
Abstract
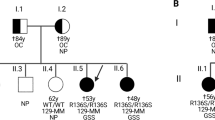
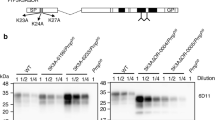
A missense variant from methionine to arginine at codon 232 (M232R) of the prion protein gene accounts for ~ 15% of Japanese patients with genetic prion diseases. However, pathogenic roles of the M232R substitution for the induction of prion disease have remained elusive because family history is usually absent in patients with M232R. In addition, the clinicopathologic phenotypes of patients with M232R are indistinguishable from those of sporadic Creutzfeldt-Jakob disease patients. Furthermore, the M232R substitution is located in the glycosylphosphatidylinositol (GPI)-attachment signal peptide that is cleaved off during the maturation of prion proteins. Therefore, there has been an argument that the M232R substitution might be an uncommon polymorphism rather than a pathogenic mutation. To unveil the role of the M232R substitution in the GPI-attachment signal peptide of prion protein in the pathogenesis of prion disease, here we generated a mouse model expressing human prion proteins with M232R and investigated the susceptibility to prion disease. The M232R substitution accelerates the development of prion disease in a prion strain-dependent manner, without affecting prion strain-specific histopathologic and biochemical features. The M232R substitution did not alter the attachment of GPI nor GPI-attachment site. Instead, the substitution altered endoplasmic reticulum translocation pathway of prion proteins by reducing the hydrophobicity of the GPI-attachment signal peptide, resulting in the reduction of N-linked glycosylation and GPI glycosylation of prion proteins. To the best of our knowledge, this is the first time to show a direct relationship between a point mutation in the GPI-attachment signal peptide and the development of disease.





Similar content being viewed by others

Data availability
The data that support the findings are available from the corresponding author upon request. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD037584 and 10.6019/PXD037584.
References
Asano M, Mohri S, Ironside JW, Ito M, Tamaoki N, Kitamoto T (2006) vCJD prion acquires altered virulence through trans-species infection. Biochem Biophys Res Commun 342:293–299. https://doi.org/10.1016/j.bbrc.2006.01.149
Baron GS, Hughson AG, Raymond GJ et al (2011) Effect of glycans and the glycophosphatidylinositol anchor on strain dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 50:4479–4490. https://doi.org/10.1021/bi2003907
Bate C, Nolan W, McHale-Owen H, Williams A (2016) Sialic acid within the glycosylphosphatidylinositol anchor targets the cellular prion protein to synapses. J Biol Chem 291:17093–17101. https://doi.org/10.1074/jbc.M116.731117
Bate C, Nolan W, Williams A (2016) Sialic acid on the glycosylphosphatidylinositol anchor regulates PrP-mediated cell signalling and prion formation. J Biol Chem 291:160–170. https://doi.org/10.1074/jbc.M115.672394
Beck J, Collinge J, Mead S (2012) Prion protein gene M232R variation is probably an uncommon polymorphism rather than a pathogenic mutation. Brain 135:e209. https://doi.org/10.1093/brain/awr294
Bellai-Dussault K, Nguyen TTM, Baratang NV, Jimenez-Cruz DA, Campeau PM (2019) Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin Genet 95:112–121. https://doi.org/10.1111/cge.13425
Braunger K, Pfeffer S, Shrimal S et al (2018) Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 360:215–219. https://doi.org/10.1126/science.aar7899
Davis EM, Kim J, Menasche BL et al (2015) Comparative haploid genetic screens reveal divergent pathways in the biogenesis and trafficking of glycophosphatidylinositolanchored proteins. Cell Rep 11:1727–1736. https://doi.org/10.1016/j.celrep.2015.05.026
Grathwohl KU, Horiuchi M, Ishiguro N, Shinagawa M (1996) Improvement of PrPSc-detection in mouse spleen early at the preclinical stage of scrapie with collagenase-completed tissue homogenization and Sarkosyl-NaCl extraction of PrPSc. Arch Virol 141:1863–1874. https://doi.org/10.1007/BF01718200
Guizzunti G, Zurzolo C (2014) The fate of PrP GPI-anchor signal peptide is modulated by P238S pathogenic mutation. Traffic 15:78–93. https://doi.org/10.1111/tra.12126
Hirata T, Kobayashi A, Tomita H et al (2022) Loss of the N-acetylgalactosamine side chain of the GPI-anchor impairs bone formation and brain functions and accelerates the prion disease pathology. J Biol Chem 298:101720. https://doi.org/10.1016/j.jbc.2022.101720
Hirata T, Mishra SK, Nakamura S et al (2018) Identification of a Golgi GPI-N-acetylgalactosamine transferase with tandem transmembrane regions in the catalytic domain. Nat Commun 9:405. https://doi.org/10.1038/s41467-017-02799-0
Hirata T, Yang J, Tomida S et al (2022) ER entry pathway and glycosylation of GPI-anchored proteins are determined by N-terminal signal sequence and C-terminal GPI-attachment sequence. J Biol Chem 298:102444. https://doi.org/10.1016/j.jbc.2022.102444
Hizume M, Kobayashi A, Teruya K et al (2009) Human prion protein (PrP) 219K is converted to PrPSc but shows heterozygous inhibition in variant Creutzfeldt-Jakob disease infection. J Biol Chem 284:3603–3609. https://doi.org/10.1074/jbc.M809254200
Jodoin J, Laroche-Pierre S, Goodyer CG, LeBlanc AC (2007) Defective retrotranslocation causes loss of anti-Bax function in human familial prion protein mutants. J Neurosci 27:5081–5091. https://doi.org/10.1523/JNEUROSCI.0957-07.2007
Kanda Y (2013) Investigation of the freely available easy-to-use software‘EZR’ for medical statistics. Bone Marrow Transplant 48:452–458. https://doi.org/10.1038/bmt.2012.244
Katorcha E, Makarava N, Savtchenko R, Baskakov IV (2015) Sialylation of the prion protein glycans controls prion replication rate and glycoform ratio. Sci Rep 5:16912. https://doi.org/10.1038/srep16912
Kinoshita T (2020) Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol 10:190290. https://doi.org/10.1098/rsob.190290
Kitamoto T, Muramoto T, Hilbich C, Beyreuther K, Tateishi J (1991) N-terminal sequence of prion protein is also integrated into kuru plaques in patients with Gerstmann-Straussler syndrome. Brain Res 545:319–321. https://doi.org/10.1016/0006-8993(91)91306-l
Kitamoto T, Nakamura K, Nakao K et al (1996) Humanized prion protein knock-in by Cre-induced site-specific recombination in the mouse. Biochem Biophys Res Commun 222:742–747. https://doi.org/10.1006/bbrc.1996.0814
Kitamoto T, Ohta M, Doh-ura K, Hitoshi S, Terao Y, Tateishi J (1993) Novel missense variants of prion protein in Creutzfeldt-Jakob disease or Gerstmann-Straussler syndrome. Biochem Biophys Res Commun 191:709–714. https://doi.org/10.1006/bbrc.1993.1275
Kitamoto T, Shin RW, Doh-ura K et al (1992) Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 140:1285–1294
Kobayashi A, Asano M, Mohri S, Kitamoto T (2007) Cross-sequence transmission of sporadic Creutzfeldt-Jakob disease creates a new prion strain. J Biol Chem 282:30022–30028. https://doi.org/10.1074/jbc.M704597200
Kobayashi A, Hirata T, Nishikaze T et al (2020) α2,3 linkage of sialic acid to a GPI anchor and an unpredicted GPI attachment site in human prion protein. J Biol Chem 295:7789–7798. https://doi.org/10.1074/jbc.RA120.013444
Kobayashi A, Sakuma N, Matsuura Y, Mohri S, Aguzzi A, Kitamoto T (2010) Experimental verification of a traceback phenomenon in prion infection. J Virol 84:3230–3238. https://doi.org/10.1128/JVI.02387-09
Koga D, Kusumi S, Shibata M, Watanabe T (2021) Applications of scanning electron microscopy using secondary and backscattered electron signals in neural structure. Front Neuroanat 15:759804. https://doi.org/10.3389/fnana.2021.759804
Kusumi S, Koga D, Watanabe T, Shibata M (2018) Combination of a cryosectioning method and section scanning electron microscopy for immuno-scanning electron microscopy. Biomed Res 39:21–25. https://doi.org/10.2220/biomedres.39.21
Minikel EV, Vallabh SM, Lek M et al (2016) Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med 8:322ra9. https://doi.org/10.1126/scitranslmed.aad5169
Murakami Y, Tawamie H, Maeda Y et al (2014) Null mutation in PGAP1 impairing Gpi-anchor maturation in patients with intellectual disability and encephalopathy. PLoS Genet 10:e1004320. https://doi.org/10.1371/journal.pgen.1004320
Nozaki I, Hamaguchi T, Sanjo N et al (2010) Prospective 10-year surveillance of human prion diseases in Japan. Brain 133:3043–3057. https://doi.org/10.1093/brain/awq216
Parchi P, Giese A, Capellari S et al (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. https://doi.org/10.1002/1531-8249(199908)46:2%3c224::AID-ANA12%3e3.0.CO;2-W
Prusiner SB, Scott MR, DeArmond SJ, Cohen FE (1998) Prion protein biology. Cell 93:337–348. https://doi.org/10.1016/s0092-8674(00)81163-0
Puig B, Altmeppen HC, Linsenmeier L et al (2019) GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog 15:e1007520. https://doi.org/10.1371/journal.ppat.1007520
Ruiz-Canada C, Kelleher DJ, Gilmore R (2009) Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136:272–283. https://doi.org/10.1016/j.cell.2008.11.047
Shibatani T, David LL, McCormack AL, Frueh K, Skach WR (2005) Proteomic analysis of mammalian oligosaccharyltransferase reveals multiple subcomplexes that contain Sec61, TRAP, and two potential new subunits. Biochemistry 44:5982–5992. https://doi.org/10.1021/bi047328f
Shiga Y, Satoh K, Kitamoto T et al (2007) J Neurol 254:1509–1517. https://doi.org/10.1007/s00415-007-0540-9
Stahl N, Borchelt DR, Hsiao K, Prusiner SB (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51:229–240. https://doi.org/10.1016/0092-8674(87)90150-4
Takeda N, Yokota O, Terada S et al (2012) Creutzfeldt-Jakob disease with the M232R mutation in the prion protein gene in two cases showing different disease courses: a clinicopathological study. J Neurol Sci 312:108–116. https://doi.org/10.1016/j.jns.2011.08.008
Takeuchi A, Mohri S, Kai H et al (2019) Two distinct prions in fatal familial insomnia and its sporadic form. Brain Commun 1:fcz045. https://doi.org/10.1093/braincomms/fcz045
Wiseman FK, Cancellotti E, Piccardo P et al (2015) The glycosylation status of PrPC is a key factor in determining transmissible spongiform encephalopathy transmission between species. J Virol 89:4738–4747. https://doi.org/10.1128/JVI.02296-14
Yang J, Hirata T, Liu YS et al (2021) Human SND2 mediates ER targeting of GPI-anchored proteins with low hydrophobic GPI attachment signals. FEBS Lett 595:1542–1558. https://doi.org/10.1002/1873-3468.14083
Acknowledgements
We thank members of the Creutzfeldt-Jakob Disease Surveillance Committee in Japan, Creutzfeldt-Jakob disease specialists in the prefectures, and Creutzfeldt-Jakob disease patients and families for providing important clinical information. We thank Hiroko Kudo, Miyuki Yamamoto, Ayumi Yamazaki, and Akinori Ninomiya for their excellent technical assistance.
Funding
This work was supported by JSPS KAKENHI Grant Number 18K05963 (AK), The Kato Memorial Trust for Nambyo Research (AK), Takeda Science Foundation (AK), The Akiyama Life Science Foundation (AK), The Ichiro Kanehara Foundation (AK), The Suhara Memorial Foundation (AK), JST ACT-X Grant Number JPMJAX201B (TH), and JSPS KAKENHI Grant Number 21H02415 (TKin).
Author information
Authors and Affiliations
Contributions
AK, TH: conceptualization; TKin, SM, TKit: methodology; AK, TH, TS, YMu, JNK, RH, AT, YMa, SK, DK, YI: investigation; AK, TH, TS, JNK, RH, SK, DK: visualization; AK, TH, TKin: funding acquisition; AK: project administration; KA, TKim, TKin, SM, TKit: Supervision; AK, TH, TS: writing—original draft; KA, TKim, TKin, SM, TKit: writing—review & editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no competing interests.
Ethical approval
This study was approved by the Institutional Ethics Committee of Hokkaido University Faculty of Veterinary Medicine (VET27-1, VET1-5). All experiments using human materials were in compliance with the Helsinki Declaration. Animal experiments were performed in strict accordance with the Regulations for Animal Experiments and Related Activities at Hokkaido University and Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions by the Ministry of Education, Culture, Sports, Science and Technology in Japan, Notice No. 71. Protocols for animal experiments were approved by the Institutional Animal Care and Use Committees of Hokkaido University (14-0170, 19-0025).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kobayashi, A., Hirata, T., Shimazaki, T. et al. A point mutation in GPI-attachment signal peptide accelerates the development of prion disease. Acta Neuropathol 145, 637–650 (2023). https://doi.org/10.1007/s00401-023-02553-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-023-02553-5