
Abstract
Background and objectives
Primary lateral sclerosis (PLS) is a motor neuron disease characterised by loss of the upper motor neurons. Most patients present with slowly progressive spasticity of the legs, which may also spread to the arms or bulbar regions. It is challenging to distinguish between PLS, early-stage amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegia (HSP). The current diagnostic criteria advise against extensive genetic testing. This recommendation is, however, based on limited data.
Methods
We aim to genetically characterize a PLS cohort using whole exome sequencing (WES) for genes associated with ALS, HSP, ataxia and movement disorders (364 genes) and C9orf72 repeat expansions. Patients fulfilling the definite PLS criteria by Turner et al. and with available DNA samples of sufficient quality were recruited from an on-going, population-based epidemiological study. Genetic variants were classified according to the ACMG criteria and assigned to groups based on disease association.
Results
WES was performed in 139 patients and the presence of repeat expansions in C9orf72 was analysed separately in 129 patients. This resulted in 31 variants of which 11 were (likely) pathogenic. (Likely) pathogenic variants resulted in 3 groups based on disease association: ALS-FTD (C9orf72, TBK1), pure HSP (SPAST, SPG7), “ALS-HSP-CMT overlap” (FIG4, NEFL, SPG11).
Discussion
In a cohort of 139 PLS patients, genetic analyses resulted in 31 variants (22%) of which 10 (7%) (likely) pathogenic associated with different diseases (predominantly ALS and HSP). Based on these results and the literature, we advise to consider genetic analyses in the diagnostic work-up for PLS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary lateral sclerosis (PLS) is a rare neurodegenerative disorder characterized by the predominant loss of upper motor neurons (UMNs) [1]. It is considered to be one of the extremes of the motor neuron disease (MND) spectrum, which in turn forms a continuum with frontotemporal dementia (FTD) [2]. Indeed, studies show that approximately 50% of PLS patients may also develop cognitive and/or behavioural changes within the FTD-spectrum [3, 4].
The exact incidence and prevalence of PLS are unknown, but it is estimated that they make up roughly 1–5% of all MND patients seen at specialized clinics [1, 5]. Diagnosing PLS is challenging, over the years several diagnostic criteria have been implemented [6,7,8], but to date there is no gold standard (including post-mortem histopathological analysis). The diagnosis is based on recognizing characteristic clinical features and ruling out potential other causes of observed symptoms. Patients most commonly present with a history of slowly progressive spasticity in the legs which over time spreads cranially or more rarely with a spastic dysarthria that spreads caudally to cause spasticity in the extremities. Subtle signs of Parkinsonism have also been reported in PLS patients [1, 7, 9].
Through the use of ancillary investigations, such as MRI-scans and laboratory testing, several conditions with similar clinical presentations can be ruled out relatively straightforward [e.g., primary progressive multiple sclerosis and X-linked adrenoleukodystrophy (X-ALD)]. However, for other conditions in the differential diagnosis, such as amyotrophic lateral sclerosis (ALS) and hereditary spastic paraplegia (HSP) this is more complex. The initial onset of ALS may be characterized by UMN signs only with lower motor neuron loss occurring later in the disease course [2]. Therefore, follow-up over a number of years is frequently required to confidently establish the absence of lower motor neuron involvement, which is considered to be a defining feature of PLS.
HSP is a heterogeneous group of genetic disorders characterized by slowly progressive spasticity of the legs (pure forms) or with additional features such as epilepsy, learning and developmental problems and hearing loss (complex forms) [1, 10, 11]. In general, onset of HSP is at younger age than in PLS, ranging from childhood to early adulthood, symptoms are symmetrical and limited to the lower extremities, progression is slower and there commonly is a positive family history for the disease [10, 12]. However, asymptomatic UMN signs in the cervical region are frequently seen in HSP cases [13], late-onset in the late fifties and early sixties has also been reported, as well as asymmetry [14,15,16]. The mode of inheritance in HSP can also be recessive or X-linked, or the cause can be de novo mutations, therefore suggesting an apparently negative family history [16]. What complicates matters even further is the fact that to date over 80 genetic HSP subtypes have been identified, but which still do not explain all cases, and that therefore negative DNA test results do not rule out HSP [11]. Lastly, recent pathology studies have shown that PSP may also present with isolated UMN signs and therefore mimic PLS [9]. PLS is a clinical syndrome and may be considered as a collection of primary pyramidal disorders with heterogeneous causes, which might be subclassified more specifically using genetics.
As our knowledge of neurodegenerative diseases has advanced over the last decades, the diagnostic criteria for PLS have been revised a number of times to incorporate these novel insights. The current consensus criteria (2020) state the following regarding genetic testing “Screening of panels for pathogenic genetic variants associated with spastic paraparesis (e.g., SPAST) is warranted in cases of progressive UMN syndromes restricted to symmetrical lower limb involvement. It is reasonable to routinely exclude the most common hereditary cause of ALS in Caucasian populations, namely an expansion in C9orf72. However, the plethora of very rare genetic variants reported in association with pedigrees containing ALS-like syndromes, including some with apparently pure UMN phenotypes, should not be considered routine tests in the diagnosis of PLS”[8]. Although this statement reflects international consensus, it must be noted that only a limited number of very small studies have been conducted on the genetics of PLS and that the data supporting this statement is quite limited. We therefore set out to perform an in-depth genetic characterization of a large cohort of primary lateral sclerosis patients from The Netherlands using whole exome sequencing with the goal of providing additional guidance to clinicians with respect to genetic testing and/or to refine the diagnostic criteria for PLS.
Methods
Study population
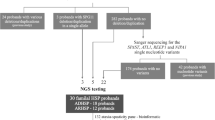
All participants were recruited through an on-going population-based, epidemiological study on MND in The Netherlands (PAN study), which started in 2006 and was approved by the medical-ethical committee of the University Medical Center Utrecht. [17] Written informed consent was obtained from all participants. In this study, patients consent to the use of data from their medical records for scientific research, are asked to fill in detailed questionnaires and provide bio-samples, including DNA. We included patients of whom a DNA sample of sufficient quality was present and which fulfilled the most recent Turner criteria for definite PLS. These criteria require age at diagnosis to be ≥ 25 years, isolated UMN signs in at least two regions, and a disease duration of more than 4 years [8].
Baseline clinical data were collected at diagnosis or within a 3-month time window after diagnosis, and included: gender, age at diagnosis, disease duration (from onset and from diagnosis), site of disease onset, regions of UMN involvement (bulbar, cervical, thoracic, lumbosacral), cognitive and/or behavioural changes (by history taking and if available Edinburgh Cognitive and behavioural ALS screen, Frontal Assessment Battery and ALS and Frontotemporal Dementia Questionnaire[18,19,20]) and family history. In cases of only one region of UMN involvement at diagnosis, follow-up data (if available) was reviewed to see if patients developed UMN dysfunction in more regions making them fulfil the diagnostic criteria (cases remaining in one region or without available follow-up data were excluded). Site of disease onset was divided into: spinal, bulbar, or respiratory. Date of symptom onset was determined at the outpatient clinic as described in our Standard Operating Procedures (SOP) for Clinical Trials 2014.
The reporting of this study conforms to the STROBE statement [21].
Genetic analyses
DNA samples were analysed at the department of human genetics of the Radboud university medical center using whole exome sequencing (WES). Capture of exons was done using an Agilent SureSelect Human All Exon 50 Mb Kit (Santa Clara, CA, USA) and WES was performed using Illumina HiSeq, both done by BGI-Europe (Copenhagen, Denmark). Median coverage depth was at least 80x. Subsequently, read mapping and variant calling were done using BWA (mapping) and GATK (calling) and copy number variant analyses were done using CoNIFER. Short tandem repeat analysis in the WES data was done using ExpansionHunter [22]. The datasets were analyzed using an in-house annotation pipeline and manual interpretation by a clinical laboratory geneticist [23]. In cases of detected short tandem repeats (STR’s) through WES, repeat-primed polymerase chain reaction (PCR’s) were performed as confirmation [24].
The presence of a C9orf72 repeat expansion was determined separately through a repeat-primed PCR for the C9orf72 hexanucleotide repeat the University Medical Centre Utrecht as described previously [25] Patients with more than 30 hexanucleotide repeats in the C9orf72 gene were considered as C9orf72 carriers [26] In case of repeat expansions additional PCR and fragment length analysis using GeneScan were performed [27].
Gene panel
Considering that the main diagnostic challenge with regards to PLS is making the distinction with ALS, HSP and perhaps to a lesser extent atypical forms of parkinsonism, we analyzed a panel consisting of genes associated with MND-FTD and “movement disorders” including HSP, dystonia, Parkinsonism and ataxias. A total of 364 genes (338 genes see Appendix A, 22 genes Appendix B and GRN, MAPT, NEK1 and C21orf2) were analysed using WES. Part of the movement disorders gene panel test is the analysis of STR’s involved in autosomal dominant spinocerebellar ataxia’s: SCA1 (ATNX1), SCA2 (AXTN2), SCA3 (ATXN3), SCA6 (CACANA1A), SCA7 (ATXN7), SCA10 (ATXN10), SCA12 (PPP2R2B), SCA17 (TBP), SCA36 (NOP36), Friedreich ataxia (FXN) and dentatorubral-pallidoluysian atrophy (ATN1).
Variants were classified using the guidelines for variant classification of the American College of Medical Genetics and Genomics (ACMG) and the Association of Molecular Pathology [28] and we assigned variants to groups based on disease association [29].
Results
Through the PAN study, we identified 221 patients with a disease duration of over 4 years, which was characterized by isolated UMN loss in at least 2 regions. After reviewing medical records, we excluded 7 cases in which there was uncertainty about the diagnosis. In 11 patients WES had already been performed in a diagnostic setting and this data was used for analysis in this study. For the remaining 202 patients DNA samples were present, of which only 128 were of sufficient quality and were analysed using WES. Therefore, WES data was available for a total of 139 cases. In 129 of these 139 cases repeat-primed PCR for C9orf72 was also performed. The baseline characteristics at the time of diagnosis for the patients that were sequenced are presented in Table 1. 14 of 139 cases had UMN dysfunction in only one region at the time of diagnosis, but this progressed to involvement in more than two regions during follow-up visits, therefore meeting the Turner criteria for PLS. In some cases minimal electromyographic (EMG) changes were identified, but these are the “‘low-grade’, non-progressive EMG signs of limited muscle denervation” that are permitted within the Turner criteria [8].
In our cohort a total of 4 patients had a positive family history of MND, and 4 patients were classified as suspect for HSP/CMT (pes cavus and tripping easily, pes cavus and hammertoes, walking problems) without a clear family history or genetic variant known in the family. In three of these cases, a pathogenic or likely pathogenic variants was identified. One patient with a family member with ALS had a C9orf72 repeat expansion (case 1) and 2 cases that had a family history suspect for HSP/CMT had a pathogenic variant in TBK1, which is associated with ALS-FTD. In the five other cases with a positive family history no relevant genetic variants were identified.
Genetic analyses (WES and repeat-primed PCR for C9orf72) revealed variants in 31 out of 139 cases (22%). There were 7 cases with one/two pathogenic variant(s), 4 cases with likely pathogenic variants and 20 cases with variants of uncertain significance (VUS) [28]. Genetic and clinical characteristics of these cases are presented in Table 2.
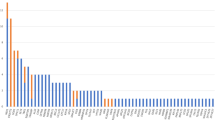
Figure 1A shows the distribution of genetics variants grouped by disease association, which includes pathogenic, likely pathogenic variants and variants of uncertain significance, whereas Fig. 1B only shows pathogenic and likely pathogenic variants. Case 8 was heterozygous for a variant in FIG4 that is likely pathogenic in a homozygous state. We therefore consider this to be a VUS.

Distribution of genetic variants found. Phenotypes were determined using the gene database of the National Centre for Biotechnology Information on 25-08-2022 [29]. A Whole cohort with all variants found; B pathogenic and likely pathogenic variants. ALS genes: FUS, NEK1, ANG, ERBB4; ALS-FTD genes: C9orf72, TBK1, CHMP2B; pure HSP genes: SPG7, SPAST, ZFVE27, FA2H, UBAP1, ALDH18A1; MSP genes: VCP, OPTN, ANXA11; “ALS-HSP-CMT overlap” genes: SPG11, DCTN1, FIG4, SETX, NIPA1, NEFL; SCA genes: ATXN2, PPP2R2B; X-ALD genes: ABCD1. ALS amyotrophic lateral sclerosis, CMT Charcot–Marie–Tooth, FTD frontotemporal dementia, HSP hereditary spastic paraplegia, MSP multisystem proteinopathy, SCA spinocerebellar ataxia, X-ALD X-linked adrenoleukodystrophy
In total, 10 cases (accounting for 7% of the total cohort) were found to a carry pathogenic or likely pathogenic variant; 5 cases had an ALS-FTD gene variant (50%), 3 cases pure HSP gene variant (33%) and 2 cases an ALS-HSP-CMT gene variant (20%). Characteristics of these groups are presented in Table 3.
In our initial cohort of patients with a disease duration longer than 4 years, we also had 9 patients that presented with isolated UMN signs in one single region (2 bulbar and 7 lower limbs). For two patients, we unfortunately had no clinical follow-up data and for the other 7 patients the symptoms remained within one region. These 9 patients did not meet the Turner criteria for PLS and are therefore not included in the results. They did have genetic analyses as mentioned above (9 WES, 8 C9orf72), which resulted in one patient with a likely pathogenic homozygous variant in SPG7 (male, onset lower extremities at 30 years old, disease duration of 36 years), one patient with a heterozygous VUS in REEP1 (male, onset lower extremities at 64 years old, disease duration of 15 years) and one with a hemizygous VUS in UBQLN2 (male, onset bulbar at 65 years old, disease duration of 8 years).
We found two cases to be compound heterozygous for variants in SPG11 (case 4) and in SPG7 (case 10).
Two patients carried identical TBK1 variants (case 6 and 7), but based on family history, family name and geographical origin we were not able to detect any family tie.
In 1 case (case 12) a variant in ABCD1 was found. According to the ACMG criteria this is classified as a VUS and the phenotype (history and MRI findings) did not match X-ALD. Unfortunately, C26:0-lysophosphatidylcholine (marker for very long-chain fatty acid accumulation in phospholipid fraction) analyses was not possible because the patients had died and therefore we were unable to definitely reject the diagnosis X-ALD on biochemical grounds [31].
In three of the eight patients that reported a positive or suspect family history for either MND or HSP/CMT phenotypes, a pathogenic or likely pathogenic variant was identified (cases 1, 6 and 7). These were a C9orf72 repeat expansions in a case where a distant relative (niece of father) had been diagnosed with ALS and two cases of a TBK1 variant (son with walking problems and foot deformities in the family). In the remaining five cases with a positive or suspect family history no relevant variants were identified.
Discussion
Current consensus criteria for PLS state that there is limited or no place for genetic testing in the work-up for PLS [8]. However, a very limited number of studies has been conducted on the genetics of PLS and as a result the data supporting this statement is sparse.
In an attempt to clarify what the contribution of genetics is to PLS susceptibility, Silani and colleagues reviewed the literature [32]. They note that in previous diagnostic criteria (Pringle) a negative family history was required [6], which was mainly meant to differentiate between PLS and HSP. However, in later criteria this requirement was dropped, as rare pedigrees with multiple PLS cases have been reported. This review identified reports of C9orf72 repeat expansions in 3 PLS patients and single cases with mutations in FIG4, UBQLN2, OPTN and DCTN1. They conclude that the finding of ALS mutations in PLS cases is rare, but also acknowledge that study sizes were limited [32].
The largest of these studies was performed by van Rheenen, et al. (n = 110), but only analyzed C9orf72 repeat expansions [25]. The only other relatively large study was performed by Mitsumoto, et al. (C9orf72 and exome sequencing), which included 34 PLS patients with disease duration of > 5 years. This study found 18% of patients to carry a variant in either ALS (C9orf72), Parkinson disease (PARK2, LRRK2) or HSP (SPG7) genes [33]. All other studies had sample sizes smaller than 10 or were case reports on single cases or pedigrees [32]. Therefore, firm conclusions regarding the frequency of pathogenic or likely pathogenic variants in PLS cannot be reached.
In our cohort, which is the largest to date, we found genetic variants in ALS or movement disorder genes in 31 out of 139 cases (22%). It must, however, be noted that a considerable portion of these (n = 21) are variants of unknown significance. It is likely that some of these variants are not clinically relevant, such as the ABCD1 variant we identified.
To provide better interpretation of variants of unknown significance, it remains crucial that they are reported in clinical and scientific reports. Only by doing so, will evidence accumulate over time and therefore allow us to accurately interpret the clinical relevance of these variants. Likely pathogenic or pathogenic variants were found in 7% of our cohort. These findings raise the complicated question of how to diagnostically classify the cases carrying (pleiotropic) ALS and HSP/CMT variants. For example, in case 9 in which a NEFL variant was identified, which is associated with CMT [34], and HSP [35]. This specific patient had bulbar and upper extremity involvement and met the criteria for definite PLS, making the clinical phenotype more compatible with PLS than HSP according to the treating physicians. According to the current consensus criteria genetic testing would not be warranted in this patient, while we believe that based on these genetic results the phenotype should be considered HSP-plus rather than PLS.
The revised El Escorial criteria for ALS state that patients with progressive upper and/or lower motor neuron signs and a family history of a defined pathogenic ALS mutation meet the criteria for “clinically definite familial ALS – Laboratory-supported” [36]. This means only progressive upper motor neuron signs in combination with a mutation in an ALS gene suffices. Therefore, it would seem reasonable to diagnose patients with PLS phenotypes carrying ALS variants as atypical or slow forms of ALS.
The Turner criteria state that testing for HSP genes should only be considered in patients with symmetrical involvement of the lower limbs [8]. Although it is uncommon, HSP patients with one-sided symptoms have been described [14, 15], and this is also seen in 2 of our cases with a (likely) pathogenic variant (“ALS-HSP-CMT overlap group”). It is even relatively common for patients with pure HSP to experience some degree of asymmetry of severity of symptoms in their legs [37, 38]. Apart from asymmetry, arm involvement (so UMN involvement in two regions) has also been documented in HSP patients [13, 39], as well as dysarthria which is reported frequently [39]. A study by Brugman et al. studied whether it was possible to differentiate sporadic HSP from PLS based on clinical presentation and concluded that this is unreliable in most cases [40]. Similarly, it seems reasonable to diagnose patients with PLS phenotypes carrying HSP variants as atypical forms of HSP.
Not all patients with a positive family history for MND or suspect for HSP/CMT in our cohort had a genetic variant identified. Despite the negative genetic findings in these cases, the family history is suggestive of a genetic cause of the disease. Indeed, it is not uncommon that in cases of HSP (also with a positive family history) the disease-causing gene is not found. Mereaux et al. performed genetic testing in a large cohort of HSP cases (1550) and pathogenic/likely pathogenic variants were found in 23.9–54.2% of the familial cases (variating between inheritance) and 26.6% of the isolated cases [16]. 5–15% of the ALS cases is familial and in 60–80% a disease-causing variant can be found (mostly C9orf72, SOD1, FUS and TARDBP) [2]. Therefore, our current knowledge of HSP and ALS genetics is incomplete. However, progress in genetic research is occurring at an ever-increasing pace and it seems highly likely that additional novel HSP and ALS genes will be discovered over the next few years. It also seems highly likely that variants in these genes will also be identified in PLS cases.
The diagnostic yield of genetic testing for HSP and ALS genes in PLS is 7% based on our findings.
We do believe that based on the results of Mitsumoto et al. [33] and our cohort, genetic analyses could have a role in the diagnostic work-up of PLS and should not be limited to patients with only symmetrical involvement of the legs.
Incorporating genetic testing in the work-up of patients with primary pyramidal disorders could lead to clarity regarding the underlying diagnosis for individual patients. Furthermore, it may also provide patients with additional information with regards to their prognosis. There are substantial differences between ALS, PLS and HSP in the rate of disease progression, degree of disability the disease will eventually cause as well as in life-expectancy. Although, this will become apparent over time, in the early stages of the disease this is frequently not clear, causing uncertainty and distress to patients. A desire for pregnancy (among relatives) may also be a reason to have clarity on whether there is a hereditary cause of the disease.
As the prices for DNA sequencing have dramatically dropped over the last couple of years, it is to be expected that genetic testing will become more widely available in the nearby future. DNA testing may, however, also yield results that are difficult to interpret, such as variants of unknown significance, and thereby provide the opposite of clarity. Genetic testing should therefore only be requested by skilled physicians with an up-to-date knowledge of the genetics of motor neuron diseases, who are capable of interpreting and adequately communicating results to patients. An infrastructure to provide genetic counselling to at risk family members should also be in place, to ensure that complicated topics such as mode of inheritance, disease penetrance and prenatal diagnostic testing are discussed appropriately. The decision to perform genetic testing in clinical practice is complex and should, in our opinion always be reached through shared decision making.
This paper offers an in-depth genetic characterization of the largest PLS cohort to date. Our findings show that patients with clinical phenotypes compatible with PLS may carry (likely) pathogenic variants in ALS and/or HSP genes. This raises the question whether PLS is a distinct disease entity or represents endophenotypes within the spectrum of different diseases such as ALS and HSP? The fact that there is no gold standard for PLS complicates this even further.
A limited number of histopathological reports on PLS patients are available, of which some predate the identification of TDP-43. Findings have been heterogeneous and perhaps these publications contain a bias towards unusual cases [41]. Of note, however are recent post-mortem studies by Mackenzie et al. that demonstrated TDP-43 pathology and degeneration of upper motor neurons, with preservation of lower motor neurons and less TDP-43 pathology in seven PLS cases [42]. The addition of a histopathological gold standard to the formal criteria for PLS would constitute a major step forward and should be focused on future efforts.
Similarly prospective data, including genetic results, on larger, international cohorts of PLS patients would also provide additional clarity. Potentially, genome-wide association studies or whole-genome sequencing studies of international PLS cohorts could lead to the identification of novel disease genes or phenotypic modifiers, that are associated with PLS. Understanding the underlying pathophysiology of PLS would be major step forward and could potentially guide therapy development, such as antisense based approaches.
References
Statland JM, Barohn RJ, Dimachkie MM, Floeter MK, Mitsumoto H (2015) Primary lateral sclerosis. Neurol Clin 33:749–760. https://doi.org/10.1016/j.ncl.2015.07.007
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH et al (2017) Amyotrophic lateral sclerosis. The Lancet 390:2084–2098. https://doi.org/10.1016/S0140-6736(17)31287-4
De Vries BS, Rustemeijer LMM, Bakker LA, Schröder CD, Veldink JH, Van Den Berg LH et al (2019) Cognitive and behavioural changes in PLS and PMA: challenging the concept of restricted phenotypes. J Neurol Neurosurg Psychiatry 90:141–147. https://doi.org/10.1136/jnnp-2018-318788
de Vries BS, Spreij LA, Rustemeijer LMM, Bakker LA, Veldink JH, van den Berg LH et al (2019) A neuropsychological and behavioral study of PLS. Amyotroph Lateral Scler Frontotemporal Degener 20:376–384. https://doi.org/10.1080/21678421.2019.1620284
Finegan E, Chipika RH, Shing SLH, Hardiman O, Bede P (2019) Primary lateral sclerosis: a distinct entity or part of the ALS spectrum? Amyotroph Lateral Scler Frontotemporal Degener 20:133–145. https://doi.org/10.1080/21678421.2018.1550518
Pringle CE, Hudson AJ, Munoz DG, Kiernan JA, Brown WF, Ebers GC (1992) Primary lateral sclerosis clinical features, neuropathology and diagnostic criteria. Brain 115:495–520
Gordon PH, Cheng B, Katz IB, Pinto M, Hays AP, Mitsumoto H et al (2006) The natural history of primary lateral sclerosis. Neurology 66:647–653. https://doi.org/10.1212/01.wnl.0000200962.94777.71
Turner MR, Barohn RJ, Corcia P, Fink JK, Harms MB, Kiernan MC et al (2020) Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry 91:373–377. https://doi.org/10.1136/jnnp-2019-322541
Krzosek P, Madetko N, Migda A, Migda B, Jaguś D, Alster P (2022) Differential diagnosis of rare subtypes of progressive supranuclear palsy and PSP-like syndromes—infrequent manifestations of the most common form of atypical parkinsonism. Front Aging Neurosci 14:1–9. https://doi.org/10.3389/fnagi.2022.804385
de Souza PVS, de Rezende Pinto WBV, de Rezende Batistella GN, Bortholin T, Oliveira ASB (2017) Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum 16:525–551. https://doi.org/10.1007/s12311-016-0803-z
Erfanian Omidvar M, Torkamandi S, Rezaei S, Alipoor B, Omrani MD, Darvish H et al (2021) Genotype–phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J Neurol 268:2065–2082. https://doi.org/10.1007/s00415-019-09633-1
McDermott CJ, White K, Bushby K, Shaw PJ (2000) Hereditary spastic paraparesis: a review of new developments. J Neurol Neurosurg Psychiatry 69:150–160. https://doi.org/10.1136/jnnp.69.2.150
Pennings M, Schouten MI, van Gaalen J, Meijer RPP, de Bot ST, Kriek M et al (2020) KIF1A variants are a frequent cause of autosomal dominant hereditary spastic paraplegia. Eur J Hum Genet 28:40–49. https://doi.org/10.1038/s41431-019-0497-z
Liu N, Chen J, Xu C, Shi T, Li J (2019) Hereditary spastic paraplegia associated with a rare IFIH1 mutation: a case report and literature review. Hereditas 156:28. https://doi.org/10.1186/s41065-019-0104-x
Rovito C, Paganoni S, Babu S, Tenforde AS (2021) Improved function in a runner with hereditary spastic paraparesis with use of extracorporeal shockwave therapy: personal clinical experience. Am J Phys Med Rehabil 100:e66–e68. https://doi.org/10.1097/PHM.0000000000001547
Mereaux JL, Banneau G, Papin M, Coarelli G, Valter R, Raymond L et al (2022) Clinical and genetic spectra of 1550 index patients with hereditary spastic paraplegia. Brain 145:1029–1037. https://doi.org/10.1093/brain/awab386
Huisman MHB, De Jong SW, Van Doormaal PTC, Weinreich SS, Schelhaas HJ, Van Der Kooi AJ et al (2011) Population based epidemiology of amyotrophic lateral sclerosis using capture-recapture methodology. J Neurol Neurosurg Psychiatry 82:1165–1170. https://doi.org/10.1136/jnnp.2011.244939
Abrahams S, Newton J, Niven E, Foley J, Bak TH (2014) Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 15:9–14. https://doi.org/10.3109/21678421.2013.805784
Dubois B, Slachevsky A, Litvan I, Pillon B (2000) The FAB: a frontal assessment battery at bedside. Neurology 55:1621. https://doi.org/10.1212/WNL.55.11.1621
Raaphorst J, Beeldman E, Schmand B, Berkhout J, Linssen WHJP, Van Den Berg LH et al. (2012) The ALS-FTD-Q A new screening tool for behavioral disturbances in ALS
von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP (2008) The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol 61:344–349. https://doi.org/10.1016/j.jclinepi.2007.11.008
van der Sanden BPGH, Corominas J, de Groot M, Pennings M, Meijer RPP, Verbeek N et al (2021) Systematic analysis of short tandem repeats in 38,095 exomes provides an additional diagnostic yield. Genet Med 23:1569–1573. https://doi.org/10.1038/s41436-021-01174-1
Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, Mensenkamp AR et al (2013) A Post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 34:1721–1726. https://doi.org/10.1002/humu.22450
Sequeiros J, Seneca S, Martindale J (2010) Consensus and controversies in best practices for molecular genetic testing of spinocerebellar ataxias. Eur J Hum Genet 18:1188–1195. https://doi.org/10.1038/ejhg.2010.10
Van Rheenen W, Van Blitterswijk M, Huisman MHB, Vlam L, Van Doormaal PTC, Seelen M et al (2012) Hexanucleotide repeat expansions in C9ORF72 in the spectrum of motor neuron diseases. Neurology 79:878–882. https://doi.org/10.1212/WNL.0b013e3182661d14
Van Mossevelde S, van der Zee J, Cruts M, Van Broeckhoven C (2017) Relationship between C9orf72 repeat size and clinical phenotype. Curr Opin Genet Dev 44:117–124. https://doi.org/10.1016/j.gde.2017.02.008
Kamsteeg EJ, Kress W, Catalli C, Hertz JM, Witsch-Baumgartner M, Buckley MF et al (2012) Best practice guidelines and recommendations on the molecular diagnosis of myotonic dystrophy types 1 and 2. Eur J Hum Genet 20:1203–1208. https://doi.org/10.1038/ejhg.2012.108
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetic Med 17:405–424. https://doi.org/10.1038/gim.2015.30
Bethesda (MD): National Library of Medicine (US) NC for BI. Gene [Internet] n.d. https://www.ncbi.nlm.nih.gov/gene. Accessed 25 Aug 2022
Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J et al (2017) disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener 18:153–174. https://doi.org/10.1080/21678421.2016.1267768.Amyotrophic
Lachtermacher MBR, Seuánez HN, Moser AB, Moser HW, Smith KD (2000) Determination of 30 X-linked adrenoleukodystrophy mutations, including 15 not previously described. Hum Mutat 15:348–353. https://doi.org/10.1002/(SICI)1098-1004(200004)15:4%3c348::AID-HUMU7%3e3.0.CO;2-N
Silani V, Corcia P, Harms MB, Rouleau G, Siddique T, Ticozzi N (2020) Genetics of primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 21:28–34. https://doi.org/10.1080/21678421.2020.1837177
Mitsumoto H, Nagy PL, Gennings C, Murphy J, Andrews H, Goetz R et al (2015) Phenotypic and molecular analyses of primary lateral sclerosis. Neurol Genet. https://doi.org/10.1212/01.NXG.0000464294.88607.dd
Stone EJ, Kolb SJ, Brown A (2021) A review and analysis of the clinical literature on Charcot–Marie–Tooth disease caused by mutations in neurofilament protein L. Cytoskeleton 78:97–110. https://doi.org/10.1002/cm.21676
Mul K, Schouten MI, van der Looij E, Dooijes D, Hennekam FAM, Notermans NC et al (2020) A hereditary spastic paraplegia predominant phenotype caused by variants in the NEFL gene. Parkinsonism Relat Disord 80:98–101. https://doi.org/10.1016/j.parkreldis.2020.09.016
Brooks BR, Miller RG, Swash M, Munsat TL (2000) El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler 1:293–299. https://doi.org/10.1080/146608200300079536
Fink JK (2001) Progressive spastic paraparesis: hereditary spastic paraplegia and its relation to primary and amyotrophic lateral sclerosis. Semin Neurol 21:199–207. https://doi.org/10.1055/s-2001-15265
Klebe S, Stolze H, Kopper F, Lorenz D, Wenzelburger R, Volkmann J et al (2004) Gait analysis of sporadic and hereditary spastic paraplegia. J Neurol 251:571–578. https://doi.org/10.1007/s00415-004-0366-7
Shi Y, Wang A, Chen B, Wang X, Niu S, Li W et al (2022) Clinical features and genetic spectrum of patients with clinically suspected hereditary progressive spastic paraplegia. Front Neurol 13:1–13. https://doi.org/10.3389/fneur.2022.872927
Brugman F, Veldink JH, Franssen H, De Visser M, Vianney-de-Jong JMB, Faber CG et al (2009) Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndromes. Arch Neurol 66:509–514. https://doi.org/10.1001/archneurol.2009.19
de Vries BS, Rustemeijer LMM, van der Kooi AJ, Raaphorst J, Schröder CD, Nijboer TCW et al (2017) A case series of PLS patients with frontotemporal dementia and overview of the literature. Amyotroph Lateral Scler Frontotemporal Degener 18:534–548. https://doi.org/10.1080/21678421.2017.1354996
Mackenzie IRA, Briemberg H (2020) TDP-43 pathology in primary lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 21:52–58. https://doi.org/10.1080/21678421.2020.1790607
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Jan Veldink reports to have sponsored research agreements with Biogen and Astra Zeneca. Leonard van den Berg declares fees to his institution from Biogen, Wave, Amylyx, Ferrer, and Cytokinetics for being on a scientific advisory board; fees to his institution from Amylyx for a lecture; an unrestricted educational grant from Takeda, and is the Chair of ENCALS and TRICALS. Michael van Es has consulted for Biogen, and has received travel grants from Shire (formerly Baxalta), performs work as a medical monitor for an on-going trial from Ferrer (NCT05178810).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Boer, E.M.J., de Vries, B.S., Pennings, M. et al. Genetic characterization of primary lateral sclerosis. J Neurol 270, 3970–3980 (2023). https://doi.org/10.1007/s00415-023-11746-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-023-11746-7