
Abstract
Coronavirus Disease 2019 is predominantly a disorder of the respiratory system, but neurological complications have been recognised since early in the pandemic. The major pathophysiological processes leading to neurological damage in COVID-19 are cerebrovascular disease, immunologically mediated neurological disorders and the detrimental effects of critical illness on the nervous system. It is still unclear whether direct invasion of the nervous system by the Severe Acute Respiratory Syndrome Coronavirus 2 occurs; given the vast numbers of people infected at this point, this uncertainty suggests that nervous system infection is unlikely to represent a significant issue if it occurs at all. In this review, we explore what has been learnt about the neurological complications of COVID-19 over the course of the pandemic, and by which mechanisms these complications most commonly occur.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coronavirus Disease 2019 (COVID-19), the clinical disorder caused by infection with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is characterised predominantly by pneumonitis with subsequent severe hypoxaemic respiratory failure [1]. In addition to the respiratory manifestations, multiple organ dysfunction such as acute kidney injury and myocarditis can occur [2]; only a few months into the pandemic, attention was drawn to the occurrence of neurological symptoms occurring in patients with COVID-19 [3]. Whilst many of these were simply symptoms commonly seen in febrile illnesses, such as headache and myalgia, a vast breadth of neurological disorders resulting from COVID-19 began to appear in the literature, including prolonged disorders of consciousness, immunologically mediated neurological diseases and cerebrovascular insults. At a group-wise level, patients with COVID-19 display elevated levels of brain-injury biomarkers such as glial fibrillary acidic protein (GFAP) and neurofilament light chain (NfL) in a severity-dependent manner, both in the presence and absence of neurological symptoms, highlighting that structural neurological damage occurs even in comparatively mild disease [4,5,6].
As the pandemic progressed, and clinical trials resulted, it became apparent that a substantial burden of the morbidity caused by the disease comes not from the virus itself, but rather the host’s response to the infection, with immunomodulatory interventions showing substantial survival benefit compared with modest effects from the use of antiviral agents [7,8,9]. In a similar vein, convincing reports of encephalitis caused by direct SARS-CoV-2 infection remain scant given the millions of individuals who have been infected, suggesting that the neurological sequelae of COVID-19 do not result from direct infection per se, but rather from indirect consequences such as inflammation and hypercoagulability. Knowing to what degree these complications of infection are specific to COVID-19, or even more common in COVID-19 than in other infectious diseases, remains an unresolved issue. In many regards, it would preferable if COVID-19 was not unique, as the transferrable lessons learnt from the unprecedented research effort undertaken during the pandemic may be one positive outcome from what has otherwise been a bleak period in human history.
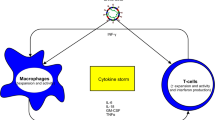
In this narrative review, we summarise what has been learnt about the acute neurological complications of COVID-19, and discuss the four main proposed pathophysiological processes which lead to their occurrence (Fig. 1). The chronic neurological and neuropsychiatric consequences of COVID-19, including both the long-term sequelae of acute neurological disease, and the less well-defined syndrome of “Long-COVID” [10], are likely to have significant public health implications, but are beyond the scope of this review.

Mechanisms by which neurological disease can occur as a result of COVID-19. Vascular disease appears to be disproportionately common in COVID-19 than in comparable infections, whereas immunologically mediated neurological conditions are similar in frequency. The evidence supporting direct central nervous system by SARS-CoV-2 as a cause of neurological disease is scant. Figure created with BioRender.com
Direct central nervous system invasion by SARS-CoV-2
In the landmark paper which first highlighted the neurological system as a potential target of COVID-19, Mao et al. concluded (without corroborating data) that the occurrence of neurological symptoms in these patients was evidence that SARS-CoV-2 could infect the nervous system [3]. Whilst no data existed to support this assertion directly at this point of the pandemic, SARS-CoV (the causative agent of the 2003 SARS outbreak) had previously been isolated from brain tissue in human autopsies, thus suggesting the potential for coronaviruses to invade the central nervous system (CNS) [11], and in light of the extensive genetic similarities between SARS-CoV and SARS-CoV-2 [12], it was reasonably hypothesized that SARS-CoV-2 may also have neuroinvasive capabilities [13].
An important factor determining the likelihood of direct cerebral invasion by SARS-CoV-2 is the expression within the CNS of the angiotensin converting enzyme 2 (ACE2) receptor, a key mediator for SARS-CoV-2 entry into multiple cell types [14]. A study using human iPSC-derived brain organoids and a transgenic mouse model demonstrated the necessity of ACE2 expression for neuroinvasion, finding that treatment of organoids with anti-ACE2 monoclonal antibodies prevented SARS-CoV-2 invasion of neurons, and intranasal inoculation of SARS-CoV-2 in mice overexpressing human ACE2 resulted in widespread viral invasion in the brain [15]. Given the extremely low expression of ACE2 in human brains and olfactory sensory neurons [16, 17], we would interpret from these results that the risk of direct brain infection is likely to be very small, although it is feasible that ACE2 expression could be upregulated by certain disease states [18], thereby facilitating entry in particular situations. In addition, other cell surface receptors are being investigated for their ability to potentiate ACE2-mediated SARS-CoV-2 cell entry. One prominent example is neuropilin-1 (NRP1), which is highly expressed in the olfactory epithelium and has been shown to facilitate SARS-CoV-2 infectivity in vitro [19, 20]; NRP1 is expressed ubiquitously throughout the body, and unlike ACE2 is well-expressed in the central nervous system [21], and so could represent a more tractable mechanism of direct neurological infection, although this has yet to be demonstrated.
The current evidence for direct viral invasion of neurons occurring in human patients is conflicting, with certain groups describing frequent viral presence [22,23,24,25], but many others finding no evidence of it at all [23, 26,27,28,29], suggesting a substantial impact of technical approach. Importantly, no descriptions of viral inclusions or reactive cellular changes typical of true infection have been reported [30]. Such reports have largely used reverse transcription PCR on brain tissue for viral detection, but the potential for contamination from blood or endothelium using this technique is high; visualisation of virus with immunohistochemistry is not only far less commonly reported [22, 30,31,32], but also presents difficulties in interpretation, as noted by Solomon et al. [31]. Of note is the distinct lack of inflammatory response at sites where viral particles were visualised; some authors have suggested that this may indicate an evasion of the immune-system, whereas our suggestion is that these particles are likely to be contaminants and not indicative of neuronal infection per se.
If SARS-CoV-2 is to reach the CNS, the virus must be able to circumvent the blood brain barrier (BBB) [33]. Frequently debated mechanisms of CNS entry include transcribial migration from olfactory sensory neurons [34], retrograde transsynaptic transmission starting from peripheral nerves [13], and hematogenous spread from the circulatory system [35].
Anosmia and ageusia are common symptoms of COVID-19 [36], and the olfactory epithelium are some of the earliest cell types to be infected [37]. Theoretically, if the virus can pass to olfactory sensory neurons, it could travel across the cribriform plate to arrive at the olfactory bulb, and subsequently access other brain regions [34]. However, this same mechanism would be open to all respiratory viruses, and it is clear that cerebral invasion, whilst it can occur with infections such as influenza, is extremely rare [38].
Similarly, should SARS-CoV-2 be able to infect other cranial or peripheral nerves, it would introduce the possibility of retrograde viral transport up the peripheral axon followed by transsynaptic migration to a neighboring neuron. Through this mechanism, a virus could hop across a series of synaptically connected neurons to ultimately arrive at neurons in the brain and spinal cord [39]. Multiple viral strains, such as herpes simplex virus-1 and rabies virus, exhibit a retrograde CNS infection strategy [40]; however, whilst trigeminal, vagus, and enteric neurons have been proposed as entry points for SARS-CoV-2 CNS infection based on the presence of certain symptomatologies [41], there is no direct evidence of peripheral nerve infection, let alone as a precursor to CNS neuroinvasion.
Whereas the prior two hypothesized routes for CNS entry require infection of peripheral or cranial nerves, the haematogenous route entails either transcytosis across the vascular endothelium of the BBB, or a Trojan horse-like reliance on infected host immune cells to bypass the BBB [42,43,44]. ACE2 is highly expressed on arterial and venous endothelial cells [14], and endothelial invasion by virus is well described [45], as is leukocyte infection [43], but neither of these explanations make the final leap to explaining passage into neurons themselves.
Immunologically mediated neurological disorders
Aberrant immunological responses to infection are a well recognised cause of both peripheral and central nervous system disease, and are broadly categorised as either parainfectious (the result of bystander damage from innate immune responses occurring at the time of acute infection) or post-infectious (erroneous adaptive autoimmune responses typically occurring in the weeks following acute infection) disorders. The most common of these conditions is Guillain-Barré syndrome (GBS), a post-infectious neuropathy which is thought to occur as a result of molecular mimicry between epitopes on the pathogen and peripheral nerve structures. There have now been numerous reports and case-series of GBS and related disorders such as Miller-Fisher Syndrome occurring following COVID-19 [46,47,48,49,50,51], but unlike in the Zika virus pandemic, there does not seem to be a disproportionately high risk of developing GBS following COVID-19 compared with other viruses. Indeed, in a large United Kingdom paired epidemiological and cohort study, the incidence of GBS in the UK was lower during the pandemic than in previous years, presumably because of reduced transmission of higher risk pathogens such as campylobacter jejuni [52]. Whilst no definite evidence of molecular mimicry has been demonstrated to explain the occurrence of GBS following COVID, comparison of SARS-CoV-2 sequencing data with primary amino acid sequences of human proteins known to be associated with immune-mediated neuropathies identified two immunologically relevant hexapeptides which were present in both datasets [53], raising the possibility of this mechanism.
The rarer, but immunologically related demyelinating central nervous system diseases of acute disseminated encephalomyelitis and transverse myelitis have also been reported following COVID-19 [54,55,56,57,58], but again the relatively scarce reports in the literature would suggest that these phenomena are not happening disproportionately frequently in patients with COVID-19.
With regards to parainfectious immunological diseases of the nervous system, a case of acute necrotising encephalopathy was reported early in the pandemic [59]. This is a rare condition characterised by symmetrical necrotising lesions in the thalami which usually occurs in children (often, but not invariably with a mutation in the RANBP2 gene) in the context of influenza infection, and is thought to be the result of a dysregulated cytokine response in conjunction with blood–brain barrier breakdown [59, 60]. Given the contemporaneous interest in the role of the “cytokine storm” (an excessive production of proinflammatory cytokines leading to widespread tissue damage and subsequent multiple organ failure) as a potentially important component of COVID-19 morbidity [61], acute necrotising encephalopathy stood to reflect an extension of the cytokine-driven systemic illness into the CNS, but comparatively few further cases have been forthcoming given the total number of infections [50, 59, 62,63,64,65,66]. Whilst true acute necrotising encephalopathy is rare, encephalopathy more broadly is common in COVID-19, and ubiquitous in very severe disease [67]. A role of a hyperinflammatory state and the “cytokine storm” in the pathogenesis of this encephalopathy has been suggested, and homology with Immune effector cell-associated neurotoxicity syndrome (ICANS) and cytokine-release syndrome (CRS) seen after Chimeric Antigen Receptor (CAR) T-cell therapy has been drawn [68, 69]. Marked upregulation of systemic inflammation induces permeability of the blood brain barrier [70, 71], with subsequent microglial and astrocytic activation which sets in motion a positive feedback loop of local inflammation [72,73,74]; this local inflammation, in conjunction with a multitude of other contributory factors, including neurotransmitter imbalances, brain energy metabolism dysfunction and the deleterious effects of medications and toxic levels of endogenously produced compounds such as urea, compromises brain function leading to encephalopathy [74].
A number of neuropathological studies of COVID-19 patients, even without a distinct neurological syndrome, have demonstrated CNS inflammatory changes, with microglial activation, astrocytosis and perivascular T-cell cuffing, most commonly in the medulla oblongata [22, 28, 75], however, similar changes have been described in other severe inflammatory states [76, 77], and it is unclear whether these changes have any specificity to COVID-19, or are simply indicative of severe infectious disease.
A host of other individual cases of autoimmune neurological diseases (such as myasthenia gravis, neuromyelitis optica spectrum disorder and antibody-mediated encephalitis) occurring in the context of COVID-19 have been published [78,79,80,81], but whether these represent coincidental disease processes (given the ubiquity of COVID-19), or the common result of an immune dyscrasia, such as the lymphopoenia often seen in COVID-19 [82, 83], is not established.
Neurovascular and thromboembolic disease
The relationship between vascular disease and COVID-19 appears to be complex; from early in the pandemic it became clear that venous thrombosis was a common occurrence [84], and that even in the absence of overt venous thromboembolism, D-dimer levels were markedly raised [85], suggesting activation of the coagulation factors in the microvasculature.
Regarding macrovascular neurological disease, it appears that COVID-19 may increase the risk of stroke greater than influenza infection [86]. Particularly, there appears to be a predilection towards large vessel occlusions, with some series even describing a reduction in the occurrence of small vessel infarction [87], although the possibility that patients with milder strokes did not seek medical attention given the health-system situation at the time should not be underestimated as a cause for this shift in pattern of disease [88]. The exact mechanism by which COVID-19 increases the risk of stroke is uncertain; there is a disproportionately high rate of cryptogenic stroke [89,90,91], i.e. not explained by large vessel atherosclerosis or cardioembolism, suggesting that the increase is not simply due to a decompensation of these processes. In those with severe respiratory involvement, and subsequently high right-heart pressures, right to left shunting of venous thromboemboli via patent foramen ovale is possible [92], but would not explain the incidence in lower severity COVID-19. More likely would be the contributions of both coagulopathy and endotheliopathy. Whilst severe illness is well known to predispose to hypercoagulability via a number of mechanisms such as immobility and dehydration, COVID-19 appears to disproportionately increase thrombotic tendency [85], and often patients display a frank coagulopathy on routine laboratory testing, with prolonged prothrombin and activated partial thromboplastin times, raised fibrinogen degradation products and d-dimer [93]. Furthermore, the occurrence of immunologically mediated thrombosis has been mooted by the comparatively frequent presence of lupus anticoagulant, as well as of anticardiolipin and antiphospholipid antibodies in patients with COVID-19 [94, 95], although the relevance of these autoantibodies in this setting is not clear.
In addition to the “haematological” predisposition toward thrombosis, endothelial disruption appears to be of particular importance in COVID-19, and would further contribute to the risk of thrombosis and subsequent stroke [96]. Post-mortem studies have demonstrated viral inclusions within endothelial cells (which richly express the ACE2 receptor [97]), with a significant associated inflammatory response leading to endotheliitis detectable in many organs including lung, heart, kidney and intestine, far in excess than the comparable condition of influenza [45, 98, 99].
Whilst these disorders of coagulation and endothelial integrity represent potential drivers of large vessel cerebrovascular disease, they also raise the possibility that impairment of the microvasculature might be involved in some of the less clear-cut neurological presentations, such as prolonged disorders of consciousness (including akinetic-mutism states) [63, 100,101,102], myoclonus (a relatively common complication with over 50 cases reported in the literature, which is seemingly unrelated to hypoxic ischaemic brain injury [103]), or even on the long-term cognitive outcomes of those who recover. Indeed, neuroimaging studies have commonly described cerebral microhaemorrhages occurring in those with severe COVID-19, reminiscent of microangiopathic disorders such as amyloid and hypertensive angiopathies [100, 104, 105], a phenomenon borne out in pathology series [32, 75, 100, 106]. As would be expected in a disorder of the endothelium, similar microvascular changes have been described in multiple organ systems [107], and it has even been suggested that this process might be the cause of the mononeuritis multiplex seen in a substantial minority of survivors of severe COVID-19 [108].
It should be noted that similar cerebral microvascular processes have been described in other relevant disease states, such as hypoxic ischaemic brain injury and acute respiratory distress syndrome from other causes [109, 110]. Whilst studies undertaking direct contemporaneous comparison between COVID-19 and similar conditions are extremely rare, there is a suggestion that systemic vascular complications are more common in COVID-19 [86, 99, 111], and of all the neurological complications of COVID-19, it may well be this aspect which is the most pathognomonic.
Neurological sequelae of critical illness
Aside from those aspects of COVID-19 which may specifically predispose to neurological disease, a substantial burden of disability will occur simply by way of the prolonged intensive care treatment experienced by so many patients. Hypoxic-ischaemic encephalopathy resulting from persistent hypoxaemia will undoubtedly leave a legacy of neurological impairment, and yet the literature has yet to delineate the prevalence of this. Delirium is commonplace in critically ill patients [112], and will have been exacerbated during the pandemic with the necessary use of older sedation agents given medication shortages [113]; the occurrence of delirium is well recognised to have detrimental long-term consequences on cognition [114]. Whilst the true incidence of encephalopathy in COVID-19 has not been fully delineated, it appears in the majority of patients requiring ICU admission [67], and EEG studies suggest that is likely to represent one of the most common acute neurological disturbances, with altered mental status being the most frequent indication for an EEG request, and diffuse slowing being the most frequently detected abnormality [115]. Interestingly, there appears to be a predominance of frontal lobe EEG abnormalities in COVID-19, which has been raised as a potentially pathognomonic feature of encephalopathy in COVID-19; other described abnormalities, such as epileptiform discharges, are commonly the result of underlying or pre-existing neurological pathology [115]. Prolonged periods of immobility, and particularly the use of prone-positioning for the amelioration of refractory hypoxaemia are associated with compressive and traction neuropathies and plexopathies [50, 116, 117]. Common risk factors for the development of critical illness polyneuromyopathy, such a prolonged ventilation, systemic inflammatory response syndrome, acute respiratory distress syndrome and multiorgan failure [118] are all frequently seen in COVID-19 [63], leading to a high occurrence of this condition [119]. Lastly, ECMO, the salvage therapy for those who can not be adequately oxygenated with invasive mechanical ventilation, is associated both with intracranial haemorrhage (largely as a result of the necessary anticoagulation) and ischaemic stroke [120, 121].
Conclusions
The COVID-19 pandemic has resulted in a substantial burden of neurological disease through various mechanisms, most notably through a preponderance towards vasculopathy and thromboembolism, the consequences of critical illness and immunological diseases triggered by infection. Whilst direct comparison with other infectious disease is challenging, it is not apparent that autoimmune complications are more common than following other similar infections, but there is a suggestion that the vascular and thrombotic manifestations are more pronounced in COVID-19 than in similar infections. Direct central nervous system infection by SARS-CoV-2 does not appear to occur with any significant frequency, if indeed it happens at all. Whilst hopefully an event which will not recur in our lifetimes, the COVID-19 pandemic has provided an unparalleled opportunity to study the consequences of severe illness on the nervous system, and it is our hope that in time the lessons learnt from this crisis may stand to improve the management and subsequent neurological health of critically unwell patients more broadly.
References
Zhou M, Zhang X, Qu J (2020) Coronavirus disease 2019 (COVID-19): a clinical update. Front Med. https://doi.org/10.1007/s11684-020-0767-8
Singh A, Zaheer S, Kumar N et al (2021) Covid19, beyond just the lungs: a review of multisystemic involvement by Covid19. Pathol Res Pract. https://doi.org/10.1016/j.prp.2021.153384
Mao L, Jin H, Wang M, et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. 8
Ameres M, Brandstetter S, Toncheva AA et al (2020) Association of neuronal injury blood marker neurofilament light chain with mild-to-moderate COVID-19. J Neurol. https://doi.org/10.1007/s00415-020-10050-y
Virhammar J, Nääs A, Fällmar D et al (2020) Biomarkers for central nervous system injury in cerebrospinal fluid are elevated in COVID-19 and associated with neurological symptoms and disease severity. Eur J Neurol. https://doi.org/10.1111/ene.14703
Kanberg N, Ashton NJ, Andersson L-M et al (2020) Neurochemical evidence of astrocytic and neuronal injury commonly found in COVID-19. Neurology 95:e1754–e1759. https://doi.org/10.1212/WNL.0000000000010111
RECOVERY Collaborative Group, Horby P, Lim WS et al (2021) Dexamethasone in hospitalized patients with Covid-19. N Engl J Med 384:693–704. https://doi.org/10.1056/NEJMoa2021436
Rezagholizadeh A, Khiali S, Sarbakhsh P, Entezari-Maleki T (2021) Remdesivir for treatment of COVID-19; an updated systematic review and meta-analysis. Eur J Pharmacol 897:173926. https://doi.org/10.1016/j.ejphar.2021.173926
Investigators REMAP-CAP, Gordon AC, Mouncey PR et al (2021) Interleukin-6 receptor antagonists in critically Ill patients with Covid-19. N Engl J Med 384(16):1491–1502. https://doi.org/10.1056/NEJMoa2100433
Nalbandian A, Sehgal K, Gupta A et al (2021) Post-acute COVID-19 syndrome. Nat Med 27:601–615. https://doi.org/10.1038/s41591-021-01283-z
Gu J, Gong E, Zhang B et al (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202:415–424. https://doi.org/10.1084/jem.20050828
Lu R, Zhao X, Li J et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Li Y, Bai W, Hashikawa T (2020) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol. 92(6):552–555. https://doi.org/10.1002/jmv.25728
Hamming I, Timens W, Bulthuis MLC et al (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637. https://doi.org/10.1002/path.1570
Song E, Zhang C, Israelow B et al (2021) Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med. https://doi.org/10.1084/jem.20202135
Chen R, Wang K, Yu J et al (2021) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front Neurol. https://doi.org/10.3389/fneur.2020.573095
Brann DH, Tsukahara T, Weinreb C et al (2020) Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv 6:eabc5801. https://doi.org/10.1126/sciadv.abc5801
Choi J-Y, Lee H-K, Park JH et al (2020) Altered COVID-19 receptor ACE2 expression in a higher risk group for cerebrovascular disease and ischemic stroke. Biochem Biophys Res Commun 528:413–419. https://doi.org/10.1016/j.bbrc.2020.05.203
Mayi BS, Leibowitz JA, Woods AT et al (2021) The role of Neuropilin-1 in COVID-19. PLoS Pathog 17:e1009153. https://doi.org/10.1371/journal.ppat.1009153
Cantuti-Castelvetri L, Ojha R, Pedro LD et al (2020) Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 370:856–860. https://doi.org/10.1126/science.abd2985
Raimondi C, Brash JT, Fantin A, Ruhrberg C (2016) NRP1 function and targeting in neurovascular development and eye disease. Prog Retin Eye Res 52:64–83. https://doi.org/10.1016/j.preteyeres.2016.02.003
Matschke J, Lütgehetmann M, Hagel C et al (2020) Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol 19:919–929. https://doi.org/10.1016/S1474-4422(20)30308-2
Fabbri VP, Foschini MP, Lazzarotto T et al (2021) Brain ischemic injury in COVID-19-infected patients: a series of 10 post-mortem cases. Brain Pathol 31:205–210. https://doi.org/10.1111/bpa.12901
Paniz-Mondolfi A, Bryce C, Grimes Z et al (2020) Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol 92:699–702. https://doi.org/10.1002/jmv.25915
Remmelink M, De Mendonça R, D’Haene N et al (2020) Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit Care. https://doi.org/10.1186/s13054-020-03218-5
Younger DS (2020) Postmortem neuropathology in Covid-19. Brain Pathol. https://doi.org/10.1111/bpa.12915
Kantonen J, Mahzabin S, Mäyränpää MI et al (2020) Neuropathologic features of four autopsied COVID-19 patients. Brain Pathol. https://doi.org/10.1111/bpa.12889
Schurink B, Roos E, Radonic T et al (2020) Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1:e290–e299. https://doi.org/10.1016/S2666-5247(20)30144-0
Bryce C, Grimes Z, Pujadas E et al (2020) Pathophysiology of SARS-CoV-2: targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. medRxiv. https://doi.org/10.1101/2020.05.18.20099960
Lou JJ, Movassaghi M, Gordy D et al (2021) Neuropathology of COVID-19 (neuro-COVID): clinicopathological update. Free Neuropathol. https://doi.org/10.17879/freeneuropathology-2021-2993
Solomon IH, Normandin E, Bhattacharyya S et al (2020) Neuropathological features of Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMc2019373
Pajo AT, Espiritu AI, Apor ADAO, Jamora RDG (2021) Neuropathologic findings of patients with COVID-19: a systematic review. Neurol Sci. https://doi.org/10.1007/s10072-021-05068-7
Koyuncu OO, Hogue IB, Enquist LW (2013) Virus infections in the nervous system. Cell Host Microbe 13:379–393. https://doi.org/10.1016/j.chom.2013.03.010
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11:995–998. https://doi.org/10.1021/acschemneuro.0c00122
Bodnar B, Patel K, Ho W et al (2021) Cellular mechanisms underlying neurological/neuropsychiatric manifestations of COVID-19. J Med Virol 93:1983–1998. https://doi.org/10.1002/jmv.26720
Kacem I, Gharbi A, Harizi C et al (2021) Characteristics, onset, and evolution of neurological symptoms in patients with COVID-19. Neurol Sci 42:39–46. https://doi.org/10.1007/s10072-020-04866-9
Gamage AM, Tan KS, Chan WOY et al (2020) Infection of human nasal epithelial cells with SARS-CoV-2 and a 382-nt deletion isolate lacking ORF8 reveals similar viral kinetics and host transcriptional profiles. PLoS Pathog 16:e1009130. https://doi.org/10.1371/journal.ppat.1009130
Hjalmarsson A, Blomqvist P, Brytting M et al (2009) Encephalitis after influenza in Sweden 1987–1998: a rare complication of a common infection. Eur Neurol 61:289–294. https://doi.org/10.1159/000206854
Cain MD, Salimi H, Diamond MS, Klein RS (2019) Mechanisms of pathogen invasion into the central nervous system. Neuron 103:771–783. https://doi.org/10.1016/j.neuron.2019.07.015
Taylor MP, Enquist LW (2015) Axonal spread of neuroinvasive viral infections. Trends Microbiol 23:283–288. https://doi.org/10.1016/j.tim.2015.01.002
DosSantos MF, Devalle S, Aran V et al (2020) Neuromechanisms of SARS-CoV-2: a review. Front Neuroanat. https://doi.org/10.3389/fnana.2020.00037
Keyhanian K, Umeton RP, Mohit B et al (2021) SARS-CoV-2 and nervous system: From pathogenesis to clinical manifestation. J Neuroimmunol 350:577436. https://doi.org/10.1016/j.jneuroim.2020.577436
Reza-Zaldívar EE, Hernández-Sapiéns MA, Minjarez B et al (2021) Infection mechanism of SARS-COV-2 and its implication on the nervous system. Front Immunol. https://doi.org/10.3389/fimmu.2020.621735
Engelhardt B, Vajkoczy P, Weller RO (2017) The movers and shapers in immune privilege of the CNS. Nat Immunol 18:123–131. https://doi.org/10.1038/ni.3666
Varga Z, Flammer AJ, Steiger P et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Fernández-Domínguez J, Ameijide-Sanluis E, García-Cabo C et al (2020) Miller–Fisher-like syndrome related to SARS-CoV-2 infection (COVID 19). J Neurol 267:2495–2496. https://doi.org/10.1007/s00415-020-09912-2
Padroni M, Mastrangelo V, Asioli GM et al (2020) Guillain-Barré syndrome following COVID-19: new infection, old complication? J Neurol 267:1877–1879. https://doi.org/10.1007/s00415-020-09849-6
Llorente Ayuso L, Torres Rubio P, do Beijinho Rosário RF et al (2020) Bickerstaff encephalitis after COVID-19. J Neurol. https://doi.org/10.1007/s00415-020-10201-1
Pfefferkorn T, Dabitz R, von Wernitz-Keibel T et al (2020) Acute polyradiculoneuritis with locked-in syndrome in a patient with Covid-19. J Neurol 267:1883–1884. https://doi.org/10.1007/s00415-020-09897-y
Rifino N, Censori B, Agazzi E et al (2020) Neurologic manifestations in 1760 COVID-19 patients admitted to Papa Giovanni XXIII Hospital, Bergamo, Italy. J Neurol. https://doi.org/10.1007/s00415-020-10251-5
Abu-Rumeileh S, Abdelhak A, Foschi M et al (2020) Guillain-Barré syndrome spectrum associated with COVID-19: an up-to-date systematic review of 73 cases. J Neurol. https://doi.org/10.1007/s00415-020-10124-x
Keddie S, Pakpoor J, Mousele C et al (2020) Epidemiological and cohort study finds no association between COVID-19 and Guillain-Barré syndrome. Brain. https://doi.org/10.1093/brain/awaa433
Lucchese G, Flöel A (2020) SARS-CoV-2 and Guillain-Barré syndrome: molecular mimicry with human heat shock proteins as potential pathogenic mechanism. Cell Stress Chaperones. https://doi.org/10.1007/s12192-020-01145-6
Zachariadis A, Tulbu A, Strambo D et al (2020) Transverse myelitis related to COVID-19 infection. J Neurol 267:3459–3461. https://doi.org/10.1007/s00415-020-09997-9
Munz M, Wessendorf S, Koretsis G et al (2020) Acute transverse myelitis after COVID-19 pneumonia. J Neurol 267:2196–2197. https://doi.org/10.1007/s00415-020-09934-w
Parsons T, Banks S, Bae C et al (2020) COVID-19-associated acute disseminated encephalomyelitis (ADEM). J Neurol 267:2799–2802. https://doi.org/10.1007/s00415-020-09951-9
Novi G, Rossi T, Pedemonte E et al (2020) Acute disseminated encephalomyelitis after SARS-CoV-2 infection. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000000797
Escobar M, Kataria S, Khan E et al (2021) Acute transverse myelitis with Dysautonomia following SARS-CoV-2 infection: a case report and review of literature. J Neuroimmunol 353:577523. https://doi.org/10.1016/j.jneuroim.2021.577523
Poyiadji N, Shahin G, Noujaim D et al (2020) COVID-19–associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology 296:E119–E120. https://doi.org/10.1148/radiol.2020201187
Wang GF, Li W, Li K (2010) Acute encephalopathy and encephalitis caused by influenza virus infection. Curr Opin Neurol 23:305–311. https://doi.org/10.1097/wco.0b013e328338f6c9
Ragab D, Salah Eldin H, Taeimah M et al (2020) The COVID-19 cytokine storm; what we know so far. Front Immunol 11:1446. https://doi.org/10.3389/fimmu.2020.01446
Zuhorn F, Omaimen H, Ruprecht B et al (2020) Parainfectious encephalitis in COVID-19: “The Claustrum Sign.” J Neurol. https://doi.org/10.1007/s00415-020-10185-y
Nersesjan V, Amiri M, Lebech A-M et al (2021) Central and peripheral nervous system complications of COVID-19: a prospective tertiary center cohort with 3-month follow-up. J Neurol. https://doi.org/10.1007/s00415-020-10380-x
Forestier G, de Beaurepaire I, Bornet G, Boulouis G (2020) Cytotoxic lesion of the corpus callosum as presenting neuroradiological manifestation of COVID-2019 infection. J Neurol. https://doi.org/10.1007/s00415-020-10166-1
Chaumont H, San-Galli A, Martino F et al (2020) Mixed central and peripheral nervous system disorders in severe SARS-CoV-2 infection. J Neurol 267:3121–3127. https://doi.org/10.1007/s00415-020-09986-y
Moreau A, Ego A, Vandergheynst F et al (2020) Cytotoxic lesions of the corpus callosum (CLOCCs) associated with SARS-CoV-2 infection. J Neurol. https://doi.org/10.1007/s00415-020-10164-3
Helms J, Kremer S, Merdji H et al (2020) Delirium and encephalopathy in severe COVID-19: a cohort analysis of ICU patients. Crit Care. https://doi.org/10.1186/s13054-020-03200-1
Lee DW, Santomasso BD, Locke FL et al (2019) ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 25:625–638. https://doi.org/10.1016/j.bbmt.2018.12.758
Pensato U, Muccioli L, Cani I et al (2021) Brain dysfunction in COVID-19 and CAR-T therapy: cytokine storm-associated encephalopathy. Ann Clin Transl Neurol 8:968–979. https://doi.org/10.1002/acn3.51348
Huang X, Hussain B, Chang J (2020) Peripheral inflammation and blood–brain barrier disruption: effects and mechanisms. CNS Neurosci Ther 27:36–47. https://doi.org/10.1111/cns.13569
Varatharaj A, Galea I (2017) The blood-brain barrier in systemic inflammation. Brain Behav Immun 60:1–12. https://doi.org/10.1016/j.bbi.2016.03.010
Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224. https://doi.org/10.1038/nrneurol.2014.38
Yang T, Velagapudi R, Terrando N (2020) Neuroinflammation after surgery: from mechanisms to therapeutic targets. Nat Immunol 21:1319–1326. https://doi.org/10.1038/s41590-020-00812-1
Wilson JE, Mart MF, Cunningham C et al (2020) Delirium. Nat Rev Dis Primers 6:1–26. https://doi.org/10.1038/s41572-020-00223-4
von Weyhern CH, Kaufmann I, Neff F, Kremer M (2020) Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet 395:e109. https://doi.org/10.1016/S0140-6736(20)31282-4
Westhoff D, Engelen-Lee JY, Hoogland ICM et al (2019) Systemic infection and microglia activation: a prospective postmortem study in sepsis patients. Immun Ageing. https://doi.org/10.1186/s12979-019-0158-7
Mazeraud A, Righy C, Bouchereau E et al (2020) Septic-associated encephalopathy: a comprehensive review. Neurotherapeutics 17:392–403. https://doi.org/10.1007/s13311-020-00862-1
Sriwastava S, Tandon M, Kataria S et al (2020) New onset of ocular myasthenia gravis in a patient with COVID-19: a novel case report and literature review. J Neurol. https://doi.org/10.1007/s00415-020-10263-1
Assini A, Gandoglia I, Damato V et al (2021) Myasthenia gravis associated with anti-MuSK antibodies developed after SARS-CoV-2 infection. Eur J Neurol. https://doi.org/10.1111/ene.14721
Ghosh R, De K, Roy D et al (2020) A case of area postrema variant of neuromyelitis optica spectrum disorder following SARS-CoV-2 infection. J Neuroimmunol 350:577439. https://doi.org/10.1016/j.jneuroim.2020.577439
Panariello A, Bassetti R, Radice A et al (2020) Anti-NMDA receptor encephalitis in a psychiatric Covid-19 patient: a case report. Brain Behav Immun 87:179–181. https://doi.org/10.1016/j.bbi.2020.05.054
Merayo-Chalico J, Rajme-López S, Barrera-Vargas A et al (2016) Lymphopenia and autoimmunity: a double-edged sword. Hum Immunol 77:921–929. https://doi.org/10.1016/j.humimm.2016.06.016
Khosroshahi LM, Rezaei N (2020) Dysregulation of the immune response in COVID-19. Cell Biol Int. https://doi.org/10.1002/cbin.11517
Klok FA, Kruip MJHA, van der Meer NJM et al (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191:145–147. https://doi.org/10.1016/j.thromres.2020.04.013
Helms J, Tacquard C, Severac F et al (2020) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46:1089–1098. https://doi.org/10.1007/s00134-020-06062-x
Merkler AE, Parikh NS, Mir S et al (2020) Risk of ischemic stroke in patients with coronavirus disease 2019 (COVID-19) vs patients with influenza. jaMA Neurol 77:1366. https://doi.org/10.1001/jamaneurol.2020.2730
de Sousa DA, Sandset EC, Elkind MSV (2020) The curious case of the missing strokes during the covID-19 pandemic. Stroke 51:1921–1923. https://doi.org/10.1161/STROKEAHA.120.030792
Perry R, Banaras A, Werring DJ, Simister R (2020) What has caused the fall in stroke admissions during the COVID-19 pandemic? J Neurol 267:3457–3458. https://doi.org/10.1007/s00415-020-10030-2
Sierra-Hidalgo F, Muñoz-Rivas N, Torres Rubio P et al (2020) Large artery ischemic stroke in severe COVID-19. J Neurol 267:3441–3443. https://doi.org/10.1007/s00415-020-09967-1
Yaghi S, Ishida K, Torres J et al (2020) SARS2-CoV-2 and stroke in a New York healthcare system. Stroke. https://doi.org/10.1161/STROKEAHA.120.030335
Tsivgoulis G, Palaiodimou L, Zand R et al (2020) COVID-19 and cerebrovascular diseases: a comprehensive overview. Ther Adv Neurol Disord. https://doi.org/10.1177/1756286420978004
Bang OY, Lee MJ, Ryoo S et al (2015) Patent foramen ovale and stroke-current status. J Stroke 17:229–237. https://doi.org/10.5853/jos.2015.17.3.229
Klok FA, Kruip MJHA, van der Meer NJM et al (2020) Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 191:148–150. https://doi.org/10.1016/j.thromres.2020.04.041
Beyrouti R, Adams ME, Benjamin L et al (2020) Characteristics of ischaemic stroke associated with COVID-19. J Neurol Neurosurg Psychiatry 91:889–891. https://doi.org/10.1136/jnnp-2020-323586
Tung ML, Tan B, Cherian R, Chandra B (2021) Anti-phospholipid syndrome and COVID-19 thrombosis: connecting the dots. Rheumatol Adv Pract. https://doi.org/10.1093/rap/rkaa081
Goshua G, Pine AB, Meizlish ML et al (2020) Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7:e575–e582. https://doi.org/10.1016/S2352-3026(20)30216-7
Ferrario CM, Jessup J, Chappell MC et al (2005) Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 111:2605–2610. https://doi.org/10.1161/CIRCULATIONAHA.104.510461
Calabretta E, Moraleda JM, Iacobelli M et al (2021) COVID-19-induced endotheliitis: emerging evidence and possible therapeutic strategies. Br J Haematol. 193(1):43–51https://doi.org/10.1111/bjh.17240
Ackermann M, Verleden SE, Kuehnel M et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med 383:120–128. https://doi.org/10.1056/NEJMoa2015432
Conklin J, Frosch MP, Mukerji SS et al (2021) Susceptibility-weighted imaging reveals cerebral microvascular injury in severe COVID-19. J Neurol Sci 421:117308. https://doi.org/10.1016/j.jns.2021.117308
Edlow BL, Claassen J, Victor JD et al (2020) Delayed reemergence of consciousness in survivors of severe COVID-19. Neurocrit Care 33:627–629. https://doi.org/10.1007/s12028-020-01133-9
Cani I, Barone V, D’Angelo R et al (2021) Frontal encephalopathy related to hyperinflammation in COVID-19. J Neurol 268:16–19. https://doi.org/10.1007/s00415-020-10057-5
Chan JL, Murphy KA, Sarna JR (2021) Myoclonus and cerebellar ataxia associated with COVID-19: a case report and systematic review. J Neurol. https://doi.org/10.1007/s00415-021-10458-0
Lersy F, Willaume T, Brisset J-C et al (2020) Critical illness-associated cerebral microbleeds for patients with severe COVID-19: etiologic hypotheses. J Neurol. https://doi.org/10.1007/s00415-020-10313-8
Choi Y, Lee MK (2020) Neuroimaging findings of brain MRI and CT in patients with COVID-19: a systematic review and meta-analysis. Eur J Radiol 133:109393. https://doi.org/10.1016/j.ejrad.2020.109393
Kirschenbaum D, Imbach LL, Rushing EJ et al (2020) Intracerebral endotheliitis and microbleeds are neuropathological features of COVID-19. Neuropathol Appl Neurobiol. https://doi.org/10.1111/nan.12677
Vinci R, Pedicino D, Andreotti F et al (2021) From angiotensin-converting enzyme 2 disruption to thromboinflammatory microvascular disease: a paradigm drawn from COVID-19. Int J Cardiol 326:243–247. https://doi.org/10.1016/j.ijcard.2020.11.016
Needham E, Newcombe V, Michell A et al (2020) Mononeuritis multiplex: an unexpectedly frequent feature of severe COVID-19. J Neurol. https://doi.org/10.1007/s00415-020-10321-8
Fanou EM, Coutinho JM, Shannon P et al (2017) Critical illness-associated cerebral microbleeds. Stroke 48:1085–1087. https://doi.org/10.1161/STROKEAHA.116.016289
Riech S, Kallenberg K, Moerer O et al (2015) The pattern of brain microhemorrhages after severe lung failure resembles the one seen in high-altitude cerebral edema. Crit Care Med 43:e386-389. https://doi.org/10.1097/CCM.0000000000001150
Burkhard-Koren NM, Haberecker M, Maccio U et al (2021) Higher prevalence of pulmonary macrothrombi in SARS-CoV-2 than in influenza A: autopsy results from ‘Spanish flu’ 1918/1919 in Switzerland to Coronavirus disease 2019. J Pathol Clin Res 7:135–143. https://doi.org/10.1002/cjp2.189
Brummel NE, Girard TD (2019) Delirium in the critically ill patient. Handb Clin Neurol 167:357–375. https://doi.org/10.1016/B978-0-12-804766-8.00019-4
Ammar MA, Sacha GL, Welch SC et al (2021) Sedation, analgesia, and paralysis in COVID-19 patients in the setting of drug shortages. J Intensive Care Med 36:157–174. https://doi.org/10.1177/0885066620951426
Müller A, von Hofen-Hohloch J, Mende M et al (2020) Long-term cognitive impairment after ICU treatment: a prospective longitudinal cohort study (Cog-I-CU). Sci Rep. https://doi.org/10.1038/s41598-020-72109-0
Antony AR, Haneef Z (2020) Systematic review of EEG findings in 617 patients diagnosed with COVID-19. Seizure 83:234–241. https://doi.org/10.1016/j.seizure.2020.10.014
Miller C, O’Sullivan J, Jeffrey J, Power D (2021) Brachial plexus neuropathies during the COVID-19 pandemic: a retrospective case series of 15 patients in critical care. Phys Ther. https://doi.org/10.1093/ptj/pzaa191
Malik GR, Wolfe AR, Soriano R et al (2020) Injury-prone: peripheral nerve injuries associated with prone positioning for COVID-19-related acute respiratory distress syndrome. Br J Anaesth 125:e478–e480. https://doi.org/10.1016/j.bja.2020.08.045
Bednarík J, Vondracek P, Dusek L et al (2005) Risk factors for critical illnesspolyneuromyopathy. J Neurol 252:343–351. https://doi.org/10.1007/s00415-005-0654-x
Frithiof R, Rostami E, Kumlien E et al (2020) Critical illness polyneuropathy and myopathy in COVID-19 patients: a prospective observational intensive care unit cross-sectional cohort study. https://doi.org/10.21203/rs.3.rs-78038/v1, https://www.researchsquare.com. Accessed 2 Mar 2021 (Preprint)
Nasr DM, Rabinstein AA (2015) Neurologic complications of extracorporeal membrane oxygenation. J Clin Neurol 11:383–389. https://doi.org/10.3988/jcn.2015.11.4.383
Notz Q, Lotz C, Herrmann J et al (2020) Severe neurological complications in critically ill COVID-19 patients. J Neurol. https://doi.org/10.1007/s00415-020-10152-7
Funding
The Cambridge COVID-19 BioResource is funded by the National Institute of Health Research.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the drafting of this manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ren, A.L., Digby, R.J. & Needham, E.J. Neurological update: COVID-19. J Neurol 268, 4379–4387 (2021). https://doi.org/10.1007/s00415-021-10581-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-021-10581-y