
Abstract
This study aimed to re-evaluate the prognostic impact of TP53 mutations and to identify specific chromosomal aberrations as possible prognostic markers in WNT-activated medulloblastoma (WNT-MB). In a cohort of 191 patients with WNT-MBs, mutations in CTNNB1, APC, and TP53 were analyzed by DNA sequencing. Chromosomal copy-number aberrations were assessed by molecular inversion probe technology (MIP), SNP6, or 850k methylation array hybridization. Prognostic impact was evaluated in 120 patients with follow-up data from the HIT2000 medulloblastoma trial or HIT registries. CTNNB1 mutations were present in 92.2%, and APC mutations in 6.8% of samples. One CTNNB1 wild-type tumor gained WNT activation due to homozygous FBXW7 deletion. Monosomy 6 was present in 78.6%, and more frequent in children than adults. 16.1% of tumor samples showed TP53 mutations, of those 60% with nuclear positivity for the p53 protein. Loss of heterozygosity at the TP53 locus (chromosome 17p13.1) was found in 40.7% (11/27) of TP53 mutant tumor samples and in 12.6% of TP53 wild-type cases (13/103). Patients with tumors harboring TP53 mutations showed significant worse progression-free survival (PFS; 5-year-PFS 68% versus 93%, p = 0.001), and were enriched for chromosomes 17p (p = 0.001), 10, and 13 losses. Gains of OTX2 (14q22.3) occurred in 38.9% of samples and were associated with poor PFS and OS (5-year-PFS 72% versus 93%, p = 0.017 resp. 5-year-OS 83% versus 97%, p = 0.006). Multivariable Cox regression analysis for PFS/OS identified both genetic alterations as independent prognostic markers. Our data suggest that patients with WNT-MB carrying TP53 mutations or OTX2 gains (58.1%) are at higher risk of relapse. Eligibility of these patients for therapy de-escalation trials needs to be debated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Activation of the WNT signaling pathway is present in about 10% of all medulloblastomas (MB). These are now considered an independent MB entity with favorable outcome [11, 30]. WNT-pathway activation was first identified by immunohistochemical demonstration of nuclear β-catenin accumulation [11]. However, nuclear β-catenin accumulation alone is believed to be insufficient to detect all WNT-MBs as some cases lack detectable nuclear accumulation [12, 16, 26], and current neuropathological guidelines recommend the combination of different methods to identify WNT-MB.
In most WNT-MBs, pathway activation is a result of activating mutations in exon 3 of CTNNB1, leading to stabilization and subsequent nuclear accumulation of its product β-catenin [10, 13, 23, 47]. However, other members of the WNT signaling pathway like the tumor suppressor genes APC, Axin1, or Axin2/Conductin were shown to harbor mutations as alternative genetic alterations [9, 19, 22, 23], while only CTNNB1 and APC mutations were found in a substantial fraction of WNT-MBs [33, 35, 38, 43, 44]. The favorable prognosis of WNT-MBs seems independent of the activating mutation [40, 43]. A substantial proportion of APC-mutant WNT-MB occur in the context of an APC germline mutation (either familial or de novo) [43]. In these cases, the MB is part of the tumor spectrum of familial adenomatous polyposis (FAP), and usually occurs as first manifestation of the disease before multiple colorectal adenomas are present.
In addition to CTNNB1 and APC mutations that are considered the main drivers of WNT activation, several further recurrent mutations have been identified in WNT-MBs, including DDX3X (36%), SMARCA4 (19%), and TP53 (14%) [21]. Whereas in Sonic-Hedgehog (SHH)-MBs, TP53 mutations were associated with very poor outcome, this was not the case in WNT-MBs [46]. Zhukova et al. showed a 5-year overall survival (OS) of 90% resp. 86% in discovery and validation cohorts in TP53 mutant cases versus 97% and 94% in TP53 wild-type WNT-MBs [46]. Lindsey et al. found TP53 mutations in 3 of 15 CTNNB1-mutated MBs and all three patients showed long-term survival [29]. However, in a recent publication by Richardson et al., four of five relapsing WNT-MBs were described to carry TP53 mutations [37].
Loss of chromosome 6 (monosomy 6) was found in the majority of WNT-MBs, with about 85% of pediatric WNT-MBs presenting monosomy 6 [6, 16, 20, 24, 30], but only 30–50% of adolescent and adult WNT-MB [5, 15]. Otherwise, WNT-MB samples present mainly stable genomes [21].
Given the very favorable prognosis with current standard therapy, patients with WNT-MB are enrolled in clinical studies aiming at a reduction of treatment intensity and hereby spare treatment-related side effects. Common to most of these trials is the reduction of radiotherapy dose to the whole brain and whole spine, either to 15 Gy (NCT01878617, St. Jude), or 18 Gy (NCT02724579, Children's Oncology Group, NCI, and NCT02066220, SIOP-Europe), which is very likely to reduce cognitive sequelae for the patients [31]. However, most patients with WNT-MB cannot be rescued when the disease recurs after standard therapy [32]. Therefore, it is of major importance to find biomarkers for identification of patients in which intensity of treatment should not be reduced, and of those patients qualifying for reduction of treatment intensity. In this large retrospective cohort, we aimed to re-evaluate the TP53 status as prognostic factor in WNT-MBs, and to screen for chromosomal aberrations and other biomarkers as possible prognostic factors.
Materials and methods
Patients
Over a period of 20 years (2000–2020), 191 MB samples with confirmed WNT activation were identified. WNT-pathway activation was defined by the presence of at least two of the following features: nuclear positivity of β-catenin, mutation of a WNT-pathway component, e.g., CTNNB1 (exon 3) or APC, and/or monosomy 6.
One-hundred and twenty patients participated in either the HIT-2000 trial (NCT00303810), the HIT-2000interim registry (NCT02238899), or the I-HIT-MED registry (NCT02417324), so detailed information on the treatment and follow-up was available (clinical cohort). The HIT2000 trial and both registries have been approved by the responsible ethics committees. Informed consent for exploratory research on tumor tissue had been given by the patients or their legal representatives.
Histopathological and molecular analyses
According to the WHO classification 2016, the tumor samples were classified after conventional (HE and reticulin) and immunohistochemical stainings including markers for pathway activation [8, 34].
Molecular inversion probe array (MIP)
A MIP array including 330,000 inversion probes (Version v2.0, Affymetrix, Santa Clara, CA, USA) was used as previously described to identify copy-number gains and losses in 108 samples [42]. The raw MIP data file was analyzed using the Nexus Copy Number 8.0 Discovery Edition software (BioDiscovery, El Segundo, CA, USA). BioDiscovery’s SNP-FASST2-Segmentation algorithm was used to make copy number and loss of heterozygosity calls. Further details are given in Online Resource Methods.
Next-generation sequencing (NGS) and Sanger sequencing
Exon 3 of CTNNB1 and exons 4–8 of TP53 were directly analyzed by Sanger sequencing using specific primers in all resp. 186 samples. Furthermore, hotspot mutations at E17 in AKT1 (exon 3), AKT2 (exon 3), and AKT3 (exon 2) were screened. Primer sequences are shown in Online Resource Table 1.
Forty WNT-MBs, including 13 CTNNB1-wild-type cases, were screened for APC mutations by one of two DNA-NGS panels as further described in Online Resource Methods and Online Resource Table 2.
450k/850k methylation bead arrays
Genome-wide methylation profiles were generated by hybridization to Illumina 450 k/850 k methylation bead arrays in 94 cases. The Heidelberg Brain Tumor classifier v11b4 was used to annotate the profiles to a methylation class [4, 18]; scores > 0.9 were considered as match. In 14 cases, copy-number information for numerical aberrations was retrieved from methylation bead arrays.
Statistical analysis
Progression-free survival (PFS) and overall survival (OS) were defined as time from first tumor surgery to (a) first tumor progression or death of any cause (PFS) or (b) to death of any cause (OS). PFS and OS rates were estimated using the Kaplan–Meier method. Univariable survival rates were compared using the log-rank test. The most relevant risk factors were identified by a multivariable Cox regression analysis with variable selection independently for PFS and OS based on minimization of Akaike’s information criterion. For variable selection, each combination of the 12 covariates (therapy protocol, clinical risk, histology, OTX2 status, TP53 copy number, TP53 mutation, chr6, chr10, chr13, chrX, WNT signaling activating mutation, and age at diagnosis) were considered (4096 different covariate combinations, including an intercept only model). Robustness of the results was investigated by repeatedly applying a subsampling approach independently for PFS and OS. Each of the 100 subsampling data sets consists of 80% of the original cohort whereby maintaining the ratio of events and non-events as in the complete cohort for PFS and OS, respectively. On each subsampling data set, Kaplan–Meier analyses were repeated and p value ranges for the log-rank test were obtained. Multivariable Cox regression analysis and variable selection procedure for the complete cohort were performed on each subsampling data set to investigate robustness of the derived model. The analyses have been performed in R version 4.1.1 with packages survival version 3.2–11 and survminer 0.4.9. All analyses were hypothesis-generating. P values were adjusted figurewise by the Bonferroni–Holm (BH) method to account for multiple testing. A (corrected) p value less than 0.05 was considered to be statistically significant.
Results
Cohort parameters
In our cohort of 191 WNT-MBs, 125 patients were children (age 3–15 years), 36 were adolescents (16–20 years), and 30 were adults (≥ 21 years). The median age was 13 years. A female predominance was found (ratio 1.4:1 (Table 1, Online Resource Table 3). The majority of tumor samples had classic histology (n = 181; 94.8%); only nine cases presented with large cell/anaplastic (LC/A) histology (4.7%; Fig. 1, Online Resource Fig. 1 and Online Resource Table 4). Among the 120 patients with clinical data, 32 (26.7%) were clinically high-risk (HR); of these, 16 patients because of metastatic disease and the remaining because of large postoperative residual tumor or LC/A histology (eight patients each). Eighty-four patients received “standard-risk” medulloblastoma therapy with 23.4 Gy CSI with a boost to the posterior fossa or tumor bed of 54 Gy followed by maintenance chemotherapy or hyperfractionated radiotherapy with a CSI-dose of 36 Gy and boosts to posterior fossa and tumor bed according to the HIT-SIOP-PNET4 trial [27]. The remaining patients received “high-risk” medulloblastoma therapies, usually with 35.2 Gy CSI followed by a boost to the posterior fossa and maintenance chemotherapy according to the HIT’91 trial in the maintenance chemotherapy arm [17] or according to the MET-HIT2000-AB4 regimen (Table 1, Online Resource Table 5) [41].

Clinical and molecular features of the WNT medulloblastoma cohort. A total of 191 patient samples with their mutational status (CTNNB1, APC, TP53) and most frequent focal and numerical chromosomal alterations are shown as well as nuclear accumulation of β-catenin and p53 protein. Survival data were available from 120 patients. Missing data are shown in gray. wt, wild-type; mut., mutation; IHC, immunohistochemistry; cnLOH, copy-neutral loss of heterozygosity; M0, no metastatic disease; M + , metastatic disease (M1-3); R0, gross-total resection (< 1.5 cm2); R + , subtotal resection; i17q, isochromosome 17q; Chr., chromosome; WCA, whole chromosomal aberrations; S, i17q and WCA data from SNP6 array instead of Molecular Inversion Probe array
Of the three major hallmarks of WNT-MBs, hotspot mutations in exon 3 of CTNNB1 were most frequent (92.2%; 176 of 191), followed by nuclear accumulation of β-catenin protein (91.5% with ≥ 5% of nuclei; Fig. 2a, b). Monosomy 6 was present in 78.6% of analyzed cases (Fig. 1). The latter hallmark was more frequent in WNT-MBs of children (87.2%) compared with adolescents (70.4%) or adults (50%).

a Distribution of the 176 CTNNB1 mutations in exon 3 at or adjacent to the 4 phosphorylation sites (purple color), including 4 short in-frame deletions. b Immunohistochemical staining of nuclear β-catenin accumulation in a WNT-MB (ID149) harboring two heterozygous APC mutations; normal cells with blue nuclei serve as internal control; scalebar = 20 µM. c, d Copy-number (upper) and allele ratio (lower) plots from molecular inversion probe array of a sibling pair (ID183 (c), ID154 (d)); the copy-neutral loss of heterozygosity within chromosome arm 5q in ID183 (c) is enlarged and the APC locus is highlighted by the gray vertical line; of note, ID154 (d) has additional mosaic losses of chromosomes 10, 12 and Y. e Distribution of APC mutations in 13 CTNNB1 wild-type WNT-MBs; the homozygous R414C is the only missense mutation and likely benign according to the ACMG/AMP classification rules, but this case (ID151) has a second homozygous stop mutation (R283*) and a copy-neutral loss of heterozygosity, so that the R414C mutation will not be translated to protein and has no tumorigenic impact anyway. One CTNNB1-mutant WNT-MB (ID122) has a heterozygous R2226* APC mutation (not shown), which is likely pathogenic. f, g Kaplan–Meier progression-free (PFS) and overall survival (OS) plots of patients with CTNNB1-mutant versus APC-mutant WNT-MB; the green line shows ID191 without CTNNB1 or APC mutation, but with a WNT-activating homozygous loss of FBXW7. The color-filled regions of the Kaplan–Meier plots indicate pointwise 95%-confidence intervals for the Kaplan–Meier estimate. The numbers at risk refer to the complete cohort. p(adj), adjusted p value after correction for multiple testing (Bonferroni–Holm)
Of the 15 CTNNB1-wild-type tumors, 13 carried APC mutations. Of the remaining two tumor samples, one (ID191) had a homozygous deletion of the FBXW7 gene located on chromosome 4q31.3 (Online Resource Fig. 2), whereas from the other sufficient DNA for APC sequencing was not available. However, this case qualified for the diagnosis of WNT-MB because of the presence of both nuclear β-catenin accumulation and monosomy 6. 450k/850k methylation array data was available in 94 cases, confirming the assignment to the WNT-MB group in 87 cases; in 7 cases, no matching score was reached (Fig. 1).
TP53 mutations were found in 16.1% (30/186 cases; Table 1). Twenty six were missense mutations located in exons 5–8; only one was a missense variant in exon 4. Additionally, we found one splice site mutation and one frameshift deletion (both exon 6), and one case had a focal homozygous deletion of the TP53 locus (chromosome 17p13.1; Fig. 4a, Online Resource Fig. 3). An accumulation of p53 protein in ≥ 5% of tumor cells was present in 20.4% of cases (38 of 186; Figs. 1, 4b). TP53 mutations were found in 18 of 38 p53-positive tumor samples. The TP53 locus showed loss of heterozygosity in 18.5% (24/130 cases), in some only present in a fraction of tumor cells (mosaicism). Of the 30 TP53-mutated cases, allele status was available from 27 samples, and 11 of these had a copy loss on chromosome 17p13.1. A significant predominance of male patients was found in cases with TP53 mutations (18 of 30 (60%), p = 0.020, Chi-square test). TP53 mutations occurred in all age groups (6–23 years) without a significant association with one of the age groups. TP53 mutant and TP53 wild-type cases did not cluster separately in UMAP analysis (Online Resource Fig. 4). LC/A histology was significantly enriched in TP53-mutated WNT-MBs (4 of 30 versus 5 of 156, p = 0.020, Chi-square test).
The E17K hotspot regions of AKT1-3 were Sanger sequenced in 165 cases, and the E17K mutation was found 6 times each in AKT1 and AKT3 (12 of 165; 7.27%).
Next-generation panel sequencing (NGS panel II) was performed in 31 cases. Besides identification of APC variants and validation of CTNNB1 and TP53 mutations, recurrent mutations (n > 2) were found in DDX3X (n = 12, 38.7%), SMARCA4 (n = 5), KMT2D (n = 5), KMT2C (n = 4), PIK3CA (n = 3), and FBXW7 (n = 3; all mutations are shown in Online Resource Table 6).
Chromosomal aberrations
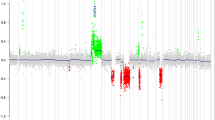
Except for monosomy 6, most WNT-MBs presented stable genomes. Genome-wide copy-number profiles were available from 128 cases with a mean of 2.0 whole chromosomal aberrations (WCAs) per case. Most frequent WCAs besides chromosome 6 were gains of chromosomes 8 and 19 in 16 samples each (12.5%), and losses of chromosomes 10 and 13 in 10 samples each (7.8%; Fig. 4e, Online Resource Fig. 5). TP53-mutated tumors were enriched for monosomy 6 (28/28 cases versus 74/101 TP53 wild-type tumors, p = 0.002, Chi-square test) and chromosome arm 17p loss (11/27 versus 13/88 TP53 wild-type tumors, p = 0.001, Chi-square test; Online Resource Fig. 6). An isochromosome 17q (i17q) was found in 8 of 130 samples (6.2%). Genomic identification of significant targets in cancer (GISTIC) analysis revealed several significantly altered focal aberrations, most notably CDK6 gain (chromosome 7q21.2), OTX2 gain (14q22.3), and AXIN2 gain (17q24.1; Online Resource Table 7 and Online Resource Fig. 7). Of note, OTX2 gain (n = 42; Table 1, Fig. 4f and Online Resource Fig. 8) was the most frequent focal aberration occurring both in TP53 mutant (8/27, 29.6%) and TP53 wild-type (33/79, 41.8%, p = 0.263, chi-square test) tumors. Cases with OTX2 gain versus balanced OTX2 locus did not cluster separately in UMAP analysis (Online Resource Fig. 4).
APC mutations
Of 13 APC-mutant cases, 8 had larger regions with copy-neutral loss of heterozygosity (cnLOH) spanning the APC locus up the telomeric region of chromosome 5q; one case had a focal loss at the APC locus (5q22.2). Eight of these nine cases with deletion carried a single mutation, and one carried a double mutation on the same allele (R283* and R414C) with allele variant fractions between 75 and 100%. The remaining four cases presented with two mutations with allele variant fractions of both mutations around 40–50% and 3 lacked LOH of the APC locus (1 × not done; Figs. 1, 2e). TP53 mutations and OTX2 gains both occurred at similar frequencies in APC-mutant WNT-MBs compared to CTNNB1-mutant tumors (TP53 mutation: 3/13 versus 27/172, p = 0.486; OTX2 gain: 4/12 versus 37/94, p = 0.686, Chi-square tests).
Occurrence of APC-mutant WNT-MB in siblings
Our WNT-MB cohort contained one sibling pair with a female patient, who developed a classic MB at the age of 11 with metastatic spread at diagnosis (ID183). Her brother was 19 when the tumor was detected, and histology was also classic, but without metastatic disease (ID154). Both were CTNNB1 wild-type, but APC sequencing revealed the same R213* mutation in both tumors, suggesting that it represented a germline mutation. The sister had an allele variant fraction of almost 100% and a large cnLOH on chromosome arm 5q, whereas the brother had a balanced APC locus, but an additional two base-pair frameshift deletion at amino acid L1129. Both APC mutations in this tumor had allele variant fractions of 40–50%.
Additionally, both siblings had no OTX2 gains, but were affected by TP53 alterations. The sister presented a monoallelic TP53 mutation (R282P) without loss of the TP53 locus, whereas the brother had a chromosome arm 17p loss, but wild-type TP53 (Fig. 2c, d). Both siblings are alive without relapse or secondary tumor.
Survival analyses
Staging and follow-up data from 120 patients were available with median follow-up of 7.4 years (range 0.1–19.6 years) in 109 patients alive at last follow-up. 5-year PFS was 88% [95% CI 82–95%] and 5-year OS was 93% [88–98%]. Univariable survival analysis did not show statistically significant associations of survival with gender and clinical HR stage (M + , residual disease; Fig. 3a–d), LC/A histology, nuclear β-catenin accumulation, i17q, AKT1-3 mutations, CDK6 gains, or monosomy 6 as well as clinical risk and therapy regimen (Online Resource Figs. 9–11). As in other studies, patients older than 16 had worse survival, reaching significance in OS (5-years OS 3–15 years 96% [92–100%] versus 16–20 years 80% [62–100%] versus ≥ 21 years 86% [63–100%], p = 0.030), but not in PFS (91% [85–98%] versus 79% [63–100%] versus 86% [63–100%], p = 0.286). After correction for multiple testing, the difference in age groups for OS did not reach significance anymore (p(adj) = 0.178; Fig. 3e, f). APC-mutated cases showed similar survival as CTNNB1-mutated cases (Fig. 2f, g). In contrast, TP53 mutations were associated with lower PFS (68% [49–93%] versus 93% [87–99%], p = 0.001, p(adj) = 0.003) and OS (88% [83–100%] versus 94% [89–99%], p = 0.105, p(adj) = 0.105; Fig. 4c, d), and TP53 copy loss also was associated with lower PFS (p = 0.004, p(adj) = 0.014), but not OS (Online Resource Fig. 12), as was loss of whole chromosome 10: PFS p < 0.001, p(adj) = 0.003; loss of chromosome 13: PFS p = 0.003, p(adj) = 0.017 (Online Resource Figs. 13–15). Importantly, gains of the OTX2 locus were associated with significant worse PFS (5-year PFS 72% [57–91%] versus 93% [85–100%], p = 0.017, p(adj) = 0.034) and OS (5-year OS 82% [69–98%] versus 97% [91–100%], p = 0.006, p(adj) = 0.019; Fig. 4g, h), which was independent and additive of the effect of TP53 mutations on univariable levels. No relapses or other events were observed among patients with tumors harboring neither TP53 mutations nor OTX2 gains (5-year PFS/OS 100%; Online Resource Table 8), whereas 5-year PFS/OS was 78% 65–93%/85% 73–98% in patients whose tumors either had a TP53 mutation or an OTX2 gain (n = 36, 48.7%) and was 50% [22–100]/83% [58–100%] among patients with both TP53 mutation and OTX2 gain (n = 7, p < 0.001, p(adj) = 0.001/p = 0.001, p(adj) = 0.010; Fig. 5a–c, Online Resource Fig. 16). The difference in patients whose tumors either had a TP53 mutation or an OTX2 gain for PFS seemed to be relatively stable under subsampling (p value ranging from 0 to 0.036 in 100 subsampling data sets; Online Resource Table 9).

Kaplan–Meier progression-free (PFS) and overall survival (OS) plots for gender (a, b), staging with clinical high-risk factors “metastatic disease” (M +) and “subtotal resected tumor” (R + ; ≥ 1.5 cm2) versus cases with non-metastatic disease and gross-total resection (M0R0; c, d), and age (e, f). e, f Log-rank p values for individual PFS analyses are: blue versus red = 0.142; red versus green = 0.894; blue versus green = 0.284. Log-rank p values for individual OS analyses are: blue versus red = 0.020; red versus green = 0.856; blue versus green = 0.036

a Distribution of TP53 mutations; colored mutations represent splice site mutation (blue), frameshift deletion (green), and nonsense mutation (red). b Immunohistochemical staining for p53 protein in a WNT-MB (ID184) with a heterozygous R248Q TP53 mutation and balanced TP53 locus; a strong nuclear staining pattern is visible focally, whereas some regions lack p53 staining (left side of the image); scalebar = 50 µM. c, d Kaplan–Meier progression-free (PFS) and overall survival (OS) plots of patients with TP53 mutant versus TP53 wild-type WNT-MB. e Summary plot of copy-number aberrations (n = 115 WNT-MBs) from molecular inversion probe array (n = 108) and SNP6 array (n = 7). The arrow indicates the OTX2 locus on chromosome arm 14q. Blue bars, gains; red bars, losses. Thickness of bars indicates frequency of alterations. f Copy-number plots from molecular inversion probe arrays of a WNT-MB case with focal OTX2 gain (upper part) and balanced OTX2 (lower part) on chromosome arm 14q. g, h Kaplan–Meier PFS and OS plots of patients with OTX2 gain versus no OTX2 gain

a, b Kaplan–Meier progression-free (PFS) and overall survival (OS) plots of patients with WNT-MBs without TP53 mutation and OTX2 gain (blue line), one of these two alterations (red line), or both alterations (green line). Log-rank p values for individual PFS analyses are: blue versus red = 0.010; red versus green = 0.050; blue versus green < 0.001. Log-rank p values for individual OS analyses are: blue versus red = 0.030; red versus green = 0.100; blue versus green < 0.001. c Pie chart showing frequency of identified risk factors in 74 WNT-MB cases with PFS/OS and genomic data from Molecular Inversion Probe array. d Venn diagram showing the distribution of TP53 mutant, OTX2 gained, and chromosome 6 lost (n = 64 with at least one of these alterations; n = 10 were wt/balanced for all 3 factors). e, f Results of the multivariable Cox regression analyses for PFS and OS; *, p < 0.05; **, p < 0.01. g Results of the subsampling approach for the multivariable Cox regression analysis with variable selection. Values in brackets refer to the IQR or the range for the subsampling approach, respectively. Values outside brackets refer to the model on the complete data (n = 74). PFS: Concordance Index [range]: 0.8 [0.76; 0.86], Median Rank: 8, Frequency Rank 1: 14%, Average AIC: 65.88; OS: Concordance Index [range]: 0.82 [0.75; 0.91], Median Rank: 3, Frequency Rank 1: 35%, Average AIC: 49.92
This effect was retained in a multivariable Cox regression analysis for PFS and OS, where OTX2 gain (HR 5.98 [1.63–22.0], p = 0.007/12.9 [1.6–103], p = 0.016), TP53 mutations (HR 10.8 [2.2–53.2], p = 0.003/6.2 [1.1–34.5], p = 0.037), and monosomy 6 (HR 0.15 [0.03–0.85], p = 0.032/0.15 [0.027–0.82], p = 0.028) were identified as significant and independent prognostic markers (Fig. 5d–f). The model selection procedure applied to the 100 subsampling data sets revealed the model containing the three mentioned covariates to have the minimal average AIC as well as the minimal median rank when ranking models according to the AIC (PFS: average AIC = 65.88 OS: average AIC = 49.92; Fig. 5g and Online Resource Fig. 17).
Discussion
Several ongoing clinical trials aim to improve quality of life for patients with WNT-MB by de-escalation of therapy, especially reduced radiation dosage. However, despite the excellent overall prognosis (of WNT-MB patients aged 15 years or younger), patients who relapse might not survive their disease. To prevent these patients from relapse, their upfront identification by suitable prognostic tumor markers is required. Currently, older age is considered the main risk factor for non-metastatic WNT-MB, but data of adolescent and adult WNT-MBs are scarce and results are not fully consistent [5, 7, 14, 15, 24, 25, 32, 36, 39, 44, 45].
Limitation of clinical parameters as risk predictors
Data on survival of adult patients (≥ 21 years) in our cohort are sparse, but our data confirmed the worse outcome for adolescents (16–20 years). Conventional clinical parameters used for stratification into standard or high risk were not suitable as predictive markers in our WNT-MB cohort (Fig. 3c, d). Although data for WNT-MB with clinical high-risk features are rare, this observation is in line with published data [32, 41].
APC-mutant MB shows similar outcomes as CTNNB1-mutated WNT-MBs
WNT-MBs with pathway-activating loss-of-function mutations in the APC gene had similar PFS/OS compared to CTNNB1-mutated cases, thus validating previous publications [40, 43]. This indicates that patients with APC-mutant WNT-MB might be included into therapy de-escalating clinical trials equally to CTNNB1-mutant WNT-MB in the absence of other risk factors.
In contrast to activating CTNNB1 mutations, which almost always occur as somatic events in the tumor, pathogenic APC mutations frequently are germline events. These cause the cancer prediposition syndrome FAP, which is associated with a very high lifetime risk for colorectal cancer. As a consequence, patients with an APC-related MB should be offered genetic counseling and screened for APC germline mutations with the aim to offer surveillance for colorectal cancer [1].
TP53 mutation correlates to increased risk of recurrent disease
We identified TP53 mutations as a robust marker for increased risk of relapse in WNT-MBs. This finding corresponds to the recent description of an over-representation of TP53 mutant tumors in relapsed WNT-MB leading to the hypothesis of a prognostic impact of TP53 mutations [37]. Zhukova et al. did not find an impact of TP53 mutations on OS, but might have missed their prognostic relevance for PFS, as this was not analyzed [46]. In line with this publication, we also could not find a significant correlation of TP53 mutations to worse OS. However, multivariable analysis also showed a prognostic impact of TP53 mutations on OS in our cohort, although this finding was less significant than for PFS and has to be validated in future trial cohorts.
TP53 loss also indicated significantly worse PFS, which was expected as loss at the TP53 locus was highly associated with TP53 mutations.
Focal gain of the master regulator OTX2 indicates poor prognosis
The master regulator gene OTX2 encodes a transcription factor essential for the normal development of CNS tissues like brain, cerebellum, pineal gland, and eye [3]. Both in the developing cerebellum and cerebellar medulloblastoma, OTX2 is highly expressed, and has been shown to possess oncogenic activity in non-WNT/non-SHH medulloblastomas [2]. Its obvious biological role in WNT-activated medulloblastoma has not been studied in detail so far. In this study, gains of OTX2 were identified as negative and independent prognosticator. Indeed, all patients with tumors lacking a TP53 mutation who died or had tumor relapse harbored an OTX2 gain. Whereas IHC positivity of OTX2 protein is visible in almost all WNT- and non-WNT/non-SHH-MB, these mostly focal gains can only be found in a subset of WNT-activated tumors and similarly in non-WNT/non-SHH-MBs, but not in SHH-MBs. OTX2 has been shown to be highly repressed in SHH-MB [28] and copy-number gains of OTX2 have not been described in large series of SHH-MB [14, 15, 39]. In standard-risk non-WNT/non-SHH-MBs, OTX2 gain was not associated with poor outcome [14]. As these gains are mostly focal, high-resolution technologies, such as MIP or gene-specific probes, should be used to detect this alteration. Of note, the copy-number information deduced from widely used DNA methylation might not be suitable to detect these OTX2 gains.
Multivariable analysis and proposal of a novel risk prediction model
In multivariable analysis, three genomic alterations were identified as independent indicators of outcome both PFS and OS, namely TP53 mutation, OTX2 gain, and monosomy 6. All relapsed patients could be identified by the first two alterations, so we suggest a risk prediction model based on these two markers. In this risk model including TP53 mutation and/or OTX2 gain, 58.1% of patients were identified having an increased risk for relapse of their disease after therapy according to the standard before the era of dose-reducing trials in WNT-MB. In this model, the remaining 41.9% of patients had WNT-MB without these genetic alterations and represented low-risk patients; no relapse occurred in these 31 patients (Fig. 5a–c). As our cohort of WNT-MB patients included patients with high-risk clinicopathological features, we also performed explorative subgroup analyses on “pure standard-risk” cohorts with patients who would had qualified for inclusion into the SIOP-PNET-5 MB (inclusion up to 16 years of age at diagnosis; n = 50) or into the SJMB12 and ACNS1422 studies (up to 22 years of age; n = 64). Of note, OTX2 gain remained prognostic in the latter standard-risk cohort, although patient numbers are substantially reduced (Online Resource Fig. 18). As the nature of the study is retrospective and the patient cohort described here was not homogeneously treated in the framework of a single controlled trial, our proposed risk model needs validation in current or future therapeutic trials of WNT-MB patients to investigate the predictive power of the model. Upon confirmation, these genetic markers might allow to more precisely identify low-risk patients eligible for dose-sparing therapeutic strategies, while for the remaining patients, dose de-escalation should be avoided, but approaches aiming to increase survival might become of relevance.
References
Achatz MI, Porter CC, Brugieres L, Druker H, Frebourg T, Foulkes WD et al (2017) Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res 23:e107–e114. https://doi.org/10.1158/1078-0432.CCR-17-0790
Adamson DC, Shi Q, Wortham M, Northcott PA, Di C, Duncan CG et al (2010) OTX2 is critical for the maintenance and progression of Shh-independent medulloblastomas. Cancer Res 70:181–191. https://doi.org/10.1158/0008-5472.CAN-09-2331
Beby F, Lamonerie T (2013) The homeobox gene Otx2 in development and disease. Exp Eye Res 111:9–16. https://doi.org/10.1016/j.exer.2013.03.007
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D et al (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B et al (2017) Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31(737–754):e736. https://doi.org/10.1016/j.ccell.2017.05.005
Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D et al (2006) Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 5:2666–2670. https://doi.org/10.4161/cc.5.22.3446
Coltin H, Sundaresan L, Smith KS, Skowron P, Massimi L, Eberhart CG et al (2021) Subgroup and subtype-specific outcomes in adult medulloblastoma. Acta Neuropathol 142:859–871. https://doi.org/10.1007/s00401-021-02358-4
Crosier S, Hicks D, Schwalbe EC, Williamson D, Leigh Nicholson S, Smith A et al (2021) Advanced molecular pathology for rare tumours: a national feasibility study and model for centralised medulloblastoma diagnostics. Neuropathol Appl Neurobiol 47:736–747. https://doi.org/10.1111/nan.12716
Dahmen RP, Koch A, Denkhaus D, Tonn JC, Sorensen N, Berthold F et al (2001) Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res 61:7039–7043
Eberhart CG, Tihan T, Burger PC (2000) Nuclear localization and mutation of beta-catenin in medulloblastomas. J Neuropathol Exp Neurol 59:333–337. https://doi.org/10.1093/jnen/59.4.333
Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE et al (2005) β-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol 23:7951–7957. https://doi.org/10.1200/JCO.2005.01.5479
Fattet S, Haberler C, Legoix P, Varlet P, Lellouch-Tubiana A, Lair S et al (2009) Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol 218:86–94. https://doi.org/10.1002/path.2514
Giles RH, van Es JH, Clevers H (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta 1653:1–24. https://doi.org/10.1016/s0304-419x(03)00005-2
Goschzik T, Schwalbe EC, Hicks D, Smith A, Zur Muehlen A, Figarella-Branger D et al (2018) Prognostic effect of whole chromosomal aberration signatures in standard-risk, non-WNT/non-SHH medulloblastoma: a retrospective, molecular analysis of the HIT-SIOP PNET 4 trial. Lancet Oncol 19:1602–1616. https://doi.org/10.1016/S1470-2045(18)30532-1
Goschzik T, Zur Muehlen A, Doerner E, Waha A, Friedrich C, Hau P et al (2021) Medulloblastoma in adults: cytogenetic phenotypes identify prognostic subgroups. J Neuropathol Exp Neurol 80:419–430. https://doi.org/10.1093/jnen/nlab020
Goschzik T, Zur Muhlen A, Kristiansen G, Haberler C, Stefanits H, Friedrich C et al (2015) Molecular stratification of medulloblastoma: comparison of histological and genetic methods to detect Wnt activated tumours. Neuropathol Appl Neurobiol 41:135–144. https://doi.org/10.1111/nan.12161
Hoff KV, Hinkes B, Gerber NU, Deinlein F, Mittler U, Urban C et al (2009) Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT’91. Eur J Cancer 45:1209–1217. https://doi.org/10.1016/j.ejca.2009.01.015
Hovestadt V, Remke M, Kool M, Pietsch T, Northcott PA, Fischer R et al (2013) Robust molecular subgrouping and copy-number profiling of medulloblastoma from small amounts of archival tumour material using high-density DNA methylation arrays. Acta Neuropathol 125:913–916. https://doi.org/10.1007/s00401-013-1126-5
Huang H, Mahler-Araujo BM, Sankila A, Chimelli L, Yonekawa Y, Kleihues P et al (2000) APC mutations in sporadic medulloblastomas. Am J Pathol 156:433–437. https://doi.org/10.1016/S0002-9440(10)64747-5
Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M et al (2012) Dissecting the genomic complexity underlying medulloblastoma. Nature 488:100–105. https://doi.org/10.1038/nature11284
Juraschka K, Taylor MD (2019) Medulloblastoma in the age of molecular subgroups: a review. J Neurosurg Pediatr 24:353–363. https://doi.org/10.3171/2019.5.PEDS18381
Koch A, Hrychyk A, Hartmann W, Waha A, Mikeska T, Waha A et al (2007) Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int J Cancer 121:284–291. https://doi.org/10.1002/ijc.22675
Koch A, Waha A, Tonn JC, Sorensen N, Berthold F, Wolter M et al (2001) Somatic mutations of WNT/wingless signaling pathway components in primitive neuroectodermal tumors. Int J Cancer 93:445–449. https://doi.org/10.1002/ijc.1342
Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA et al (2012) Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123:473–484. https://doi.org/10.1007/s00401-012-0958-8
Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H et al (2010) Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol 28:3054–3060. https://doi.org/10.1200/JCO.2009.25.7121
Korshunov A, Sahm F, Zheludkova O, Golanov A, Stichel D, Schrimpf D et al (2019) DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol 21:214–221. https://doi.org/10.1093/neuonc/noy155
Lannering B, Rutkowski S, Doz F, Pizer B, Gustafsson G, Navajas A et al (2012) Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol 30:3187–3193. https://doi.org/10.1200/JCO.2011.39.8719
Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M et al (2016) Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530:57–62. https://doi.org/10.1038/nature16546
Lindsey JC, Hill RM, Megahed H, Lusher ME, Schwalbe EC, Cole M et al (2011) TP53 mutations in favorable-risk Wnt/Wingless-subtype medulloblastomas. J Clin Oncol 29:e344–e346. https://doi.org/10.1200/JCO.2010.33.8590 (author reply e347–348)
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (2016) WHO Classification of Tumours of the Central Nervous System. International Agency for Research on Cancer, Lyon
Michalski JM, Janss AJ, Vezina LG, Smith KS, Billups CA, Burger PC et al (2021) Children’s Oncology Group Phase III trial of reduced-dose and reduced-volume radiotherapy with chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 39:2685–2697. https://doi.org/10.1200/JCO.20.02730
Nobre L, Zapotocky M, Khan S, Fukuoka K, Fonseca A, McKeown T et al (2020) Pattern of relapse and treatment response in WNT-activated medulloblastoma. Cell Rep Med 1:100038. https://doi.org/10.1016/j.xcrm.2020.100038
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al (2017) The whole-genome landscape of medulloblastoma subtypes. Nature 547:311–317. https://doi.org/10.1038/nature22973
Pietsch T, Haberler C (2016) Update on the integrated histopathological and genetic classification of medulloblastoma—a practical diagnostic guideline. Clin Neuropathol 35:344–352. https://doi.org/10.5414/NP300999
Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J et al (2012) Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488:106–110. https://doi.org/10.1038/nature11329
Remke M, Hielscher T, Northcott PA, Witt H, Ryzhova M, Wittmann A et al (2011) Adult medulloblastoma comprises three major molecular variants. J Clin Oncol 29:2717–2723. https://doi.org/10.1200/JCO.2011.34.9373
Richardson S, Hill RM, Kui C, Lindsey JC, Grabovksa Y, Keeling C et al (2022) Emergence and maintenance of actionable genetic drivers at medulloblastoma relapse. Neuro Oncol 24:153–165. https://doi.org/10.1093/neuonc/noab178
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L et al (2012) Novel mutations target distinct subgroups of medulloblastoma. Nature 488:43–48. https://doi.org/10.1038/nature11213
Shih DJ, Northcott PA, Remke M, Korshunov A, Ramaswamy V, Kool M et al (2014) Cytogenetic prognostication within medulloblastoma subgroups. J Clin Oncol 32:886–896. https://doi.org/10.1200/JCO.2013.50.9539
Surun A, Varlet P, Brugieres L, Lacour B, Faure-Conter C, Leblond P et al (2020) Medulloblastomas associated with an APC germline pathogenic variant share the good prognosis of CTNNB1-mutated medulloblastomas. Neuro Oncol 22:128–138. https://doi.org/10.1093/neuonc/noz154
von Bueren AO, Kortmann RD, von Hoff K, Friedrich C, Mynarek M, Muller K et al (2016) Treatment of children and adolescents with metastatic medulloblastoma and prognostic relevance of clinical and biologic parameters. J Clin Oncol 34:4151–4160. https://doi.org/10.1200/JCO.2016.67.2428
Wang Y, Cottman M, Schiffman JD (2012) Molecular inversion probes: a novel microarray technology and its application in cancer research. Cancer Genet 205:341–355. https://doi.org/10.1016/j.cancergen.2012.06.005
Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S et al (2018) Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 19:785–798. https://doi.org/10.1016/S1470-2045(18)30242-0
Wong GC, Li KK, Wang WW, Liu AP, Huang QJ, Chan AK et al (2020) Clinical and mutational profiles of adult medulloblastoma groups. Acta Neuropathol Commun 8:191. https://doi.org/10.1186/s40478-020-01066-6
Zhao F, Ohgaki H, Xu L, Giangaspero F, Li C, Li P et al (2016) Molecular subgroups of adult medulloblastoma: a long-term single-institution study. Neuro Oncol 18:982–990. https://doi.org/10.1093/neuonc/now050
Zhukova N, Ramaswamy V, Remke M, Pfaff E, Shih DJ, Martin DC et al (2013) Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J Clin Oncol 31:2927–2935. https://doi.org/10.1200/JCO.2012.48.5052
Zurawel RH, Chiappa SA, Allen C, Raffel C (1998) Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res 58:896–899
Acknowledgements
This study was supported by grants of the German Children Cancer Foundation, Grants DKS 2006.03, 2009.19, 2011.01, and 2014.17 to Torsten Pietsch and 2001.03, 2005.13, 2009.18, 2010.08, 2015.06 to Stefan Rutkowski, and 2017.10 and 2019.07 to Stefan Rutkowski and Martin Mynarek.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Goschzik, T., Mynarek, M., Doerner, E. et al. Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol 144, 1143–1156 (2022). https://doi.org/10.1007/s00401-022-02505-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-022-02505-5