
Abstract
This paper reviews advances in epilepsy in recent years with an emphasis on therapeutics and underlying mechanisms, including status epilepticus, drug and surgical treatments. Lessons from rarer epilepsies regarding the relationship between epilepsy type, mechanisms and choice of antiepileptic drugs (AED) are explored and data regarding AED use in pregnancy are reviewed. Concepts evolving towards a move from treating seizures to treating epilepsy are discussed, both in terms of the mechanisms of epileptogenesis, and in terms of epilepsy’s broader comorbidity, especially depression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Definitions and classification
Definitions in epilepsy have always been problematic [1–5]. The disorder is characterised by seizures but not all seizures are due to epilepsy—febrile seizures or drug induced seizures, for example. Earlier classifications sought to reconcile these difficulties by describing different electroclinical syndromes but new data from modern imaging and genetics need to be incorporated.
Diagnosis is difficult because in practice, the diagnostic electrical hallmark of epilepsy may be absent interictally, especially in adults or if seizures are infrequent and interictal epileptiform discharges may occasionally be present in those without seizures. Moreover, in some instances, an “epileptic EEG” may be associated with an epileptic encephalopathy, in which overt seizures may be few or none, such as Landau–Kleffner syndrome, and a cognitive disorder dominates the presentation.
The International League Against Epilepsy recently consulted in an attempt to synthesise a consensus view [6], whose output will be published in 2017. The result promises to be useful and pragmatic, recognizing that the syndromes are multifaceted; any one case defined by an association of clinical, electrophysiological, etiological and comorbid factors. It also accepts that it is not always known if seizures are part of focal or generalized epilepsy and that in some cases, such as tuberous sclerosis, genetic and structural causes overlap. Some terms will be dropped, for example, childhood epilepsies where the seizures remit will be called pharmacoresponsive rather than benign, recognizing that children whose seizures remit may nevertheless have significant persisting psychosocial comorbidities.
The ILAE has also pondered the question of whether a single seizure may be considered to be epilepsy [7] and concluded that it may if there is a greater than 60% chance of another seizure; a risk conferred by the presence of EEG spikes or a major structural aetiology. Epilepsy may be considered to have gone away after ten years with no seizures and with no treatment. This approach has pragmatic utility, rather than mechanistic validity and is useful in allowing driving regulatory authorities to treat those with lower risk more leniently and may be helpful in deciding when to treat medically after a single seizure [8].
Some frontal lobe epilepsies may be particularly difficult to diagnose, often with non-diagnostic ictal scalp EEGs and some were initially considered to be a movement disorder, e.g. “paroxysmal nocturnal dystonia” [9] in which its epileptic basis was shown later [9, 10]. The situation has become more complex with the discovery that patients with frontal lobe epilepsy may also have epileptic nocturnal wandering, with similarities to parasomnias and also brief nocturnal movements which are not due to seizure discharges but may be a release phenomenon of interictal discharges [11]. They may suffer also from non-epileptic parasomnias more frequently than the general population. In the new classification, the phenomenon will be renamed “Sleep-related hypermotor epilepsy (SHE)”.
Status epilepticus and limbic encephalitis
The ILAE recently defined status epilepticus as: “a condition resulting either from the failure of the mechanisms responsible for seizure termination or from the initiation of mechanisms, which lead to abnormally, prolonged seizures (after time point t 1). It is a condition, which can have long-term consequences (after time point t 2), including neuronal death, neuronal injury, and alteration of neuronal networks, depending on the type and duration of seizures” [12]. Timepoint t 1 is at 5 min after seizure onset, when it is recognized for generalized tonic–clonic status epilepticus that evolution to status is increasingly likely and when treatment should be initiated. T2 is at 30 min, after which there is increasing risk of irreversible consequences. Status is divided along four axes; semiology, aetiology, EEG correlates and age. These axes align with the prognosis of status, which when adequately treated is determined by cause and the age and gender of the patient. The electroclinical state is another prognosticator; subtle status evolving from convulsive status has a particularly poor prognosis [13, 14].
The impressive out-of-hospital randomized, double-blind RAMPART study has shown that IM midazolam is at least as effective as IV lorazepam in the early treatment of status, in adults and children [15, 16], probably because IM speed of administration of midazolam compensates for speed of IV distribution of lorazepam. It has long been known that the effect of benzodiazepines in status epilepticus wears off very rapidly [17, 18] and it has subsequently been demonstrated that GABAA receptor sensitivity is reduced, sometimes long term [18]. Receptor trafficking may be contributory [19, 20]. As well as a reduction in inhibitory neurotransmitters, within 1 h of onset of status in rats, there is an increase in surface NMDA receptors in status, associated with increased excitation [21]. Cholinergic mechanisms are also implicated, supported by the observation that in pilocarpine induced status epilepticus; the addition of scopolamine provides additional seizure control, when combined with phenobarbital and benzodiazepines, raising the possibility of the use of drug combinations in status [22].
Basic mechanisms are starting to align with clinical evidence in the initial treatment of status with benzodiazepines, but thereafter the evidence is less clear. Initial uncontrolled reports suggested a 70% success rate for the treatment of status epilepticus with levetiracetam [23], but a recent randomized controlled trial of out-of-hospital clonazepam plus either levetiracetam or placebo was abandoned because of a lack of benefit in the levetiracetam arm [24]. This mirrors the finding that diazepam plus phenytoin confers no additional benefit to lorazepam alone at 12 h [14] and raises questions around the appropriate timing of the addition of AED to benzodiazepines. It also emphasizes the importance of properly controlled studies in an area where few have been undertaken. Shorvon et al. have undertaken meta-analyses of existing therapies [25–27]. From generally poor quality studies of lacosamide, levetiracetam, phenobarbital, phenytoin or valproate in benzodiazepine resistant status, they found efficacy ranging from 50% (phenytoin), to levetiracetam (68.5%), phenobarbital (58–84%) and valproate 76%. Lacosamide treatments were too few to give figures. The conclusion remains that all these drugs may be useful but there is no clear guidance on choice. The caution with which data from uncontrolled studies must be interpreted is highlighted by a recent randomized study of valproate versus phenobarbital which showed a 44% response to valproate and an 81% response to phenobarbital. However, in children, valproate may have fewer adverse effects and better efficacy than phenobarbital [28, 29] and similar efficacy to phenytoin [30]. But children may not be comparable to adults with a greater proportion of generalized epilepsies, more responsive to valproate. Future options include derivatives of valproate such as valnactomide and butylpropylacetamide, which may be more potently antiepileptic and less teratogenic in animal studies [31].
For status epilepticus which remains refractory to a second line AED, a range of intravenous benzodiazepines or anaesthetic agents may be considered and again Shorvon et al. found that studies are of poor quality. They found that 35% of patients in these studies died and a further 13% had severe neurological deficits and 13% mild neurological deficits on recovery. Studies underway may help answer some of these questions [32, 33]. Ketamine’s role in blocking NMDA receptors [34] has led to it become increasingly popular in the treatment of refractory status, with some efficacy on the basis of uncontrolled retrospective series [35–37]. A randomized trial in children is planned [38]. A recent trial of hypothermia showed no benefit at 90 days [39].
It is increasingly recognized that some patients with refractory status epilepticus, where the cause was previously unrecognized, may be suffering from an antibody mediated encephalopathy, “limbic encephalitis”. Antibodies implicated include LGI1 and NMDA, with CASPR less associated with seizures [40, 41]. More recently, GABAB and AMPA receptors have been implicated in some cases [42]. A specific phenotype of very brief, frequent and highly focal, faciobrachial dystonic seizures is almost pathognomonic of LGI1 associated disease, often heralding a more severe encephalopathy [43] and providing an opportunity to intervene at an earlier stage. Limbic encephalitis exhibits characteristic changes on MRI in the mesial temporal structures, especially the amygdalae [44] and responds primarily to immunotherapy and treatment of any associated tumour, rather than to AED [41, 45]. Early suspicion of the diagnosis and treatment, even before definitive serological confirmation, is recommended. Many patients will recover with appropriate treatment but may be left with ongoing epilepsy and hippocampal sclerosis is a reported outcome [46]. The extent to which epilepsy in patients, who have not suffered limbic encephalitis, may be attributable to antibody-mediated disease is an area of exploration which may open new avenues of treatment for chronic epilepsy. Small cohorts suggest increased rates of antibody positivity but their significance is not yet clear [47, 48].
Pharmacological treatment of epilepsy and underlying mechanisms/genetics
In 2000, Kwan and Brodie [49] found that 63% of unselected patients in an epilepsy service were rendered seizure free with medication. Since then despite numerous antiepileptic drugs becoming available, they found that the chance of a patient, who is diagnosed in 2017 becoming seizure free, has changed little [50]. Some studies are more optimistic; refractory epilepsy may have a greater chance of 12-month remission with or without AED change [51–53] at around 5% per year and although up to 40% may relapse [51], many of these may have a second longer remission.
The broad sweep of AEDs, generally affecting ion channels or neurotransmitters is unchanged, but there is slowly increasing evidence for a differential effect in specific syndromes.
Of established epilepsy drugs, ethosuximide, often forgotten by adult neurologists, has the most specific mechanism in relation to its role in the absence epilepsy. It acts on T-type calcium channels [54], implicated in the thalamocortical disturbance believed for decades to underlie generalized epilepsies [55]. Valproate and ethosuximide have clearly demonstrated greater efficacy over lamotrigine in childhood absence epilepsy [56]. A small, non-randomized study has suggested that ethosuximide may be also associated with a greater chance of long-term remission [57]. In a mouse model of absence epilepsy, Bomben et al. [58] selectively ablated P/Q channels in the neurons of layer VI that provide the descending cortical projection to the thalamus. This produced spike-wave activity with clinical absences suppressed by ethosuximide. This very selective lesion supports the view that a highly specific cortical abnormality is necessary and sufficient to generate the thalamocortical oscillations of absence epilepsy. Not all patients respond equally to medication. A clinical imaging and EEG study, comparing those patients responsive to valproate to those who are resistant, suggested different patterns of activation may underlie the varying therapeutic responses [59].
Despite strong epidemiological evidence of a genetic basis of IGE, relevant genes remain elusive, hampering efforts to identify specific drug targets. A recent genome wide association study suggested links to SCN1A, a known cause of GEFS+, protocadherin PCDH7 and PCDH19, both known to be associated with epilepsy and learning disability [60]. An analysis of microdeletions in generalized epilepsy showed an increased burden (7.3%) compared to controls (4%) and specific involvement of a range of genes known to be important in epilepsy, psychiatry and neurodevelopment [61].
The first major application of pharmacogenetics in epilepsy, and probably still the most widely applicable, has been the identification of patients from South East Asia who are HLA-B*1502 positive, putting them at high risk for Stevens–Johnson syndrome from carbamazepine and the elimination of this life-threatening complication by pre-treatment screening [62, 63]. Genetic understanding is creeping into other areas of pharmacological therapeutics. It has been realized for a number of years that sodium channel blocking drugs may be deleterious for children with Dravet syndrome [64, 65], although this may not be so clear for adult patients [66]. It is now known that Dravet syndrome is commonly due to a genetic truncations leading to total loss of function or missense mutations causing partial loss of function of the sodium channel, usually SCN1A [67, 68], which is located on inhibitory interneurons and causes hyperexcitability and seizures as a result of loss of function. A previously empirical observation of relative AED efficacy is now underpinned by a mechanistic understanding, which can guide drug choice. Mutations of the SCN8A gene are also associated with epilepsy, sometimes with a Dravet-like syndrome [69]. However, the phenotype may depend on the pathophysiology of the mutation, which may be a gain or a loss of function [70]. In four children with epileptic encephalopathy onset in the first months of life, Boerma described a response to phenytoin [71]. One of these had been demonstrated to have a gain of function mutation.
There are a number of other instances where rare monogenic cases of epilepsy have been evaluated in detail and treatment tailored to the identified pathophysiological mechanism, with varying success. Most consistently effective is the use of ketogenic diet to switch cerebral energy metabolism away from glucose in patients with Glut-1 deficiency, which may be dramatically successful [72, 73]. Retigabine (ezogabine) increases activity at KCNQ2 channels [74] and has been used to treat the neonatal epileptic encephalopathy associated with reduced function mutations of the KCNQ2 channel with some success [75]. Unfortunately, this drug is to be withdrawn from use in 2017 because of the pigmentary changes it may induce in skin, mucosae and eyes [76]. GRIND2 mutations resulting in gain of activity of the NMDA receptor may cause balloon swelling and cell death. Children with a severe encephalopathy due to this mutation may possibly benefit from treatment with memantine, more generally used in Alzheimer’s disease which inhibits this channel [77]. KCNT1 encodes a sodium-activated potassium channel and has been implicated in the migrating partial epilepsy of childhood and in autosomal dominant frontal lobe epilepsy, both causing a gain of function [78]. Two children with this mutation and a severe epilepsy phenotype were helped by the administration of quinidine [79]. These cases illustrate the importance of not only an electroclinical and genetic diagnosis of these epilepsies but also delineation of the specific pathophysiology of the mutation to enable drug choice, which may include opportunities beyond those conventionally used in the antiepileptic armamentarium.
Epileptogenesis and inflammation
Another focus is the mechanisms of epileptogenesis; the process from initiation of pathological changes to the development of epilepsy and possibly the maintenance of epilepsy. There are changes, which involve altered gene expression, inflammation, protein production and changes in connectivity, which may all be the target for drugs to suppress epileptogenesis. One of the most studied pathways links to the rapamycin (mTOR) pathway (Fig. 1). Upregulation of mTOR, a serine/threonine protein kinase, occurs as a result of the TSC1 and TSC2 mutations of tuberous sclerosis (TS) complex. Other mutations in the pathway may be associated with overgrowth in megalencephaly [80]. mTOR has a role in protein synthesis and inhibition of mTOR, cell growth and replication by everolimus, a rapamycin analogue, has been shown to reduce overgrowth of malignantly transformed tubers [81]. Animal models have shown an antiepileptic effect of mTOR inhibition [82] but this has been more difficult to demonstrate in humans. However, a recent double-blind study of 366 patients showed a dose-related seizure reduction of up to 40% with everolimus, in patients with TS [83]. However, mTOR inhibitors may also have a direct effect on Kv1.1 ion channels, independent of epileptogenesis [84], blurring their possible mechanism in seizure suppression.

Pathway showing some of the relationships between mTOR and cellular function which may be modulated in epileptogenesis and their modulation through inflammatory pathways and by drugs. AMPK 5′ AMP-activated protein kinase, IRS1 insulin receptor substrate 1, JAK Janus kinase, MTOR mechanistic target of rapamycin, PDK1 pyruvate dehydrogenase lipoamide kinase isozyme 1, P13K PI3 kinase, PKB protein kinase B, PtdIns phosphatidylinositol, PTEN phosphatase and tensin homologue, RHEB ras homolog enriched in brain (GTP binding protein), STRADA STE20-related kinase adaptor alpha, STK11 serine/threonine kinase 11, TSC tuberous sclerosis complex
Whilst immunological mechanisms are clearly implicated in the aetiology of certain epilepsies such as limbic encephalitis [85] or Rasmussen encephalitis [86], increasing attention has been given to them in commoner forms of epilepsy. There is broad evidence for their significance, especially from animal studies and involving cytokines, changes in the blood brain barrier and pathological alterations associated with altered excitability [87–94]. Pathological examination of resected human specimens of focal cortical dysplasia [95] has also shown substantial increases in mRNA expression of Toll-like receptors 2 and 4 and associated with high-mobility group box protein 1, restricted to astrocytes and microglia in pathological tissue. These interact through interleukin IL1-β. Microglia activation appears increased more in focal cortical dysplasia (FCD) type II than in FCD I, associated with the migration of activated lymphocytes and activation of the mTOR pathway, linking inflammation to epileptogenesis [96]. A recent systematic review and meta-analysis [97] has described increased CNS levels of interleukins of the IL1 family as well as of chemocytokines CCL 3-5, which are involved in monocyte and lymphocyte migration. IL6 appears to be elevated in serum but not in CNS. A recent study of patients with moderate to severe cerebral trauma found a relationship between cerebrospinal fluid IL1-β levels and an allelic variant of the IL1-β gene to the risk of developing epilepsy [98]. This provides the first evidence of a biomarker that might be used to predict epilepsy after an epileptogenic insult and possibly a means of pharmacological intervention. These may need to be complex; a recent study suggested a single intervention was inadequate and a cocktail of anti-inflammatory drugs was required to prevent epileptogenesis [99]. A small case series of intractable childhood onset epilepsy has already been treated successfully with human recombinant IL-1 receptor antagonist (Anakinra\) [100] and it is hopeful that, as there are already many drugs affecting the immune system and some affecting the blood brain barrier, that this will prove a fertile area for development.
Recently, mutations of the DEPDC5 (DEP domain containing 5, involved in g-protein signalling) gene have been demonstrated in patients with cortical dysplasia and in up to 12% of small families of patients with familial focal epilepsy phenotypes, including ADNFLE without demonstrable lesions [101–103]. This gene is involved in the same GATOR pathway as mTOR. Although the GATOR (gap activity towards RAG’s) pathway is generally associated with protein synthesis, it appears to reduce the levels of Kv1.1 potassium channels in hippocampal pyramidal neurons increasing seizure expression, which can be reversed by inhibitors [104]. These findings link lesional and non-lesional ion channel related epilepsies to the same pathway, providing a potential opportunity for the wider use of inhibitors in treatment.
Although the scope is expanding, the relationship of these mechanisms to the majority of epilepsies, those triggered by a neurological insult (focal epilepsies) or a complex genetic trait (generalized epilepsies) remains to be established. It has long been recognized that epilepsy due to trauma is more likely in those with a family history of epilepsy [105] providing a potential to link to genetic mechanisms. But the development of epilepsy may take 20 years [105, 106]. The key will be to identify those patients at high risk and to find a low risk preventative treatment akin to aspirin in stroke and very large, long-term follow up studies, will be needed to establish efficacy. Biomarkers such as IL1-β for evolving epileptogenesis are needed to identify high risk patients and to act as drug targets.
Antiepileptic drug trials
Despite being a common disorder, the number of high quality trials of antiepileptic drugs is small. Trials of new AED are normally in the form of an add-on therapy in refractory partial epilepsy, usually with the end point of a 50% reduction of seizures. This may be realistic in showing a biological effect but does not confer the psychosocial benefits of seizure freedom, and therefore drugs enter the market with the knowledge that they will not dramatically alter the burden of refractory epilepsy. The Federal Drug Administration in the US requires monotherapy trials against placebo and the European Medicines Agency requires head-to-head trial of active agents. Consequently, results cannot cross the Atlantic, delaying introduction and increasing cost for manufacturers. Both types of trials have their merits. The result is a non-systematic hotchpotch of evidence in relation to monotherapy in epilepsy. Whilst the pragmatic study SANAD has guided many UK clinicians to lamotrigine as first line in monotherapy for focal epilepsy [107], carbamazepine remains a drug of choice in many countries and studies [108]. A recent study has shown that zonisamide is non-inferior to carbamazepine in new onset focal epilepsy in adults [109]. A large study of 1688 new onset patients compared time to withdrawal of levetiracetam in two arms to first choice carbamazepine or valproate in monotherapy in adults [110]. Overall, the drugs performed similarly but in a post hoc analysis, levetiracetam withdrawal rate was lower in those over 60, especially in comparison to carbamazepine, with fewer adverse effects rather than greater efficacy [111].
The repertoire of AED considered effective in IGE has traditionally been more restricted that for focal epilepsy. Case reports have supported the use of lacosamide [112, 113] and it is the subject of ongoing larger scale studies. Perampanel has been found to be effective as an add-on for refractory generalized epilepsy with tonic–clonic seizures [114].
Cannabis contains approximately 80 different active cannabinoids and was used in the nineteenth century as an AED [115]. It has been known for many years to be an antagonist at NMDA receptors with antiepileptic activity [116]. Δ9 tetrahydrocannabinol is the main psychoactive component of cannabis, acting on THC1 and THC2 receptors but other components, especially cannabidiol (CBD) do not act on these receptors, are not psychoactive. They may have medicinal properties through a range of other actions [117]. Clinical studies in the 1970s and 80s reviewed in [117] pointed to antiepileptic effects and recent anecdotal evidence and an open labelled trial have shown benefit in epileptic encephalopathies such as Dravet syndrome [118, 119], which have had a profound social effect in the United States, with parents moving their families to states where cannabis is legal [120]. Although their mechanisms point to a potential role for cannabinoids of relevance to epilepsy [121], there are as yet, no good studies to support their widespread use. The adverse effects of natural cannabis are widely known [122] and a particular problem for adolescents. Cannabinoids should be avoided by those with epilepsy, especially the young, who are already at risk of psychiatric problems, until good quality trials support their use.
Epilepsy and comorbid depression
Data extracted from a US population survey of 340,000 households and those with epilepsy were compared to those without [123]. Two percent had suffered with epilepsy and reported increases in a range of disorders (Table 1). A figure of approximately one third affected by depression is consistent with numerous previous studies. The relationship to epilepsy is complex. In studies of IGE, the epilepsy and its impact may be important [124] but there is often dissociation between a good seizure outcome and a poor psychosocial outcome [125]. A key factor predicting outcome relates to family environment support [126] but a biological association is supported by the observation that children and adults have an increased risk of psychiatric disturbance, even before the onset of their epilepsy [127, 128], and by a broad range of experimental studies [129]. Interactions between epilepsy and depression may include shared abnormalities in a number of neurotransmitters including 5HT1A mechanisms [130, 131] and via glutamate, where low-dose ketamine, an antiepileptic NMDA antagonist, may have an impact on depression [132]. These studies illustrate a bidirectional relationship of epilepsy and depression, involving both biological and psychosocial factors.
A common concern is that antidepressants may increase seizures. The risk of de novo seizures from the use antidepressants is 0.1% for newer drugs and 0.3% from older drugs, e.g. tricyclics [133]. Exceptions may be maprotiline, bupropion or clomipramine with a higher risk [134] but overall, those in the treatment arm of antidepressant trials had fewer seizures than those in the placebo arms [134]. In smaller studies of those with epilepsy at therapeutic doses of antidepressants, many will experience an improvement in their epilepsy [135]. A recent review has brought together the newer mechanistic evidence, showing that 5HT1A may mediate a number of actions, which have antiepileptic effects, including increasing GABA activity and reducing inflammatory cytokines and those patients with epilepsy may have reduced PET ligand binding at 5 HT1A sites [136]. In a mouse model of sudden unexplained death in epilepsy, drugs acting on 5-HT3 receptors (fluoxetine, blocked by ondansetron) reduced respiratory arrest in seizures, without affecting the seizures themselves [137], a further possible mode of benefit of antidepressants in epilepsy. Where possible, it may be appropriate to avoid those antidepressants with pharmacokinetic interactions with AED, such as fluvoxamine, paroxetine and fluoxetine. Hopefully, neurologists can now encourage the use of antidepressants, especially as psychiatric comorbidity is a greater determinant of quality of life than seizure frequency in those with refractory epilepsy [138].
Antiepileptic drugs and pregnancy
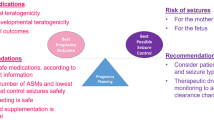
In recent years, information regarding major congenital malformation (MCM) rates has been consolidated in epilepsy and pregnancy registries. AED are divided into those with reasonably quantified risk and those with insufficient data. This becomes self-reinforcing with increased reluctance to prescribe drugs of uncertain risk to those who may conceive. The most recent data from the UK epilepsy and pregnancy register, shows a very clear dose-related effect with valproate risk 5% with <600 mg daily increasing to 11% at over 1000 mg. Carbamazepine at 2% risk when given at <500 mg daily, 3% at 500–1000 mg and 5% at >1000 mg. Lamotrigine had a less steep curve with 2% at <200 mg, increasing to 3.5% over 400 mg daily [139]. These data are similar to those published from European and US registries [140]. Oxcarbazepine, not widely used in the UK, appears to have similar low risk to lamotrigine at 2.2% [141]. The risk for levetiracetam appears similarly low at 0.7% in monotherapy, increasing in polytherapy [142]. Added to the risk of MCM are concerns over more subtle neurodevelopmental disturbances, including lower IQ, autism and ADHD, which may conceivably arise from exposure to valproate at any stage of pregnancy [143–146]. Although not widely used in pregnant women, topiramate and zonisamide may be associated with significantly lower birthweight [147]. Recent data have also shown the importance of considering genetic factors in teratogenicity. A family history of abnormalities increases the risk. The risk to a second child, where a first was affected by an AED may be as high as 17–36% [148, 149]. Clinicians must also consider the risk to the mother of epilepsy in pregnancy and data suggest a tenfold increase in mortality compared to non-epilepsy controls, largely due to SUDEP [150].
Epilepsy surgery
Given the low chance of response to medical therapy after the failure of two AED [49], this is the widely accepted yardstick for defining refractoriness and the appropriateness for consideration of resective epilepsy surgery. The proportion of patients for whom surgery may be successful is not clear, but is estimated as a maximum of around 2% of the total cohort. With an incidence of 0.5%, in the USA and a prevalence of 750,000, this translates to up to 3500 incident cases and 15,000 prevalent cases, in which surgery might be considered. The rate of epilepsy surgery has remained static at around 1500 cases per year [151, 152] for over 20 years. The pattern of cases operated may be changing with a reduction in mesial temporal sclerosis [153], perhaps due to improved outcomes of childhood febrile seizures. At the same time, the outcomes of extratemporal epilepsies are improving with new diagnostic techniques. The mortality of surgery is around 0.1–0.5% [151], similar to the annual rate of SUDEP in refractory epilepsy [154], i.e. the mortality of ongoing refractory epilepsy exceeds the post-operative risk after one year. Complication rates have reduced [155] and are around 3% for major and 7% for minor complications; one of the commonest complications is a visual field defect after temporal lobectomy [151, 156]. The treatment is cost-effective in the long term, with sustained remission and close to half of adult patients and 86% of children may be able to stop their AEDs. Two recent studies have found risk factors for seizure recurrence after post-operative drug withdrawal included pre-operative seizure frequency and post-operative EEG abnormalities [157, 158]. They also found about one third of those relapsing will not come back under control with re-introduction of medication, especially those with early recurrence, perhaps reflecting a less complete surgical remission.
Health-related quality of life often returns to normal in those who become seizure free [159]. Negative prognostic factors include high seizure frequency and long duration at baseline [160, 161]. Those with lesions such as cavernomas or benign tumours may achieve 77% seizure freedom at two years, even if surgery is undertaken after a long seizure history [162].
Advances in epilepsy surgery include alternative methods to resective surgery; improvements in techniques of case selection for surgery and neurostimulation techniques.
Radiosurgery for arteriovenous malformations may give excellent outcomes for associated epilepsy and positive prognostic factors have been reported to be presentation with haemorrhage rather than epilepsy and the absence of post-treatment haemorrhage [163–165]. A recent meta-analysis of stereotactic radiosurgery for mesial temporal sclerosis [166] showed that the total number of patients reported remains low (<200) but that half became seizure free at a median of 14 months after treatment with a complication rate of around 8% (excluding headache which was more common) and rates of visual field defects similar to open surgery. MRI-guided laser thermocoagulation has been undertaken in a few patient with initially promising results. Procedural morbidity is low and patients may be admitted for just one day. It has been suggested as appropriate particularly for older patients. [167–170]. The electrodes inserted for stereotactic EEG recording may also be used to deliver a thermocoagulation induced lesion to the surrounding brain, with a diameter of 4.5–7 mm. This has been undertaken in patients with hypothalamic hamartoma, for whom surgery is difficult and with a high success in remission of the gelastic seizures associated with these lesions [171]. Early indications are that this may be an approach which can be undertaken in cases of focal cortical dysplasia.
The identification of patients who will benefit from epilepsy surgery relies on the demonstration of a single brain region responsible for the epilepsy, which can be safely resectable. Identification of a responsible lesion has been demonstrated in numerous studies to predict a better outcome [172]. Even in those where imaging is normal, resection on the basis of an intracranial EEG abnormality is more likely to result in seizure freedom if the resected tissue is pathologically abnormal [173]. Increasing pre-operative identification of pathology through improved MRI, through higher field strengths up to 7 T in vivo and enhancing 3 T with automated measures of hippocampal volumes potentially gives a greater chance of identifying candidates who may benefit from surgery [174–176]. In those whom structural imaging remains negative, then FDG-PET can aid in the decision making, either in favour of surgery, e.g. in those thought to have non-dominant TLE or against surgery in more complex cases [177, 178]. Magnetoencephalography is not widely used [179], but a recent study demonstrated that if all MEG abnormal areas were resected, prognosis was improved and MEG can be used to target SEEG more successfully [180]. Tight clustering of MEG abnormalities predicted a better outcome than more dispersed abnormalities. High density EEG source imaging using increased electrode number may also be valuable in predicting the outcome of surgery [181]. Intravascular stent EEG, shown to be safe in sheep may be a non-invasive method of intracranial EEG recording in the future [182].
Where resective surgery is not possible, palliative stimulation techniques may be considered. The most established and widely used is vagus nerve stimulation which is safe, with a low risk of complications, such as infection, haematoma and vocal cord palsy [183]. An analysis from the VNS registry combined with pooled study data totaling 8423 patients [184] found that responder rate, defined by a 50% seizure reduction, was 47% at 0–14 months and 63% at 24–48 months with seizure free rates rising from 5–10% over the same period. Quality of life measures also improved with VNS [185], which may relate to seizure reduction, reduced AED load in association with successful antiepileptic treatment or putative effects of VNS on mood [186]. Responsive stimulation involves a closed circuit of intracranial electrodes with electrical stimuli delivered to the brain according to a seizure detection paradigm. The circuit is often installed following electrode placement in an unsuccessful attempt to identify a surgical target. In 191 patients there was a 37.9% responder rate compared to 17.3% in the sham group. [187]. Electrodes placed in the thalamus have been associated with a 69% median reduction in seizure frequency and a 35% rate of serious adverse events, including infection in 10% and lead misplacement in 8% [188]. Other targets under investigation include the nucleus accumbens [189] and the cerebellum [190]. Optogenetic methods [191], successful in animals, have not yet been applied in humans.
Summary
A new classification of epilepsies will support the integration of novel aetiological and genetic factors with the existing electroclinical classification and help identify when a single seizure might be considered epilepsy on the basis of an abnormal EEG or imaging. Midazolam IM has emerged as the benzodiazepine of choice in out-of-hospital treatment of status epilepticus and a valid alternative in hospital, but good clinical studies are lacking beyond this early stage. Limbic encephalitis is increasingly diagnosed and primary treatment is immunotherapy rather than AED. The significance of antibodies more generally in epilepsy remains unclear. Most epilepsy treatment remains without a clear evidence base but ethosuximide and valproate have been demonstrated to be the most efficacious AED in absence epilepsy. Perampanel and lacosamide are new drugs which are emerging as treatments for tonic–clonic seizures in generalized epilepsy. A small number of specific genetic epilepsies have allowed personalized treatment in specific cases but this has not yet had broader application. Epileptogenesis is a fertile area of research and everolimus, an inhibitor of the mTor pathway, has demonstrated efficacy in epilepsy associated with TS, showing the clinical potential of this avenue of research for the first time. Epilepsy and pregnancy registers are consolidating data pointing to the use of lamotrigine, levetiracetam, carbamazepine and/or oxcarbazepine as those AED with the lowest risk of major congenital malformations. New evidence has associated topiramate and zonisamide with low birth weight. Clinicians can treat comorbid depression with most modern antidepressants, reassured that there is little evidence of an adverse effect on their patient’s epilepsy. Surgical treatment of epilepsy remains under-utilised and the selection of patients for surgical treatment of epilepsy is becoming more refined with the use of functional imaging to support structural imaging. Alternative ablative treatments are being explored but are not yet widespread. Stimulation techniques other than VNS are areas of research, which remain to find their place.
Overall, recent epilepsy research has started to change our thinking and approach to patients, as we slowly move towards a more rational basis by which to treat this common condition.
References
Gastaut H, Caveness W, Landolt H et al (1964) A proposed international classification of epileptic seizures. Epilepsia 5:297–306. doi:10.1111/j.1528-1157.1964.tb03337.x
Merlis JK (1970) Proposal for an international classification of the epilepsies. Epilepsia 11:114–119. doi:10.1111/j.1528-1157.1970.tb03873.x
Commission on Classification and Terminology of the International League Against Epilepsy (1981) Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 22:489–501. doi:10.1111/j.1528-1157.1981.tb06159.x
Engel J (2001) A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: report of the ILAE task force on classification and terminology. Epilepsia 42:796–803. doi:10.1046/j.1528-1157.2001.10401.x
Berg AT, Berkovic SF, Brodie MJ et al (2010) Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 51:676–685. doi:10.1111/j.1528-1167.2010.02522.x
Scheffer IE, French J, Hirsch E et al (2016) Classification of the epilepsies: new concepts for discussion and debate-special report of the ILAE Classification Task Force of the Commission for Classification and Terminology. Epilepsia Open 1:37–44. doi:10.1002/epi4.5
Fisher RS, Acevedo C, Arzimanoglou A et al (2014) ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55:475–482. doi:10.1111/epi.12550
Krumholz A, Wiebe S, Gronseth GS et al (2015) Evidence-based guideline: management of an unprovoked first seizure in adults: Report of the Guideline Development Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology 84:1705–1713. doi:10.1212/WNL.0000000000001487
Lugaresi E, Cirignotta F, Montagna P (1986) Nocturnal paroxysmal dystonia. J Neurol Neurosurg Psychiatry 49:375–380. doi:10.1136/JNNP.49.4.375
Tinuper P, Cerullo A, Cirignotta F et al (1990) Nocturnal paroxysmal dystonia with short-lasting attacks: three cases with evidence for an epileptic frontal lobe origin of seizures. Epilepsia 31:549–556. doi:10.1111/j.1528-1157.1990.tb06105.x
Parrino L, Halasz P, Tassinari CA, Terzano MG (2006) CAP, epilepsy and motor events during sleep: the unifying role of arousal. Sleep Med Rev 10:267–285. doi:10.1016/j.smrv.2005.12.004
Trinka E, Cock H, Hesdorffer D et al (2015) A definition and classification of status epilepticus: report of the ILAE task force on classification of status epilepticus. Epilepsia 56:1515–1523. doi:10.1111/epi.13121
Treiman DM, Walton NY, Kendrick C (1990) A progressive sequence of electroencephalographic changes during generalized convulsive status epilepticus. Epilepsy Res 5:49–60. doi:10.1016/0920-1211(90)90065-4
Treiman DM, Meyers PD, Walton NY et al (1998) A comparison of four treatments for generalized convulsive status epilepticus. N Engl J Med 339:792–798. doi:10.1056/NEJM199809173391202
Welch RD, Nicholas K, Durkalski-Mauldin VL et al (2015) Intramuscular midazolam versus intravenous lorazepam for the prehospital treatment of status epilepticus in the pediatric population. Epilepsia 56:254–262. doi:10.1111/epi.12905
Silbergleit R, Durkalski V, Lowenstein D et al (2012) Intramuscular versus intravenous therapy for prehospital status epilepticus. N Engl J Med 366:591–600. doi:10.1056/NEJMoa1107494
Kapur J, Macdonald RL (1997) Rapid seizure-induced reduction of benzodiazepine and Zn 2+ sensitivity of hippocampal dentate granule cell GABA A receptors. J Neurosci 17:7532–7540
Kapur J, Stringer JL, Lothman EW (1989) Evidence that repetitive seizures in the hippocampus cause a lasting reduction of GABAergic inhibition. J Neurophysiol 61:417–426
Goodkin HP, Joshi S, Mtchedlishvili Z et al (2008) Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J Neurosci 28:2527–2538. doi:10.1523/JNEUROSCI.3426-07.2008
Naylor DE (2005) Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J Neurosci 25:7724–7733. doi:10.1523/JNEUROSCI.4944-04.2005
Naylor DE, Liu H, Niquet J, Wasterlain CG (2013) Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiol Dis 54:225–238. doi:10.1016/j.nbd.2012.12.015
Löscher W (2015) Single versus combinatorial therapies in status epilepticus: novel data from preclinical models. Epilepsy Behav 49:20–25. doi:10.1016/j.yebeh.2015.02.027
Trinka E (2011) What is the evidence to use new intravenous AEDs in status epilepticus? Epilepsia 52:35–38. doi:10.1111/j.1528-1167.2011.03232.x
Navarro V, Dagron C, Elie C et al (2016) Prehospital treatment with levetiracetam plus clonazepam or placebo plus clonazepam in status epilepticus (SAMUKeppra): a randomised, double-blind, phase 3 trial. Lancet Neurol 15:47–55. doi:10.1016/S1474-4422(15)00296-3
Shorvon S, Ferlisi M (2011) The treatment of super-refractory status epilepticus: a critical review of available therapies and a clinical treatment protocol. Brain 134:2802–2818. doi:10.1093/brain/awr215
Shorvon S, Ferlisi M (2012) The outcome of therapies in refractory and super-refractory convulsive status epilepticus and recommendations for therapy. Brain 135:2314–2328. doi:10.1093/brain/aws091
Yasiry Z, Shorvon SD (2014) The relative effectiveness of five antiepileptic drugs in treatment of benzodiazepine-resistant convulsive status epilepticus: a meta-analysis of published studies. Seizure 23:167–174. doi:10.1016/j.seizure.2013.12.007
Malamiri RA, Ghaempanah M, Khosroshahi N et al (2012) Efficacy and safety of intravenous sodium valproate versus phenobarbital in controlling convulsive status epilepticus and acute prolonged convulsive seizures in children: a randomised trial. Eur J Paediatr Neurol 16:536–541. doi:10.1016/j.ejpn.2012.01.012
Su Y, Liu G, Tian F et al (2016) Phenobarbital versus valproate for generalized convulsive status epilepticus in adults: a prospective randomized controlled trial in China. CNS Drugs 30:1201–1207. doi:10.1007/s40263-016-0388-6
Vardhan Gupta H, Kaur G, Chawla R et al (2015) comparative assessment for the efficacy of valproate and phenytoin for controlling seizures in patients of convulsive status epilepticus: a randomized controlled trial. J Adv Med Dent Sci Res 3:S15–S20
Shekh-Ahmad T, Mawasi H, McDonough JH et al (2015) The potential of sec-butylpropylacetamide (SPD) and valnoctamide and their individual stereoisomers in status epilepticus. Epilepsy Behav 49:298–302. doi:10.1016/j.yebeh.2015.04.012
Bleck T, Cock H, Chamberlain J et al (2013) The established status epilepticus trial 2013. Epilepsia 54:89–92. doi:10.1111/epi.12288
Legriel S, Pico F, Tran-Dinh Y-R et al (2016) Neuroprotective effect of therapeutic hypothermia versus standard care alone after convulsive status epilepticus: protocol of the multicentre randomised controlled trial HYBERNATUS. Ann Intensiv Care 6:54. doi:10.1186/s13613-016-0159-z
Johnson JW, Glasgow NG, Povysheva NV (2015) Recent insights into the mode of action of memantine and ketamine. Curr Opin Pharmacol 20:54–63. doi:10.1016/j.coph.2014.11.006
Synowiec AS, Singh DS, Yenugadhati V et al (2013) Ketamine use in the treatment of refractory status epilepticus. Epilepsy Res 105:183–188. doi:10.1016/j.eplepsyres.2013.01.007
Hoefler J, Rohracher A, Kalss G et al (2016) (S)-Ketamine in refractory and super-refractory status epilepticus: a retrospective study. CNS Drugs 30:869–876. doi:10.1007/s40263-016-0371-2
Gaspard N, Foreman B, Judd LM et al (2013) Intravenous ketamine for the treatment of refractory status epilepticus: a retrospective multicenter study. Epilepsia 54:1498–1503. doi:10.1111/epi.12247
Rosati A, Ilvento L, L’Erario M et al (2016) Efficacy of ketamine in refractory convulsive status epilepticus in children: a protocol for a sequential design, multicentre, randomised, controlled, open-label, non-profit trial (KETASER01). BMJ Open 6:e011565. doi:10.1136/bmjopen-2016-011565
Legriel S, Lemiale V, Schenck M et al (2016) Hypothermia for neuroprotection in convulsive status epilepticus. N Engl J Med 375:2457–2467. doi:10.1056/NEJMoa1608193
Irani SR, Alexander S, Waters P et al (2010) Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 133:2734–2748. doi:10.1093/brain/awq213
Titulaer MJ, McCracken L, Gabilondo I et al (2013) Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 12:157–165. doi:10.1016/S1474-4422(12)70310-1
Dogan Onugoren M, Deuretzbacher D, Haensch CA et al (2015) Limbic encephalitis due to GABAB and AMPA receptor antibodies: a case series. J Neurol Neurosurg Psychiatry 86:965–972. doi:10.1136/jnnp-2014-308814
Irani SR, Michell AW, Lang B et al (2011) Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 69:892–900. doi:10.1002/ana.22307
Wagner J, Schoene-Bake J-C, Malter MP et al (2013) Quantitative FLAIR analysis indicates predominant affection of the amygdala in antibody-associated limbic encephalitis. Epilepsia 54:1679–1687. doi:10.1111/epi.12320
Irani SR, Stagg CJ, Schott JM et al (2013) Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 136:3151–3162. doi:10.1093/brain/awt212
Bien CG, Urbach H, Schramm J et al (2007) Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology 69:1236–1244. doi:10.1212/01.wnl.0000276946.08412.ef
Vanli-Yavuz EN, Erdag E, Tuzun E et al (2016) Neuronal autoantibodies in mesial temporal lobe epilepsy with hippocampal sclerosis. J Neurol Neurosurg Psychiatry 87:684–692. doi:10.1136/jnnp-2016-313146
Brenner T, Sills GJ, Hart Y et al (2013) Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia 54:1028–1035. doi:10.1111/epi.12127
Kwan P, Brodie MJ (2000) Early identification of refractory epilepsy. N Engl J Med 342:314–319. doi:10.1056/NEJM200002033420503
Brodie MJ (2016) Outcomes in newly diagnosed epilepsy in adolescents and adults: insights across a generation in Scotland. Seizure. doi:10.1016/j.seizure.2016.08.010
Callaghan B, Schlesinger M, Rodemer W et al (2011) Remission and relapse in a drug-resistant epilepsy population followed prospectively. Epilepsia 52:619–626. doi:10.1111/j.1528-1167.2010.02929.x
Luciano AL, Shorvon SD (2007) Results of treatment changes in patients with apparently drug-resistant chronic epilepsy. Ann Neurol 62:375–381. doi:10.1002/ana.21064
Choi H, Heiman G, Pandis D et al (2008) Seizure remission and relapse in adults with intractable epilepsy: a cohort study. Epilepsia 49:1440–1445. doi:10.1111/j.1528-1167.2008.01601.x
Coulter DA, Huguenard JR, Prince DA (1989) Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol 25:582–593. doi:10.1002/ana.410250610
Jasper H, Kershman J, Gibbs FA et al (1941) Electroencephalographic classification of the epilepsies. Arch Neurol Psychiatry 45:903. doi:10.1001/archneurpsyc.1941.02280180015001
Glauser TA, Cnaan A, Shinnar S et al (2010) Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med 362:790–799. doi:10.1056/NEJMoa0902014
Berg AT, Levy SR, Testa FM, Blumenfeld H (2014) Long-term seizure remission in childhood absence epilepsy: might initial treatment matter? Epilepsia 55:551–557. doi:10.1111/epi.12551
Bomben VC, Aiba I, Qian J et al (2016) Isolated P/Q calcium channel deletion in layer VI corticothalamic neurons generates absence epilepsy. J Neurosci 36:405–418. doi:10.1523/JNEUROSCI.2555-15.2016
Szaflarski JP, Kay B, Gotman J et al (2013) The relationship between the localization of the generalized spike and wave discharge generators and the response to valproate. Epilepsia 54:471–480. doi:10.1111/epi.12062
International League Against Epilepsy Consortium on Complex Epilepsies (2014) Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol 13:893–903. doi:10.1016/S1474-4422(14)70171-1
Lal D, Ruppert A-K, Trucks H et al (2015) Burden analysis of rare microdeletions suggests a strong impact of neurodevelopmental genes in genetic generalised epilepsies. PLoS Genet 11:e1005226. doi:10.1371/journal.pgen.1005226
Chung W-C, Hung S-I, Hong H-S et al (2004) A marker for Stevens–Johnson syndrome. Nature 428:386. doi:10.1038/428486a
Chen P, Lin J-J, Lu C-S et al (2011) Carbamazepine-induced toxic effects and HLA-B*1502 screening in Taiwan. N Engl J Med 364:1126–1133. doi:10.1056/NEJMoa1009717
Horn CS, Ater SB, Hurst DL (1986) Carbamazepine-exacerbated epilepsy in children and adolescents. Pediatr Neurol 2:340–345. doi:10.1016/0887-8994(86)90074-3
Guerrini R, Dravet C, Genton P et al (1998) Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 39:508–512. doi:10.1111/j.1528-1157.1998.tb01413.x
Snoeijen-Schouwenaars FM, Veendrick MJBM, van Mierlo P et al (2015) Carbamazepine and oxcarbazepine in adult patients with Dravet syndrome: friend or foe? Seizure 29:114–118. doi:10.1016/j.seizure.2015.03.010
Claes L, Del-Favero J, Ceulemans B et al (2001) De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68:1327–1332. doi:10.1086/320609
Oliva M, Berkovic SF, Petrou S (2012) Sodium channels and the neurobiology of epilepsy. Epilepsia 53:1849–1859. doi:10.1111/j.1528-1167.2012.03631.x
Morgan LA, Millichap JJ (2015) Spectrum of SCN8A-related epilepsy. Pediatr Neurol Briefs 29:16. doi:10.15844/pedneurbriefs-29-2-7
Blanchard MG, Willemsen MH, Walker JB et al (2015) De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. J Med Genet 52:330–337. doi:10.1136/jmedgenet-2014-102813
Boerma RS, Braun KP, van de Broek MPH et al (2016) Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 13:192–197. doi:10.1007/s13311-015-0372-8
Klepper J (2008) Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia 49:46–49. doi:10.1111/j.1528-1167.2008.01833.x
Leen WG, Taher M, Verbeek MM et al (2014) GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol 261:589–599. doi:10.1007/s00415-014-7240-z
Wickenden AD, Yu W, Zou A et al (2000) Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol 58:591–600. doi:10.1124/mol.58.3.591
Millichap J, Park K, Tsuchida T et al (2016) KCNQ2 encephalopathy. Neurol Genet. doi:10.1212/NXG.0000000000000096
Garin Shkolnik T, Feuerman H, Didkovsky E et al (2014) Blue-gray mucocutaneous discoloration: a new adverse effect of ezogabine. JAMA Dermatol 150:984–989. doi:10.1001/jamadermatol.2013.8895
Li D, Yuan H, Ortiz-Gonzalez XR et al (2016) GRIN2D recurrent de novo dominant mutation causes a severe epileptic encephalopathy treatable with NMDA receptor channel blockers. Am J Hum Genet. doi:10.1016/j.ajhg.2016.07.013
Milligan CJ, Li M, Gazina EV et al (2014) KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol 75:581–590. doi:10.1002/ana.24128
Mikati MA, Jiang Y, Carboni M et al (2015) Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol 78:995–999. doi:10.1002/ana.24520
Guerrini R, Dobyns WB (2014) Malformations of cortical development: clinical features and genetic causes. Lancet Neurol 13:710–726. doi:10.1016/S1474-4422(14)70040-7
Krueger DA, Care MM, Holland K et al (2010) Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363:1801–1811. doi:10.1056/NEJMoa1001671
Russo E, Citraro R, Donato G et al (2013) mTOR inhibition modulates epileptogenesis, seizures and depressive behavior in a genetic rat model of absence epilepsy. Neuropharmacology 69:25–36. doi:10.1016/j.neuropharm.2012.09.019
French JA, Lawson JA, Yapici Z et al (2016) Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 6736:1–11. doi:10.1016/S0140-6736(16)31419-2
Sosanya NM, Brager DH, Wolfe S et al (2015) Rapamycin reveals an mTOR-independent repression of Kv1.1 expression during epileptogenesis. Neurobiol Dis 73:96–105. doi:10.1016/j.nbd.2014.09.011
Dalmau J, Gleichman AJ, Hughes EG et al (2008) Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 7:1091–1098. doi:10.1016/S1474-4422(08)70224-2
Ramaswamy V, Walsh JG, Sinclair DB et al (2013) Inflammasome induction in Rasmussen’s encephalitis: cortical and associated white matter pathogenesis. J Neuroinflammation 10:918. doi:10.1186/1742-2094-10-152
Aronica E, Ravizza T, Zurolo E, Vezzani A (2012) Astrocyte immune responses in epilepsy. Glia 60:1258–1268. doi:10.1002/glia.22312
Balosso S, Ravizza T, Aronica E, Vezzani A (2013) The dual role of TNF-α and its receptors in seizures. Exp Neurol 247:267–271. doi:10.1016/j.expneurol.2013.05.010
Prabowo A, Iyer A, Anink J et al (2013) Differential expression of major histocompatibility complex class I in developmental glioneuronal lesions. J Neuroinflammation 10:12. doi:10.1186/1742-2094-10-12
Hoda U, Agarwal NB, Vohora D et al (2016) Resveratrol suppressed seizures by attenuating IL-1β, IL1-Ra, IL-6, and TNF-α in the hippocampus and cortex of kindled mice. Nutr Neurosci. doi:10.1080/1028415X.2016.1189057
Vezzani A, French J, Bartfai T, Baram TZ (2011) The role of inflammation in epilepsy. Nat Rev Neurol 7:31–40. doi:10.1038/nrneurol.2010.178
Maroso M, Balosso S, Ravizza T et al (2011) Interleukin-1β biosynthesis inhibition reduces acute seizures and drug resistant chronic epileptic activity in mice. Neurotherapeutics 8:304–315. doi:10.1007/s13311-011-0039-z
Janigro D (2012) Are you in or out? Leukocyte, ion, and neurotransmitter permeability across the epileptic blood-brain barrier. Epilepsia 53:26–34. doi:10.1111/j.1528-1167.2012.03472.x
Janigro D, Iffland PH, Marchi N, Granata T (2013) A role for inflammation in status epilepticus is revealed by a review of current therapeutic approaches. Epilepsia 54:30–32. doi:10.1111/epi.12271
Zurolo E, Iyer A, Maroso M et al (2013) Activation of TLR, RAGE and HMGB1 signaling in malformations of cortical development. Brain 134:1015–1032. doi:10.1093/brain/awr032
Iyer A, Zurolo E, Spliet WGM et al (2010) Evaluation of the innate and adaptive immunity in type I and type II focal cortical dysplasias. Epilepsia 51:1763–1773. doi:10.1111/j.1528-1167.2010.02547.x
de Vries EE, van den Munckhof B, Braun KPJ et al (2016) Inflammatory mediators in human epilepsy: a systematic review and meta-analysis. Neurosci Biobehav Rev 63:177–190. doi:10.1016/j.neubiorev.2016.02.007
Diamond ML, Ritter AC, Failla MD et al (2014) IL-1-β associations with posttraumatic epilepsy development: a genetics and biomarker cohort study. Epilepsia 55:1109–1119. doi:10.1111/epi.12628
Kwon YS, Pineda E, Auvin S et al (2013) Neuroprotective and antiepileptogenic effects of combination of anti-inflammatory drugs in the immature brain. J Neuroinflammation 10:30. doi:10.1186/1742-2094-10-30
Jyonouchi H, Geng L (2016) Intractable epilepsy (IE) and responses to anakinra, a human recombinant IL-1 receptor agonist (IL-1ra): case reports. J Clin Cell Immunol 7:1–5. doi:10.4172/2155-9899.1000456
Dibbens LM, de Vries B, Donatello S et al (2013) Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 45:546–551. doi:10.1038/ng.2599
Scheffer IE, Heron SE, Regan BM et al (2014) Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 75:782–787. doi:10.1002/ana.24126
Baulac S, Ishida S, Marsan E et al (2015) Familial focal epilepsy with focal cortical dysplasia due to DEPDC 5 mutations. Ann Neurol 77:675–683. doi:10.1002/ana.24368
Weckhuysen S, Marsan E, Lambrecq V et al (2016) Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia 57:994–1003. doi:10.1111/epi.13391
Christensen J, Pedersen MG, Pedersen CB et al (2009) Long-term risk of epilepsy after traumatic brain injury in children and young adults: a population-based cohort study. Lancet 373:1105–1110. doi:10.1016/S0140-6736(09)60214-2
Annegers JF, Hauser A, Rocca WA (1998) A population-based study of seizures brain injuries. N Engl J Med 338:20–24. doi:10.1056/NEJM199801013380104
Marson AG, Al-Kharusi AM, Alwaidh M et al (2007) The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet (London, England) 369:1000–1015. doi:10.1016/S0140-6736(07)60460-7
Glauser T, Ben-Menachem E, Bourgeois B et al (2013) Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia 54:551–563. doi:10.1111/epi.12074
Baulac M, Brodie MJ, Patten A et al (2012) Efficacy and tolerability of zonisamide versus controlled-release carbamazepine for newly diagnosed partial epilepsy: a phase 3, randomised, double-blind, non-inferiority trial. Lancet Neurol 11:579–588. doi:10.1016/S1474-4422(12)70105-9
Leitinger M, Trinka E, Gardella E et al (2016) Diagnostic accuracy of the Salzburg EEG criteria for non-convulsive status epilepticus: a retrospective study. Lancet Neurol 15:1054–1062. doi:10.1016/S1474-4422(16)30137-5
Pohlmann-Eden B, Marson AG, Noack-Rink M et al (2016) Comparative effectiveness of levetiracetam, valproate and carbamazepine among elderly patients with newly diagnosed epilepsy: subgroup analysis of the randomized, unblinded KOMET study. BMC Neurol 16:149. doi:10.1186/s12883-016-0663-7
Afra P, Adamolekun B (2012) Lacosamide treatment of juvenile myoclonic epilepsy. Seizure 21:202–204. doi:10.1016/j.seizure.2011.12.010
Yates S, Wechsler R, Beller C (2014) Lacosamide for uncontrolled primary generalized tonic-clonic seizures: an open-label extension study (P3.276). Neurology 82(P3):276
French JA, Krauss GL, Wechsler RT et al (2015) Perampanel for tonic-clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology 85:950–957. doi:10.1212/WNL.0000000000001930
Gowers WR (1881) Epilepsy and other chronic convulsive disorders. Churchill, London
Feigenbaum JJ, Bergmann F, Richmond SA et al (1989) Nonpsychotropic cannabinoid acts as a functional N-methyl-d-aspartate receptor blocker. Proc Natl Acad Sci USA 86:9584–9587
Devinsky O, Cilio MR, Cross H et al (2014) Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 55:791–802. doi:10.1111/epi.12631
Porter BE, Jacobson C (2013) Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav 29:574–577. doi:10.1016/j.yebeh.2013.08.037
Devinsky O, Marsh E, Friedman D et al (2016) Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 15:270–278. doi:10.1016/S1474-4422(15)00379-8
Young S (2013) Marijuana stops child’s severe seizures-CNN.com. In: CNN. http://edition.cnn.com/2013/08/07/health/charlotte-child-medical-marijuana/. Accessed 18 Dec 2016
Soltesz I, Alger BE, Kano M et al (2015) Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat Rev Neurosci 16:264–277. doi:10.1038/nrn3937
Volkow ND, Baler RD, Compton WM, Weiss SRB (2014) Adverse health effects of marijuana use. N Engl J Med 370:2219–2227. doi:10.1056/NEJMra1402309
Ottman R, Lipton RB, Ettinger AB et al (2011) Comorbidities of epilepsy: results from the epilepsy comorbidities and health (EPIC) survey. Epilepsia 52:308–315. doi:10.1111/j.1528-1167.2010.02927.x
Caplan R, Siddarth P, Stahl L et al (2008) Childhood absence epilepsy: behavioral, cognitive, and linguistic comorbidities. Epilepsia 49:1838–1846. doi:10.1111/j.1528-1167.2008.01680.x
Camfield CS, Camfield PR (2007) Long-term social outcomes for children with epilepsy. Epilepsia 48:3–5. doi:10.1111/j.1528-1167.2007.01390.x
Ferro MA, Camfield CS, Levin SD et al (2013) Trajectories of health-related quality of life in children with epilepsy: a cohort study. Epilepsia 54:1889–1897. doi:10.1111/epi.12388
Jones JE, Watson R, Sheth R et al (2007) Psychiatric comorbidity in children with new onset epilepsy. Dev Med Child Neurol 49:493–497. doi:10.1111/j.1469-8749.2007.00493.x
Hesdorffer DC, Ishihara L, Mynepalli L et al (2012) Epilepsy, suicidality, and psychiatric disorders: a bidirectional association. Ann Neurol 72:184–191. doi:10.1002/ana.23601
Mazarati A, Sankar R (2016) Common mechanisms underlying epileptogenesis and the comorbidities of epilepsy. Cold Spring Harb Perspect Med 6:1–18. doi:10.1101/cshperspect.a022798
Lothe A, Didelot A, Hammers A et al (2008) Comorbidity between temporal lobe epilepsy and depression: a [18 F] MPPF PET study. Brain 131:2765–2782. doi:10.1093/brain/awn194
Martinez A, Finegersh A, Cannon DM et al (2013) The 5-HT1A receptor and 5-HT transporter in temporal lobe epilepsy. Neurology 80:1465–1471. doi:10.1212/WNL.0b013e31828cf809
Larkin GL, Beautrais AL (2011) A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol 14:1127–1131. doi:10.1017/S1461145711000629
Schmitz B (2002) Antidepressant drugs: indications and guidelines for use in epilepsy. Epilepsia 43:14–18. doi:10.1046/j.1528-1157.2002.043s2014.x
Harden CL, Goldstein MA (2002) Mood disorders in patients with epilepsy. CNS Drugs 16:291–302. doi:10.2165/00023210-200216050-00002
Cardamone L, Salzberg M, O’Brien T, Jones N (2013) Antidepressant therapy in epilepsy: can treating the comorbidities affect the underlying disorder? Br J Pharmacol 168:1531–1554. doi:10.1111/bph.12052
Kanner AM (2016) Most antidepressant drugs are safe for patients with epilepsy at therapeutic doses: a review of the evidence. Epilepsy Behav 61:282–286. doi:10.1016/j.yebeh.2016.03.022
Faingold CL, Randall M, Zeng C et al (2016) Serotonergic agents act on 5-HT3 receptors in the brain to block seizure-induced respiratory arrest in the DBA/1 mouse model of SUDEP. Epilepsy Behav 64:166–170. doi:10.1016/j.yebeh.2016.09.034
Boylan LS, Flint LA, Labovitz DL et al (2004) Depression but not seizure frequency predicts quality of life in treatment-resistant epilepsy. Neurology 62:258–261. doi:10.1212/01.WNL.0000103282.62353.85
Campbell E, Kennedy F, Russell A et al (2014) Malformation risks of antiepileptic drug monotherapies in pregnancy: updated results from the UK and Ireland Epilepsy and Pregnancy Registers. J Neurol Neurosurg Psychiatry 85:1029–1034. doi:10.1136/jnnp-2013-306318
Patel SI, Pennell PB (2016) Management of epilepsy during pregnancy: an update. Ther Adv Neurol Disord 9:118–129. doi:10.1177/1756285615623934
Hernandez S, Shen A, Holmes LB (2012) Comparative safety of antiepileptic drugs during pregnancy For the North American. Neurology 78:1692–1699. doi:10.1212/WNL.0b013e3182574f39
Mawhinney E, Craig J, Morrow J et al (2013) Levetiracetam in pregnancy. Neurology 80:400–405. doi:10.1212/WNL.0b013e31827f0874
Meador KJ, Baker GA, Browning N et al (2013) Fetal antiepileptic drug exposure and cognitive outcomes at age 6 years (NEAD study): a prospective observational study. Lancet Neurol 12:244–252. doi:10.1016/S1474-4422(12)70323-X
Christensen J (2013) Prenatal valproate exposure and risk of autism spectrum disorders. JAMA 309:1696–1703. doi:10.1001/jama.2013.2270
Bromley RL, Mawer GE, Briggs M et al (2013) The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J Neurol Neurosurg Psychiatry 84:637–643. doi:10.1136/jnnp-2012-304270
Veiby G, Engelsen BA, Gilhus NE et al (2013) Early child development and exposure to antiepileptic drugs prenatally and through breastfeeding. JAMA Neurol 70:1367. doi:10.1001/jamaneurol.2013.4290
Hernández-Díaz S, Mittendorf R, Smith CR et al (2014) Association between topiramate and zonisamide use during pregnancy and low birth weight. Obstet Gynecol 123:21–28. doi:10.1097/AOG.0000000000000018
Campbell E, Devenney E, Morrow J et al (2013) Recurrence risk of congenital malformations in infants exposed to antiepileptic drugs in utero. Epilepsia 54:165–171. doi:10.1111/epi.12001
Vajda FJE, O’Brien TJ, Lander CM et al (2013) Teratogenesis in repeated pregnancies in antiepileptic drug-treated women. Epilepsia 54:181–186. doi:10.1111/j.1528-1167.2012.03625.x
Edey S, Moran N, Nashef L (2014) SUDEP and epilepsy-related mortality in pregnancy. Epilepsia 55:72–74. doi:10.1111/epi.12621
Jobst BC, Cascino GD, R K et al (2015) Resective epilepsy surgery for drug-resistant focal epilepsy. JAMA 313:285. doi:10.1001/jama.2014.17426
Englot DJ, Ouyang D, Garcia PA et al (2012) Epilepsy surgery trends in the United States, 1990–2008. Neurology 78:1200–1206. doi:10.1212/WNL.0b013e318250d7ea
Helmstaedter C, May TW, von Lehe M et al (2014) Temporal lobe surgery in Germany from 1988 to 2008: diverse trends in etiological subgroups. Eur J Neurol 21:827–834. doi:10.1111/ene.12322
Shorvon S, Tomson T (2011) Sudden unexpected death in epilepsy. Lancet 378:2028–2038. doi:10.1016/S0140-6736(11)60176-1
Tebo CC, Evins AI, Christos PJ et al (2014) Evolution of cranial epilepsy surgery complication rates: a 32-year systematic review and meta-analysis. J Neurosurg 120:1415–1427. doi:10.3171/2014.1.JNS131694
Bjellvi J, Flink R, Rydenhag B, Malmgren K (2015) Complications of epilepsy surgery in Sweden 1996–2010: a prospective, population-based study. J Neurosurg 122:519–525. doi:10.3171/2014.9.JNS132679
Menon R, Rathore C, Sarma SP, Radhakrishnan K (2012) Feasibility of antiepileptic drug withdrawal following extratemporal resective epilepsy surgery. Neurology 79:770–776. doi:10.1212/WNL.0b013e3182644f7d
Yardi R, Irwin A, Kayyali H et al (2014) Reducing versus stopping antiepileptic medications after temporal lobe surgery. Ann Clin Transl Neurol 1:115–123. doi:10.1002/acn3.35
Taft C, Sager Magnusson E, Ekstedt G, Malmgren K (2014) Health-related quality of life, mood, and patient satisfaction after epilepsy surgery in Sweden—a prospective controlled observational study. Epilepsia 55:878–885. doi:10.1111/epi.12616
Picot M-C, Jaussent A, Neveu D et al (2016) Cost-effectiveness analysis of epilepsy surgery in a controlled cohort of adult patients with intractable partial epilepsy: a 5-year follow-up study. Epilepsia 57:1669–1679. doi:10.1111/epi.13492
Edelvik A, Rydenhag B, Olsson I et al (2013) Long-term outcomes of epilepsy surgery in Sweden: a national prospective and longitudinal study. Neurology 81:1244–1251. doi:10.1212/WNL.0b013e3182a6ca7b
Rydenhag B, Flink R, Malmgren K (2013) Surgical outcomes in patients with epileptogenic tumours and cavernomas in Sweden: good seizure control but late referrals. J Neurol Neurosurg Psychiatry 84:49–53. doi:10.1136/jnnp-2012-302449
Ding D, Quigg M, Starke R et al (2015) Predictors of seizure improvement following stereotactic radiosurgery for cerebral arteriovenous malformations in a prospective cohort of 229 patients with AVM-associated epilepsy (P1.001). Neurology 84(P1):001
Przybylowski CJ, Ding D, Starke RM et al (2015) Seizure and anticonvulsant outcomes following stereotactic radiosurgery for intracranial arteriovenous malformations. J Neurosurg 122:1299–1305. doi:10.3171/2014.11.JNS141388
Ditty BJ, Omar NB, Foreman PM et al (2016) Seizure outcomes after stereotactic radiosurgery for the treatment of cerebral arteriovenous malformations. J Neurosurg. doi:10.3171/2015.12.JNS152461
Feng E-S, Sui C-B, Wang T-X, Sun G-L (2016) Stereotactic radiosurgery for the treatment of mesial temporal lobe epilepsy. Acta Neurol Scand 134:442–451. doi:10.1111/ane.12562
Waseem H, Osborn KE, Schoenberg MR et al (2015) Laser ablation therapy: an alternative treatment for medically resistant mesial temporal lobe epilepsy after age 50. Epilepsy Behav 51:152–157. doi:10.1016/j.yebeh.2015.07.022
Ribot R, Jagid J, Serrano E et al (2015) MRI-guided stereotactic laser ablation of mesial temporal structures for the treatment of refractory temporal lobe epilepsy (S31.008). Neurology 84(S31):008
Gonzalez-Martinez J, Vadera S, Mullin J et al (2014) Robot-assisted stereotactic laser ablation in medically intractable epilepsy. Neurosurgery 10:167–173. doi:10.1227/NEU.0000000000000286
Willie JT, Laxpati NG, Drane DL et al (2014) Real-time magnetic resonance-guided stereotactic laser amygdalohippocampotomy for mesial temporal lobe epilepsy. Neurosurgery 74:569–584. doi:10.1227/NEU.0000000000000343
Wellmer J, Voges J, Parpaley Y (2016) Lesion guided radiofrequency thermocoagulation (L-RFTC) for hypothalamic hamartomas, nodular heterotopias and cortical dysplasias: review and perspective. Seizure 41:206–210. doi:10.1016/j.seizure.2016.05.013
de Tisi J, Bell GS, Peacock JL et al (2011) The long-term outcome of adult epilepsy surgery, patterns of seizure remission, and relapse: a cohort study. Lancet 378:1388–1395. doi:10.1016/S0140-6736(11)60890-8
Gonzalez-Martinez J, Bulacio J, Alexopoulos A et al (2013) Stereoelectroencephalography in the “difficult to localize” refractory focal epilepsy: early experience from a North American epilepsy center. Epilepsia 54:323–330. doi:10.1111/j.1528-1167.2012.03672.x
Coan AC, Kubota B, Bergo FPG et al (2014) 3T MRI quantification of hippocampal volume and signal in mesial temporal lobe epilepsy improves detection of hippocampal sclerosis. AJNR Am J Neuroradiol 35:77–83. doi:10.3174/ajnr.A3640
Mellerio C, Labeyrie M-A, Chassoux F et al (2014) 3T MRI improves the detection of transmantle sign in type 2 focal cortical dysplasia. Epilepsia 55:117–122. doi:10.1111/epi.12464
De Ciantis A, Barba C, Tassi L et al (2016) 7 T MRI in focal epilepsy with unrevealing conventional field strength imaging. Epilepsia 57:445–454. doi:10.1111/epi.13313
Rathore C, Dickson JC, Teotónio R et al (2014) The utility of 18F-fluorodeoxyglucose PET (FDG PET) in epilepsy surgery. Epilepsy Res 108:1306–1314. doi:10.1016/j.eplepsyres.2014.06.012
Yang P-F, Pei J-S, Zhang H-J et al (2014) Long-term epilepsy surgery outcomes in patients with PET-positive, MRI-negative temporal lobe epilepsy. Epilepsy Behav 41:91–97. doi:10.1016/j.yebeh.2014.09.054
Bagić A (2016) Look back to leap forward: the emerging new role of magnetoencephalography (MEG) in nonlesional epilepsy. Clin Neurophysiol 127:60–66. doi:10.1016/j.clinph.2015.05.009
Murakami H, Wang ZI, Marashly A et al (2016) Correlating magnetoencephalography to stereo-electroencephalography in patients undergoing epilepsy surgery. Brain 139:2935–2947. doi:10.1093/brain/aww215
Lascano AM, Perneger T, Vulliemoz S et al (2016) Yield of MRI, high-density electric source imaging (HD-ESI), SPECT and PET in epilepsy surgery candidates. Clin Neurophysiol 127:150–155. doi:10.1016/j.clinph.2015.03.025
Oxley TJ, Opie NL, John SE et al (2016) Minimally invasive endovascular stent-electrode array for high-fidelity, chronic recordings of cortical neural activity. Nat Biotechnol 34:320–327. doi:10.1038/nbt.3428
Révész D, Rydenhag B, Ben-Menachem E (2016) Complications and safety of vagus nerve stimulation: 25 years of experience at a single center. J Neurosurg Pediatr 18:97–104. doi:10.3171/2016.1.PEDS15534
Englot DJ, Rolston JD, Wright CW et al (2016) Rates and predictors of seizure freedom with vagus nerve stimulation for intractable epilepsy. Neurosurgery 79:345–353. doi:10.1227/NEU.0000000000001165
Ryvlin P, Gilliam FG, Nguyen DK et al (2014) The long-term effect of vagus nerve stimulation on quality of life in patients with pharmacoresistant focal epilepsy: the PuLsE (open prospective randomized long-term effectiveness) trial. Epilepsia 55:893–900. doi:10.1111/epi.12611
Martin JLR, Martín-Sánchez E (2012) Systematic review and meta-analysis of vagus nerve stimulation in the treatment of depression: variable results based on study designs. Eur Psychiatry 27:147–155. doi:10.1016/j.eurpsy.2011.07.006
Heck CN, King-Stephens D, Massey AD et al (2014) Two-year seizure reduction in adults with medically intractable partial onset epilepsy treated with responsive neurostimulation: final results of the RNS system pivotal trial. Epilepsia 55:432–441. doi:10.1111/epi.12534
Salanova V, Witt T, Worth R et al (2015) Long-term efficacy and safety of thalamic stimulation for drug-resistant partial epilepsy. Neurology 84:1017–1025. doi:10.1212/WNL.0000000000001334
Kowski AB, Voges J, Heinze H-J et al (2015) Nucleus accumbens stimulation in partial epilepsy-A randomized controlled case series. Epilepsia 56:e78–e82. doi:10.1111/epi.12999
Kros L, Eelkman Rooda OHJ, De Zeeuw CI, Hoebeek FE (2015) Controlling cerebellar output to treat refractory epilepsy. Trends Neurosci 38:787–799. doi:10.1016/j.tins.2015.10.002
Krook-Magnuson E, Armstrong C, Oijala M et al (2013) On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat Commun 4:1376. doi:10.1038/ncomms2376
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Dr. Manford has no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Manford, M. Recent advances in epilepsy. J Neurol 264, 1811–1824 (2017). https://doi.org/10.1007/s00415-017-8394-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-017-8394-2