
Abstract
Purpose
This study aimed to evaluate the hypothesis that active smoking impacts upon mediators and abundance of circulating fibrocyte cells in smoking-related disease characterised by fibrosis.
Methods
Flow cytometry and enzyme-linked immunosorbent assays were used to investigate blood from five patient groups: healthy never-smokers, healthy current smokers, stable chronic obstructive pulmonary disease (COPD) active smokers, idiopathic pulmonary fibrosis (IPF) never-smokers, and IPF active smokers.
Results
A significant inverse dose–response relationship was observed in healthy smokers among cumulative smoking burden (pack-years) and fibrocyte abundance (p = 0.006, r = −0.86). Among serum profibrotic fibrocyte chemokines measured, CCL18 rose significantly alongside fibrocyte numbers in all five subject groups, while having an inverse dose–response relationship with pack-year burden in healthy smokers (p = 0.003, r = −0.89). In IPF, CCL2 rose in direct proportion to fibrocyte abundance irrespective of smoking status but had lower serum levels in those currently smoking (p = < 0.001). For the study population, CXCL12 was decreased in pooled current smokers versus never-smokers (p = 0.03).
Conclusion
The suppressive effect of current, as distinct from former, chronic smoking on circulating fibrocyte abundance in healthy smokers, and modulation of regulatory chemokine levels by active smoking may have implications for future studies of fibrocytes in smoking-related lung diseases as a potential confounding variable.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fibrocytes are fibroblast-like peripheral blood cells that migrate to the regions of tissue injury [1]. These hematopoietic cells comprise 0.1 – 0.5 percent of the circulating leukocyte population and are variably defined by the expression of type I collagen (Col-1), CXCR4, CD11b, CD13, CD34, CR45, MHC II, and CD86 [2,3,4,5]. Fibrocytes constitute bone marrow precursor cells believed to transform into fibroblasts or myofibroblasts, under the influence of different molecules including cytokines, growth factors, and components of the extracellular matrix (ECM) [6,7,8,9,10,11,12].
Chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) are two chronic, severe smoking-related fibrogenic lung diseases. The role of circulating fibrocytes in the pathogenesis of IPF is unclear, though several studies have implicated the CXCL12-CXCR4 chemokine axis in murine models and enhanced expression of CXCL12 in human lung tissue and plasma [4, 13]. An unanswered question concerns the origin of the peribronchial fibrogenesis observed in COPD airway walls [14].
Circulating fibrocyte levels are upregulated in stable IPF and further so in acute exacerbations of IPF. There are limited data on fibrocyte biology in COPD, though smoking/COPD have a depressive effect on bone marrow-derived precursor cell abundance in general, within which fibrocytes could be differentially affected [15]. Furthermore, opposing effects of cigarette smoke extract and nicotine on bone marrow-derived mesenchymal stem-cell (MSC) function and their progeny [16,17,18,19]. In that context, it was hypothesised in the present study that smoking burden impacts circulating fibrocyte levels and in a dose-dependent manner. Furthermore, it was postulated that smoking status may discordantly influence the abundance of circulating fibrocytes in stable IPF, COPD smokers, ostensibly “healthy” smokers and healthy non-smokers. Finally, it was hypothesised that relevant blood markers of fibrocyte chemotaxis/signalling would be differentially impacted in smoking-related phenotypes.
Materials and Methods
Study Population
Seventeen age-matched healthy volunteers, i.e. nine healthy non-smokers (H–NS), and eight healthy smokers (H–S) were studied as controls. Ten actively smoking patients diagnosed with COPD (COPD–S) as per standard Global Initiative for Obstructive Lung Disease (GOLD) criteria [20], 7 IPF non-smokers, and 4 current smokers with IPF (IPF–NS and IPF–S, respectively) as defined per standard ATS/ERS diagnostic criteria [21] were recruited from respiratory outpatient clinics, the acute medical assessment unit and emergency department of the host institution, i.e. St Vincent’s University Hospital, Dublin, Ireland. The research protocol was approved by the host institute’s research and ethics committee and informed consent was obtained (For inclusion and exclusion criteria, see supplementary file).
Self-reported smoking status was confirmed objectively by urinary tobacco metabolite—cotinine assays using commercial kits (GFC Diagnostics, Oxfordshire, UK) [22].
Experimental Techniques
Peripheral blood mononuclear cells (PBMC) and serum were isolated from peripheral blood of all subjects using a gradient with ficoll-paque. 1 ml aliquots of cells were placed at −80 °C in a freezer overnight before transferring to long-term liquid nitrogen storage pending their use for flow cytometry. Fibrocytes were identified as cells positive for anti-CD45, Collagen-1, and CXCR4 antibodies and fibrocyte concentrations were measured relative to total leucocyte counts and as absolute numbers (For details, see supplementary file). The serum sample from each subject was stored at −80 °C until chemokine/cytokine analysis. The concentrations of CXCL12, CCL2, CCL18, interleukin 12 (IL–12), interferon Gamma (IFN–γ), and transforming growth factor beta 1 (TGF–β1) in the serum were determined by enzyme-linked immunosorbent assays (ELISA) using commercial kits according to the manufacturer’s instructions (For manufacturer details, see supplementary file).
Statistical analyses were conducted using SPSS version 29. To account for small sample sizes and non-normally distributed outcomes, nonparametric tests were used to compare independent samples. Categorical variables were compared between phenotype groups using Fisher’s exact test and continuous variables with the Mann–Whitney U test (if two groups compared) or the Kruskal–Wallis H test (if three or more groups compared). Spearman’s rank order correlation (ρ) was used to assess the relation between continuous and/or discrete variables. To account for family-wise error rate associated with multiple comparisons, a Bonferroni correction was applied, and the significance threshold was set at p < 0.01. (for details see supplementary file).
Results
All individuals in each study group were matched for gender distribution (p = 0.56) and age (p = 0.09). The subjects in all three current-smoker groups were matched for their respective smoking pack-year history (p = 0.69). Only the ratio of FEV1 to FVC (H(2) = 10.85; p = 0.004) and urine cotinine levels (H(4) = 32.21; p < 0.001)) differed significantly across the phenotype groups, in keeping with the expected airflow obstruction in COPD-S subjects and confirmed smoking status (Table 1).
Fibrocytes were quantified by flow cytometry using the gating strategy as shown in Fig. 1 in all subjects. Given that the central hypothesis of the current study was that smoking impacts upon circulating fibrocyte levels, the relative prevalence of these cells was analysed as a function of cumulative smoking burden, initially in the absence of clinical disease. Among these H–S subjects, the analysis revealed a significant negative correlation or an inverse dose–response relationship between the smoking pack-year history and percentages of circulating fibrocytes (Spearman’s ρ = -0.868, 99% CI (2-tailed) −0.987 to −0.136; p = 0.005, Fig. 2A). Smoking pack-year was noted to vary from as low as 8 pack year to as high as 60. Despite the low number of subjects studied, the inversely proportional relationship between fibrocyte level and corresponding pack-year value did not appear to be driven by one or two outlying values, and therefore was felt to be consistent with a tight relationship of cumulative smoking burden to circulating fibrocyte level in ostensibly “healthy” smokers. The next analysis was to assess if this inverse dose–response would lead to the expected finding of fewer fibrocytes in healthy smokers than in non-smokers. There was a numerically lower mean percentage of circulating fibrocytes in the H–S group (0.61 ± 0.17%) as compared with the H-NS group (0.78 ± 0.19%) but this difference was not statistically significant (U = 33.5, p = 0.81, Fig. 2B). To see if current smoker status might account for a concordant depressive effect in another phenotypic context, the prevalence of circulating fibrocytes was next assessed against an IPF background.

Expression of CD45, collagen I (Col I), and CXCR4 in peripheral circulating leucocytes—Representative flow cytometric analysis from a patient with COPD. A The total cell population was analysed for PerCP stained CD45 expression. B This gate was then applied to the CD45 positive cells which are live, i.e. 99.9%. C Live CD45 positive cell population was further evaluated for APC-stained Col I positive cells. D Live CD45, Col-I positive cell population was further gated down to BV450 stained CXCR4 positive cells, i.e. 3776 events of 957,460 CD45+ cells are alive Col1+ and CXCR4+ (0.39%) called fibrocytes. E The isotope control for APC-stained Col I cells. F The isotype control for BV450 stained CXCR4 cells

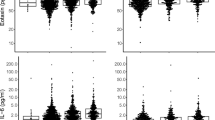
Effect of chronic smoke exposure on circulating fibrocyte abundance. A Relationship between cumulative burden of tobacco (expressed as cigarette pack-years) and fibrocyte abundance (% of total circulating leukocytes) in healthy smokers. B Prevalence of fibrocytes (% of total circulating leukocytes) in healthy subjects assessed by smoking status. C Prevalence of fibrocytes (% of total circulating leukocytes) in IPF non-smokers and IPF smokers
The data once more revealed a numerically lower mean level of circulating fibrocytes in IPF smokers compared to non-smoker IPF subjects which did not attain statistical significance (U = 8.0, p = 0.31, Fig. 2C), likely impacted by the very small number (n = 4) of IPF current smokers that could be found for this study.
Considering the observed inverse dose–response relationship of smoking burden and fibrocyte level, an exploratory analysis was conducted of potential associations among plausible chemokine markers of circulating fibrocyte activation in serum and smoking status in healthy individuals. CCL18, derived from alveolar macrophages (a cell type exposed directly to smoke) is a known mediator of fibrogenesis implicated in the smoking-related lung disease IPF, and in the ILD associated with systemic sclerosis, another disease where fibrocytes play a role [23]. The data indicated that there was a statistically insignificant, directly proportional relationship among CCL18 levels and fibrocyte levels, consistent with possible candidate status for CCL18 in fibrocyte regulation, observed in the H–NS (ρ = 0.75, 99% CI 0.086–1.0; p = 0.02, Fig. 3A) and H–S cohorts (ρ = 0.78, 99% CI 0.0151–1.0, p = 0.02, Fig. 3B). The mean levels of CCL18 were similar in both groups (U = 27.0, p = 0.39, Fig. 3C). There was a statistically significant inverse relationship among pack-year value and CCL18 level in the H–S group, consistent with (though not proving) the potential for CCL18 to be involved in mediating the inverse dose–response relationship observed among circulating fibrocyte levels and the burden of chronic smoke exposure (ρ = 0.89, 99% CI 0.123–1.0; p = 0.003, Fig. 3D). The same directly proportional relationship of serum CCL18 with fibrocyte abundance seen in each healthy group was observed in the larger group of COPD smokers (p = 0.75, 99% CI −0.086 to 1.0; p = 0.01, Fig. 3E).

Role of Serum CCL18 in smoking-induced fibrocyte regulation in healthy subjects. A Relationship between serum CCL18 and circulating fibrocytes as a percentage of total leukocytes among healthy non-smokers. B Relationship between serum CCL18 and circulating fibrocytes as a percentage of total leukocytes among healthy smokers. C Serum CCL18 levels in healthy non-smokers versus healthy smokers. D Relationship of serum CCL18 levels to cigarette pack-year burden in the healthy smoker group. E Relationship between serum CCL18 and circulating fibrocytes as a percentage of total leukocytes among COPD smokers
The chemokine CCL2/MCP-1, implicated in fibrocyte recruitment via its cognate receptors CCR2 and CCR4 has been shown in IPF to be at elevated concentrations in serum and BALF [24]. The data herein confirmed a significantly higher serum CCL2 level in IPF smoker subjects compared to healthy smokers and compared to COPD smokers (H(2) = 16.476, p < 0.001, Fig. 4A). There was a direct correlation of serum CCL2 levels and percent prevalence of circulating fibrocytes among IPF non-smokers (ρ = 0.89, Fig. 4B) and among IPF smokers (ρ = 1.0, Fig. 4B).

Association of serum CCL2 with circulating fibrocyte abundance and smoking status in idiopathic pulmonary fibrosis. A Concentration of serum CCL2 among healthy smokers, IPF smokers, and COPD smokers. B Relationship of serum CCL2 to circulating fibrocyte prevalence among IPF non-smokers and IPF smokers. C Concentration of serum CCL2 in IPF non-smokers and IPF smokers
We next sought to extend these findings by analysing the impact of smoking status in IPF on the enhanced level of CCL2 production. The data showed a trend towards a significant reduction in serum CCL2 levels in IPF smokers versus IPF non-smokers, consistent with a possible depressive effect of current smoking on a fibrocyte-associated profibrotic chemokine in IPF although the finding needs to be interpreted with caution in view of the low numbers of IPF smokers (U = 2.0, p = 0.02, Fig. 4C).
CXCL12 has attracted interest as the ligand interacting with CXCR4 in the purported migration of circulating fibrocytes to sites of injury, such as has been postulated to occur in IPF and COPD [4, 25]. Others have shown higher blood concentrations of CXCL12 in IPF, with levels directly related to levels of circulating fibrocytes [13]. In the present study, there was a significantly higher level of serum CXCL12 observed in the IPF group versus the non-IPF cohorts (U = 71, p = 0.01, Fig. 5A). Additionally, in IPF never-smokers the directly proportional relationship in blood of CXCL12 levels and fibrocyte percent prevalence was demonstrated (Spearman’s ρ = -0.86, 99% CI −0.677 to 1.0; p = 0.01, Fig. 5B) with a weaker, similar relationship observed in the small number of IPF smokers (data not shown). The same relationship was observed in the COPD smoker group (Spearman’s ρ = 0.84, 99% CI 0.073–1.0; p = 0.002, Fig. 5C). To extend upon these findings and link them back to the central hypothesis regarding how smoking impacts fibrocyte biology, CXCL12 levels were analysed by smoking status. While the observations of concordant, numerically reduced mean levels of serum CXCL12 in the smoker as opposed to non-smoker subgroups of IPF subjects (Fig. 5D) and healthy subjects (Fig. 5E) were not statistically significant, by pooling all non-smokers (healthy and IPF) compared to all pooled smokers (healthy, IPF, and COPD), there was evidence to suggest a lowering of CXCL12 levels in the current smokers (U = 104, p = 0.033, Fig. 5F).

Role of Serum CXCL12 in smoking-induced fibrocyte regulation in healthy subjects, IPF and COPD patients. A Concentration of serum CXCL12 in IPF and non-IPF groups. B Relationship between serum CXCL12 and circulating fibrocytes in IPF-Non Smokers. C Relationship between serum CXCL12 and circulating fibrocytes in COPD smokers. D Concentration of serum CXCL12 among IPF non-smokers VS IPF smokers. E Concentration of serum CXCL12 in healthy non-smokers and healthy smokers. F Concentration of serum CXCL12 in all smokers VS all non-smokers
Discussion
In the present study, data was gathered on the influence of chronic tobacco smoke consumption on measurable features of circulating blood fibrocytes in otherwise healthy controls and the smoking-related fibrogenic lung disease states of IPF and COPD. For the first time to our understanding, a significant inverse association of cumulative self-reported tobacco burden and numbers of circulating fibrocytes in otherwise healthy smokers was seen, suggesting a depressive effect of chronic smoking on fibrocyte abundance in the absence of clinical disease. The numerically lower mean abundance of fibrocytes observed in healthy smokers versus healthy non-smokers in the current work was not significantly different, which we postulate is due to underpowering. The direction of the observed inverse dose–response relationship among circulating fibrocytes and cigarette pack-year (lowest cell abundance in longest-smoking individuals) is of central importance as it is incompatible with an inference that merely advancing age explains the higher fibrocyte levels in disease-free smokers, even though increasing age is well described as being directly associated with higher circulating fibrocyte levels [23, 26]. Although underpowered, the numerically lower mean number of fibrocytes for current smokers was concordantly seen where each of the two smoker phenotypic groups was compared with their non-smoker group (H–S vs H–NS, and separately for IPF–S vs IPF–NS). The largest, published, relevant comparator dataset we could find had mixed smoking status histories (current/former/never smoker) within each of two cohorts; COPD and age-matched controls, but interestingly, the group enriched with current smokers (COPD) had a numerically lower mean number of CD45+/Col1+ circulating fibrocytes versus the control group who were enriched for never-smokers, a result concordant with the current study findings, and deserving of investigation in an appropriately powered study [25]. No published studies have compared circulating fibrocyte proportions in COPD versus IPF. IPF has a far greater tissue burden of fibrotic lesions in the pulmonary parenchyma versus the comparatively modest peribronchial fibrosis observed in COPD pathologic specimens [14]. The current study showed concordant relationships for observed pro-fibrotic chemokines/ligands having positive correlation with circulating fibrocyte abundance irrespective of whether the phenotype was COPD or IPF. The fact that smoking has been shown to exert a depressive effect in general on bone marrow-derived progenitor cells (which could include fibrocytes) is in keeping with the current study which observed a reduction by active smoking of the elevated blood fibrocyte levels otherwise found in IPF, a depressive effect of smoking on fibrocyte abundance as was similarly observed for healthy subjects [15].
Little is currently known about the role of chemokines in any potential trafficking of fibrocytes in COPD. Tissue fibrocytes have been shown to be present at greater density in (mainly former smoker) COPD airways than in controls, and correlated with blood fibrocytes [27]. The chemokine CCL18 has some similarities with TGF-β in that it up-regulates collagen production by lung fibroblasts. To our knowledge, the current study is the first to show an association of serum CCL18 levels with increasing fibrocyte abundance in COPD, and has relevance in light of published data that emerged in the literature after the current study was undertaken, showing recruitment of blood fibrocytes during acute exacerbations of COPD, linked to worsening of clinically important endpoints including lung function and mortality [25]. In keeping with the hypothesis that smoking downregulates circulating fibrocytes, the pro-fibrotic chemokine CCL18 had levels that were inversely proportional to cumulative smoking burden in healthy smokers.
The mechanistic and prognostic role of fibrocytes is more established in IPF, a disease characterised by marked pulmonary parenchymal fibrosis and where circulating fibrocytes are strongly upregulated versus controls, and more so in acute IPF exacerbations [4, 13, 28]. Importantly, previous investigators have used human control blood samples to demonstrate in vitro a stimulatory effect of CCL2 exposure on CCR2 receptors expressed on fibrocytes in these samples resulting in proliferation of fibrocytes, migration, and differentiation into a myofibroblast phenotype [29]. Data from Ekert and co-workers supports a mechanism for the CCL2/CCR2 axis in fibrocyte pathobiology and predicts a linear relationship in blood among CCL2 and fibrocyte levels in IPF but was only tested in blood from BALB/c mice / healthy human donors and not tested in IPF subjects. To the best of our knowledge, the current data provides the first direct evidence of such a relationship in IPF subjects. There was a statistically significant direct correlation in blood of IPF non-smokers among CCL2 levels and percent prevalence of circulating fibrocytes providing external validation to the relevant work of Ekert et al. In IPF, we postulate that in a higher powered study where current smokers with IPF may have lower levels of circulating fibrocytes, CCL2 may, in part, mediate this effect based on the significant reduction in CCL2 levels observed in the current work in IPF smokers versus their non-smoker counterparts.
CXCL12 is well-implicated in fibrocyte trafficking in fibrotic lung disease [4, 13, 25]. The current study confirmed the previously reported higher levels of CXCL12 in IPF, an association of CXCL12 serum levels with fibrocyte levels in IPF and separately in COPD and showed for the first time this association in a pure population of current smokers with COPD [25]. CXCL12 is postulated herein to play a role in the hypothesised down-regulation of fibrocyte abundance in current smokers, based on the present study’s finding of significantly downregulated CXCL12 in a pooled analysis of all smokers compared to all non-smokers.
Taken as a whole, the findings are consistent with the hypothesis that chronic smoke exposure may lead to a reduction in circulating fibrocytes, bone marrow-derived cells, and that in IPF, this putative effect may be mediated by the CCL2 axis and likely also by the CXCL12 axis. The present study has important limitations however, chief of which is the low number of samples studied, and future studies would need greater sample size to generate more reliable results. A particular limitation of the study was the low numbers of IPF smokers, with a resultant failure to show statistical significance for the numerically lower mean number of fibrocytes in IPF smokers, likely due to underpowering rather than no effect of smoke on IPF fibrocytes in the authors’ view. In neither the analyses of IPF nor healthy subjects were numerically higher mean fibrocyte numbers observed in the smoker subgroups, and others have shown a reduction in bone marrow-derived cells in chronic smokers, that rebounds with smoking cessation [15, 30, 31]. The abrupt rise of circulating progenitor cells upon smoking cessation may explain the reported lack of difference in circulating fibrocyte levels in control subjects parsed by ever-smoking status (current/former smokers combined as one group versus never-smokers) in a larger study than the current one [31]. Proof of the present hypothesis will require higher numbers of subjects than were recruited herein. Future work examining how smoking affects circulating fibrocytes could evaluate isolated cell populations to measure gene expression and translation profiles in addition to functional assays that include an evaluation of chemotactic alterations.
Conclusion
Bone marrow-derived circulating progenitor cells called fibrocytes are implicated in causation/prognosis of prevalent smoking-related lung diseases but little is known about the impact of ongoing chronic smoking on these cells. The present data shows a significant suppressive effect of ongoing cumulative smoking burden on the abundance of these cells in a dose-responsive manner in preclinical smoke-exposed subjects that may confound interpretation of data in studies of circulating fibrocytes where current as opposed to former smoking status is uncontrolled for. Larger studies are needed in this area to better understand the impact of smoking on fibrocytes in health and disease.
Data Availability
No datasets were generated or analysed during the current study.
References
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A (1994) Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1:71–81
Aiba STH (1997) Inverse correlation between CD34 expression and proline-4-hydroxylase immunoreactivity on spindle cells noted in hypertrophic scars and keloids. J Cutan Pathol 24:65–69
Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R (2004) Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol 36:598–606
Phillips RJ, Burdick MD, Hong K, Lutz MA, Murray LA, Xue YY, Belperio JA, Keane MP, Strieter RM (2004) Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J Clin Invest 114(3):438–446
Sun L, Louie MC, Vannella KM, Wilke CA, LeVine AM, Moore BB, Shanley TP (2011) New concepts of IL-10-induced lung fibrosis: fibrocyte recruitment and M2 activation in a CCL2/CCR2 axis. Am J Physiol Lung Cell Mol Physiol 300(3):L341–L353
Zandvoort A, Postma DS, Jonker MR, Noordhoek JA, Vos JT, van der Geld YM, Timens W (2006) Altered expression of the Smad signalling pathway: implications for COPD pathogenesis. Eur Respir J 28:533–541
Roberts AB, McCune BK, Sporn MB (1992) TGF-beta: regulation of extracellular matrix. Kidney Int 41:557–559
Schiller M, Javelaud D, Mauviel A (2004) TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci 35:83–92
Morty RE, Konigshoff M, Eickelberg O (2009) Transforming growth factor-beta signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc 6:607–613
Sugiura H, Ichikawa T, Liu X, Kobayashi T, Wang XQ, Kawasaki S, Togo S, Kamio K, Mao L, Ann Y, Ichinose M, Rennard SI (2009) N-acetyl-L-cysteine inhibits TGF-beta1-induced profibrotic responses in fibroblasts. Pulm Pharmacol Ther 22:487–491
Araya J, Nishimura SL (2010) Fibrogenic reactions in lung disease. Annu Rev Pathol 5:77–98
Camara J, Jarai G (2010) Epithelial–mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair 3(1):2
Mehrad B, Burdick MD, Zisman DA, Keane MP, Belperio JA, Strieter RM (2007) Circulating peripheral blood fibrocytes in human fibrotic interstitial lung disease. Biochem Biophys Res Commun 353(1):104–108
Barnes PJ (2019) Small airway fibrosis in COPD. Int J Biochem Cell Biol. https://doi.org/10.1016/j.biocel.2019.105598
Huertas A, Testa U, Riccioni R, Petrucci E, Riti V, Savi D, Serra P, Bonsignore MR, Palange P (2010) Bone marrow-derived progenitors are greatly reduced inpatients with severe COPD and low-BMI. Respir Physiol Neurobiol 170:23–31
Fang MA, Frost PJ, Iida-Klein A, Hahn TJ (1991) Effects of nicotine on cellular function in UMR 106–01 osteoblast-like cells. Bone 12:283–286
Gullihorn L, Karpman R, Lippiello L (2005) Differential effects of nicotine and smoke condensate on bone cell metabolic activity. J Orthop Trauma 19:17–22
Ramp WK, Lenz LG, Galvin RJ (1991) Nicotine inhibits collagen synthesis and alkaline phosphatase activity, but stimulates DNA synthesis in osteoblast-like cells. Proc Soc Exp Biol Med 197:36–43
Liu XD, Zhu YK, Umino T, Spurzem JR, Romberger DJ, Wang H, Reed E, Rennard SI (2001) Cigarette smoke inhibits osteogenic differentiation and proliferation of human osteoprogenitor cells in monolayer and three-dimensional collagen gel culture. J Lab Clin Med 137:208–219
Agustí A, Celli BR, Criner GJ, Halpin D, Anzueto A, Barnes P, Bourbeau J, Han MK, Martinez FJ, de Oca MM, Mortimer K, Papi A, Pavord I, Roche N, Salvi S, Sin DD, Singh D, Stockley R, Victorina López Varela M, Wedzicha JA, Vogelmeier CF (2023) Global initiative for chronic obstructive lung disease 2023 report: GOLD executive summary. Eur Respir J 61:2300239. https://doi.org/10.1183/13993003.00239-2023
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr, Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183:788–824
Cope G, Nayyar P, Holder R, Gibbons J, Bunce R (1996) A simple near-patient test for nicotine and its metabolites in urine to assess smoking habit. Clin Chim Acta 256:135–149. https://doi.org/10.1016/s0009-8981(96)06417-0
Mathai SK, Gulati M, Peng X, Russell TR, Shaw AC, Rubinowitz AN, Murray LA, Siner JM, Antin-Ozerkis DE, Montgomery RR, Reilkoff RA, Bucala RJ, Herzog EL (2010) Circulating monocytes from systemic sclerosis patients with interstitial lung disease show an enhanced profibrotic phenotype. Lab Invest 90(6):812–823
Suga M, Iyonaga K, Ichiyasu H, Saita N, Yamasaki H, Ando M (1999) Clinical significance of MCP-1 levels in BALF and serum in patients with interstitial lung diseases. Eur Respir J 14(2):376–382
Dupin I, Allard B, Ozier A, Maurat E, Ousova O, Delbrel E, Trian T, Bui HN, Dromer C, Guisset O, Blanchard E, Hilbert G, Vargas F, Thumerel M, Marthan R, Girodet PO, Berger P (2016) Blood fibrocytes are recruited during acute exacerbations of chronic obstructive pulmonary disease through a CXCR4-dependent pathway. J Allergy Clin Immunol 137(4):1036–1042
Hartlapp I, Abe R, Saeed RW, Peng T, Voelter W, Bucala R, Metz CN (2001) Fibrocytes induce an angiogenic phenotype in cultured endothelial cells and promote angiogenesis in vivo. FASEB J 15:2215–2224
Dupin I, Thumerel M, Maurat E, Coste F, Eyraud E, Begueret H, Trian T, Montaudon M, Marthan R, Girodet PO, Berger P (2019) Fibrocyte accumulation in the airway walls of COPD patients. Eur Respir J 54(3):1802173. https://doi.org/10.1183/13993003.02173-2018
Moeller A, Gilpin SE, Ask K, Cox G, Cook D, Gauldie J, Margetts PJ, Farkas L, Dobranowski J, Boylan C, O’Byrne PM, Strieter RM, Kolb M (2009) Circulating fibrocytes are an indicator of poor prognosis in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 179:588–594
Ekert JE, Murray LA, Das AM, Sheng H, Giles-Komar J, Rycyzyn MA (2011) Chemokine (C-C motif) ligand 2 mediates direct and indirect fibrotic responses in human and murine cultured fibrocytes. Bio Med Central - Fibrogenesis Tissue Repair. https://doi.org/10.1186/1755-1536-4-23
Bonsignore MR, Palange P, Testa U, Huertas A, Antonucci R, Serra P, Bonsignore G (2006) Circulating CD34+ cells are decreased inchronic obstructive pulmonary disease. Proc Am Thorac Soc 3:537–538
Kondo T, Hayashi M, Takeshita K, Numaguchi Y, Kobayashi K, Iino S, Inden Y, Murohara T (2004) Smoking cessation rapidly increases circulating progenitor cells in peripheral blood in chronic smokers. Arterioscler Thromb Vasc Biol 24:1442–1447
Acknowledgements
Dr Alfonso Fernandez, Director of Flow Cytometry, UCD Conway Institute of Biomolecular and Biomedical Research.
Funding
Open Access funding provided by the IReL Consortium. Funding support was provided by the UCD OBRSS Research Support Scheme.
Author information
Authors and Affiliations
Contributions
The research is the primary project of Dr Faheem Khan MD research by thesis. We identified patients, drew their blood samples, processed the PBMCs, flow cytometry, ELISA and data analysis. Mr White, Dr Judge and Dr Das did initial preliminary research to identify the potential pro-fibrotic biomarkers. We attended weekly research meetings presided by Dr Butler and Prof Keane. Dr Carolyn Ingram provided statistical expertise to bring this project to completion.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, F., Judge, E.P., Das, J.P. et al. Effects of Active Chronic Cigarette-Smoke Exposure on Circulating Fibrocytes. Lung (2024). https://doi.org/10.1007/s00408-024-00720-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00408-024-00720-3