyotrophic lateral sclerosis: An update for clinicians [Internet]. Vol. 19, Genetics in Medicine. [cited 2016 Oct 24]. p. 267–74. http://www.ncbi.nlm.nih.gov/pubmed/27537704
Jones CT, Swingler RJ, Simpson SA, Brock DJ (1995) Superoxide dismutase mutations in an unselected cohort of Scottish amyotrophic lateral sclerosis patients. J Med Genet [Internet] 32(4):290–2. http://www.ncbi.nlm.nih.gov/pubmed/7643359. Accessed Apr 1995
Leighton D, Newton J, Colville S, Bethell A, Craig G, Cunningham L, et al (2019) Clinical audit research and evaluation of motor neuron disease (CARE-MND): a national electronic platform for prospective, longitudinal monitoring of MND in Scotland. Amyotroph Lateral Scler Front Degener [Internet] 20(3–4):242–50. http://www.ncbi.nlm.nih.gov/pubmed/30889975. Accessed 3 Apr 2019
Leighton DJ, Newton J, Stephenson LJ, Colville S, Davenport R, Gorrie G et al (2019) Changing epidemiology of motor neurone disease in Scotland. J Neurol [Internet] 266(4):817–825. https://doi.org/10.1007/s00415-019-09190-7. Accessed 25 Apr 2019
UK Genetic Testing Network (2019) NHS UK Genetic Testing Network. [cited 2019 Jul 17]. London Institute of Neurology - UK Genetic Testing Network. https://ukgtn.nhs.uk/find-a-test/search-by-laboratory/laboratory/london-institute-of-neurology-47/
UK Genetic Testing Network (2019) Sheffield RGC - UK Genetic Testing Network [Internet]. [cited 2019 Jul 17]. https://ukgtn.nhs.uk/find-a-test/search-by-laboratory/laboratory/sheffield-rgc-40/
National Centre for Biotechnology Information (2017) Gene Testing Registry. ALS panel - Tests - GTR - NCBI [Internet]. [cited 2019 Jul 17]. https://www.ncbi.nlm.nih.gov/gtr/tests/503152/
Abel, Olubunmi; Al-Chalabi A (2015) Institute of Psychiatry, Psychology & Neuroscience, Kings College London. 2015. The Amyotrophic Lateral Sclerosis Online Genetics Database (ALSoD). http://alsod.iop.kcl.ac.uk/
Abel, O (2016) ALSoD (6.0) [Internet]. http://alsod.iop.kcl.ac.uk/
Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA (2000) Online Mendelian Inheritance in Man (OMIM). Hum Mutat [Internet] 15(1):57–61. https://www.omim.org/
Taylor AM, Pattie A, Deary IJ (2018) Cohort Profile Update: The Lothian Birth Cohorts of 1921 and 1936. Int J Epidemiol [Internet] 47(4):1042–1042r. https://academic.oup.com/ije/article/47/4/1042/4931207. Accessed 1 Aug 2018
Cleary EM, Pal S, Azam T, Moore DJ, Swingler R, Gorrie G, et al (2016) Improved PCR based methods for detecting C9orf72 hexanucleotide repeat expansions. Mol Cell Probes [Internet] 30(4):218–24. http://www.ncbi.nlm.nih.gov/pubmed/27288208. Accessed Aug 2016
Ellard S, Baple EL, Callaway A, Berry I, Forrester N, Turnbull C, et al (2020) ACGS best practice guidelines for variant classification in rare disease 2020. Assoc Clin Genomic Sci [Internet]. https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf
Fox BI, Hollingsworth JC, Gray MD, Hollingsworth ML, Gao J, Hansen RA (2013) Developing an expert panel process to refine health outcome definitions in observational data. J Biomed Inform [Internet]. 46(5):795–804. http://www.ncbi.nlm.nih.gov/pubmed/23770041. Accessed Oct 2013
Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, et al (2014) Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet [Internet] 46(2):107–15. http://www.ncbi.nlm.nih.gov/pubmed/24362816. Accessed Feb 2014
Niven E, Newton J, Foley J, Colville S, Swingler R, Chandran S et al (2015) Validation of the Edinburgh Cognitive and Behavioural Amyotrophic Lateral Sclerosis Screen (ECAS): a cognitive tool for motor disorders. Amyotroph Lateral Scler Front Degener [Internet]. 16(3–4):172–179. https://doi.org/10.3109/21678421.2015.1030430. Accessed 27 Apr 2015
Elamin M, Bede P, Montuschi A, Pender N, Chio A, Hardiman O (2015) Predicting prognosis in amyotrophic lateral sclerosis: a simple algorithm. J Neurol [Internet] 262(6):1447–54. http://www.ncbi.nlm.nih.gov/pubmed/25860344. Accessed Jun 2015
R Core Team (2017) R: A Language and Environment for Statistical Computing [Internet]. R Foundation for Statistical Computing. Vienne, Austria: R Foundation for Statistical Computing, Vienna, Austria. http://www.r-project.org/
Krippendorff K (2011) Agreement and Information in the Reliability of Coding. Commun Methods Meas [Internet]. 5(2):93–112. https://doi.org/10.1080/19312458.2011.568376. Accessed 1 Apr 2011
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al (2016) Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet [Internet] 98(6):1067–76. http://www.cell.com/ajhg/pdf/S0002-9297(16)30059-3.pdf
Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S et al (2019) C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol Commun [Internet]. 7(1):115. https://doi.org/10.1186/s40478-019-0724-4. Accessed 17 Dec 2019
Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, et al (2012) Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol [Internet]. 11(4):323–30. http://www.ncbi.nlm.nih.gov/pubmed/22406228. Accessed Apr 2012
Chiò A, Mazzini L, D’Alfonso S, Corrado L, Canosa A, Moglia C, et al (2018) The multistep hypothesis of ALS revisited. Neurology [Internet] 91(7):e635–42. http://www.ncbi.nlm.nih.gov/pubmed/30045958. Accessed 25 Jul 2018
Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci [Internet] 17(1):17–23. http://www.ncbi.nlm.nih.gov/pubmed/24369373. Accessed Jan 2014
Nguyen HP, Van Mossevelde S, Dillen L, De Bleecker JL, Moisse M, Van Damme P, et al (2018) NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol Aging [Internet] 61:255.e1–255.e7. https://www.sciencedirect.com/science/article/pii/S0197458017302774?via%3Dihub. Accessed 1 Jan 2018
Kenna KP, van Doormaal PTC, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, et al (2016) NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet [Internet] 48(9):1037–42. http://www.ncbi.nlm.nih.gov/pubmed/27455347
Watson A, Pribadi M, Chowdari K, Clifton S, Joel Wood, Miller BL, et al (2016) C9orf72 repeat expansions that cause frontotemporal dementia are detectable among patients with psychosis. Psychiatry Res [Internet] 235:200–2. http://www.ncbi.nlm.nih.gov/pubmed/26723138. Accessed 8 Jan 2016
Vats A, Gourie-Devi M, Suroliya V, Verma S, Faruq M, Sharma A, et al (2017) Analysis of C9orf72 repeat expansion in amyotrophic lateral sclerosis patients from North India. J Neurol Sci [Internet]. 373:55–7. http://www.ncbi.nlm.nih.gov/pubmed/28131227. Accessed 15 Feb 2017
Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, et al (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. Lancet Neurol [Internet] 14(3):291–301. http://www.ncbi.nlm.nih.gov/pubmed/25638642. Accessed 28 Mar 2015
Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, et al (2012) Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol [Internet] 11(3):232–40. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3315021&tool=pmcentrez&rendertype=abstract. Accessed Mar 2012
Murphy NA, Arthur KC, Tienari PJ, Houlden H, Chiò A, Traynor BJ (2017) Age-related penetrance of the C9orf72 repeat expansion. Sci Rep [Internet] 7(1):2116. http://www.nature.com/articles/s41598-017-02364-1. Accessed 18 Dec 2017
O’Brien M, Burke T, Heverin M, Vajda A, McLaughlin R, Gibbons J et al (2017) Clustering of neuropsychiatric disease in first-degree and second-degree relatives of patients with amyotrophic lateral sclerosis. JAMA Neurol [Internet] 74(12):1425–1430. https://doi.org/10.1001/jamaneurol.2017.2699. Accessed 1 Dec 2017
Miltenberger-Miltenyi G, Conceição VA, Gromicho M, Pronto-Laborinho AC, Pinto S, Andersen PM, et al (2019) C9orf72 expansion is associated with accelerated decline of respiratory function and decreased survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry [Internet] 90(1):118–20. http://www.ncbi.nlm.nih.gov/pubmed/29661924. Accessed 16 Jan 2019
Yamashita S, Ando Y (2015) Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl Neurodegener [Internet] 4(1):13. http://www.ncbi.nlm.nih.gov/pubmed/26213621. Accessed 24 Dec 2015
Domi T, Schito P, Sferruzza G, Russo T, Pozzi L, Agosta F, et al (2024) Unveiling the SOD1-mediated ALS phenotype: insights from a comprehensive meta-analysis. J Neurol [Internet] 271(3):1342–54. http://www.ncbi.nlm.nih.gov/pubmed/37930481
Vance C, Rogelj B, Hortobágyi T, De Vos KJ, Nishimura AL, Sreedharan J, et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science [Internet]. 323(5918):1208–11. http://www.ncbi.nlm.nih.gov/pubmed/19251628. Accessed 27 Feb 2009
Riva N, Pozzi L, Russo T, Pipitone GB, Schito P, Domi T et al (2022) NEK1 variants in a cohort of Italian Patients With Amyotrophic Lateral Sclerosis. Front Neurosci [Internet]. https://doi.org/10.3389/fnins.2022.833051/full. Accessed 14 Apr 2022
Mu W, Lu HM, Chen J, Li S, Elliott AM (2016). Sanger confirmation is required to achieve optimal sensitivity and specificity in next-generation sequencing panel testing. J Mol Diagnostics [Internet] 18(6):923–32. http://www.ncbi.nlm.nih.gov/pubmed/27720647. Accessed Nov 2016
Udine E, Jain A, van Blitterswijk M (2023) Advances in sequencing technologies for amyotrophic lateral sclerosis research. Mol Neurodegener [Internet] 18(1):4. https://doi.org/10.1186/s13024-022-00593-1. Accessed 13 Jan 2023
Acknowledgements
We are grateful to all the people with MND who participated in these studies. We continue to devote all efforts to ensure that the information they donate is maximised to help patients of the future.
For the purpose of open access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript version arising from this submission
CARE-MND Consortium members: Andrew Bethell, Susan Byrne, Siddharthan Chandran, Myles Connor, Gillian Craig, Richard Davenport, Ondrej Dolezal, Callum Duncan, Moira Flett, Louise Gardiner, Jessica Gill, Isaac Chau, Janice Hatrick, Micheala Johnson, Katja Lassak, Juan Larraz, Helen Lennox, Pauline MacDonald, Laura Marshall, Dympna McAleer, Alison McEleney, Kitty Millar, Ian Morrison, Louise Murrie, Judith Newton, Suvankar Pal, David Perry, Gowri Saravanan, David Simpson, Susan Stewart, Dorothy Storey, Gill Stott, Robert Swingler, David Thompson, Carol Thornton, Carolyn Webber and Michael Wong.
Lothian birth cohorts group members: Sarah Harris, James Prendergast, Tom Russ, Adele Taylor, and Ian Deary.
Funding
DL received funding for PhD study at the inception of this study from the Chief Scientist Office for Scotland, the Motor Neuron Disease Association and Motor Neuron Disease Scotland (CAF/MND/15/01). This work is also supported by the UK Dementia Research Institute which receives its funding from UK DRI Ltd, funded by the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflicts of interest
The authors report no competing interests.
Ethics approval
The Scottish MND Register/CARE-MND Platform is approved by MREC/98/0/56 1989–2010, 10/MRE00/78 2011–2015, and the Scotland A Research Ethics Committee 15/SS/0126 2015 onwards. The Scottish MND DNA Bank and the Scottish Regenerative Neurology Tissue Bank are approved by MREC/98/0/56 1989–2010, 10/MRE00/77 2011 to 2013, 13/ES/0126 2013–2015, 15/ES/0094 2015-present. Ethical permission for the LBC1936 study protocol was obtained from the Multi-Centre Research Ethics Committee for Scotland (Wave 1: MREC/01/0/56), the Lothian Research Ethics Committee (Wave 1: LREC/2003/2/29), and the Scotland A Research Ethics Committee (Waves 2, 3,4 and 5: 07/MRE00/58). Ethical permission for the LBC1921 study protocol was obtained from the Lothian Research Ethics Committee (Wave 1: LREC/1998/4/183; Wave 2: LREC/2003/7/23; Wave 3: LREC1702/98/4/183), the Scotland A Research Ethics Committee (Waves 4 and 5: 10/MRE00/87).
Consent to participate
All participants provided informed consent to before contributing to the Scottish MND Register/CARE-MND Platform, Scottish MND DNA Bank and the Scottish Regenerative Neurology Tissue Bank and Lothian Birth Cohorts.
Additional information
The members of the Lothian Birth Cohorts Group and the CARE-MND Consortium are mentioned in Acknowledgments section.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Leighton, D.J., Ansari, M., Newton, J. et al. Genotypes and phenotypes of motor neuron disease: an update of the genetic landscape in Scotland. J Neurol (2024). https://doi.org/10.1007/s00415-024-12450-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00415-024-12450-w