
Abstract
Electrocardiographic findings and arrhythmias are common in cardiomyopathies. Both may be an early indication of a specific diagnosis or may occur due to myocardial fibrosis and/or reduced contractility. Brady- and tachyarrhythmias significantly contribute to increased morbidity and mortality in patients with cardiomyopathies. Antiarrhythmic therapy including risk stratification is often challenging and plays a major role for these patients. Thus, an “electrophysiological” perspective on guidelines on cardiomyopathies may be warranted. As the European Society of Cardiology (ESC) has recently published a new guideline for the management of cardiomyopathies, this overview aims to present key messages of these guidelines. Innovations include a new phenotype-based classification system with emphasis on a multimodal imaging approach for diagnosis and risk stratification. The guideline includes detailed chapters on dilated and hypertrophic cardiomyopathy and their phenocopies, arrhythmogenic right ventricular cardiomyopathy, and restrictive cardiomyopathy as well as syndromic and metabolic cardiomyopathies. Patient pathways guide clinicians from the initial presentation to diagnosis. The role of cardiovascular magnetic resonance imaging and genetic testing during diagnostic work-up is stressed. Concepts of rhythm and rate control for atrial fibrillation have led to new recommendations, and the role of defibrillator therapy in primary prevention is discussed in detail. Whilst providing general guidelines for management, the primary objective of the guideline is to ascertain the disease etiology and disease-specific, individualized management.
Zusammenfassung
Arrhythmien und EKG(Elektrokardiographie)-Auffälligkeiten sind bei Kardiomyopathien häufig. Sie können ein erster Hinweis auf eine spezifische Diagnose sein oder im Verlauf einer Erkrankung als Folge von Fibrosierung und/oder reduzierter Herzfunktion auftreten. Brady- und Tachyarrhythmien tragen dabei signifikant zu einer erhöhten Morbidität und Mortalität bei Patienten mit Kardiomyopathien bei. Eine antiarrhythmische Therapie einschließlich einer Risikostratifizierung ist oft eine Herausforderung und bei der Behandlung dieser Patienten von besonderer Bedeutung. Daher ist eine elektrophysiologische Sicht auf Empfehlungen zu Kardiomyopathien sinnvoll. Da die ESC (European Society of Cardiology) kürzlich eine neue Leitlinie zum Management von Kardiomyopathien veröffentlichte, werden in der vorliegenden Arbeit deren wichtigsten Empfehlungen mit besonderem Fokus auf der kardialen Elektrophysiologie vorgestellt. Innovationen umfassen eine neue phänotypbasierte Klassifikation mit Schwerpunkt auf multimodaler Bildgebung zur Diagnostik und Risikostratifizierung. Die aktuelle Leitlinie enthält ausführliche Kapitel zur dilatativen und hypertrophen Kardiomyopathie und deren Phänokopien, der arrhythmogenen rechtsventrikulären Kardiomyopathie, der restriktiven Kardiomyopathie sowie syndromalen und metabolischen Kardiomyopathien. Pfade leiten von der Erstvorstellung bis zur Diagnose und Therapie. Die Rolle der kardiovaskulären Magnetresonanztomographie und der genetischen Diagnostik erfährt einen besonderen Stellenwert. Konzepte zur Rhythmus- und Frequenzkontrolle bei Vorhofflimmern führten zu neuen Empfehlungen, und die Rolle der Defibrillatortherapie in der Primärprävention wird ausführlich diskutiert. Das Hauptziel der neuen Leitlinie ist, die Krankheitsursachen bestmöglich zu ermitteln, um eine spezifische, individualisierte Behandlung zu ermöglichen.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Electrocardiographic findings and arrhythmias are closely related to cardiomyopathies (CM). Both may be an early indication of a specific diagnosis or are (just) the consequence of structural heart disease with myocardial fibrosis and impaired contractility. Arrhythmias significantly contribute to increased morbidity and mortality, and CM are the leading cause of exercise induced syncope and sudden cardiac death (SCD) in young people (Fig. 1). Antiarrhythmic therapy including risk stratification in CM is often challenging and plays a major role in cardiac electrophysiology. Thus, an “electrophysiological” perspective on CM guidelines may be warranted. As the European Society of Cardiology (ESC) has recently published a new guideline for the management of CM [1] this overview aims to present key aspects of this guideline.

Characteristics of cardiomyopathies focusing on history of arrhythmias, electrocardiographic and electrophysiological findings with selected therapeutic options. Fx family history, ICD implantable cardioverter defibrillator, RBBB right bundle branch block, CRT cardiac resynchronization therapy, OAC oral anticoagulation, VEs ventricular extrasystoles, AAD antiarrhythmic drugs, VT/VF ventricular tachycardia/fibrillation
Brief overview of the new guidelines on cardiomyopathies
Cardiomyopathies (CM) are common and may lead to heart failure and life-threatening ventricular arrhythmias (VA). Prevalence and incidence of arrhythmias are difficult to assess, as disease expression varies throughout life. Geographical distribution of genetic variants also influences the prevalence in different populations, ethnicities, regions, and countries. Hypertrophic cardiomyopathy (HCM) for example has got an estimated overall prevalence of 0.2%, while for dilated cardiomyopathy (DCM) estimates are wider ranging from 0.036% to 0.4% [1]. In the past, CM were mainly classified by anatomical and clinical characteristics, using echo- and electrocardiography, invasive angiography, and histology. Nowadays, knowledge about CM has expanded exponentially by utilizing new imaging approaches and molecular technologies. For the first time, the new ESC guideline addresses CM other than HCM. It includes detailed chapters on HCM, DCM, arrhythmogenic right ventricular cardiomyopathy (ARVC), restrictive cardiomyopathy (RCM) including cardiac amyloidosis (CA) as well as syndromic and metabolic CM including Anderson-Fabry disease but also discusses important differential diagnoses such as cardiac sarcoidosis. The new term “non-dilated left ventricular cardiomyopathy” (NDLVC) replaces “hypokinetic non-dilated cardiomyopathy.” It is defined “as the presence of non-ischemic LV scarring or fatty replacement regardless of the presence of global or regional wall motion abnormalities, or isolated global LV hypokinesia without scarring.” Thus, the diagnosis is predominantly based on cardiac magnetic resonance imaging (CMR).
General aspects of arrhythmias in cardiomyopathies
The spectrum of arrhythmias ranges from bradyarrhythmia, atrial and/or ventricular extrasystoles (VEs) to atrial fibrillation/flutter (Fig. 2) and/or life-threatening VA [2]. Atrial fibrillation (AF) represents the most common arrhythmia and is associated with an increased risk of cardioembolic events, heart failure, and death in patients with CM. In parallel to AF patients without CM, there is increasing evidence for AF ablation in an era of a steady increase in catheter ablations [3, 4]. The prevention of SCD plays an important role in the new recommendations. The guideline includes five class I indications for ICD therapy in CM (all secondary prevention) and is in line with the 2022 ESC Guidelines for the management of patients with VA and the prevention of SCD [2, 5, 6]. It expands the previously established concept of individualized decision-making and supports etiology-specific individual management and risk stratification. Recommendations for genetic testing have increased (seven class I recommendations!) and the guideline acknowledges the growing evidence on certain high-risk genotypes regardless of LV morphology and function. This includes, for example, Class I recommendations for genetic testing in all patients fulfilling diagnostic criteria for CM as well as genetic cascade testing of relatives in case of a pathological or likely pathological variant in the index patient (Class I). Next to genetic testing, the role of CMR has increased in recent years [7] but still lacks randomized trials to guide ICD therapy. Thus, the ESC guideline stresses the importance of a tailored, etiology-based individual management approach incorporating the patient’s history, electro- and echocardiography, CMR, laboratory analysis, genetics, as well as electrophysiological findings/studies.

Typical atrial flutter in a 69-year-old male with apical hypertrophic cardiomyopathy (septal thickness of 16 mm). Please note the electrocardiographic signs of left ventricular hypertrophy with high R waves going along with prominent negative T‑waves in V4–V6
A standardized pathway after initial presentation and the role of electrocardiography in cardiomyopathies
The proposed systematic diagnostic approach includes characterization of a phenotype with multimodality imaging. In addition to routine echo- and electrocardiography [8], CMR characterizes myocardial tissue and identifies edema and/or scarring [9,10,11,12,13,14]. The distribution and severity of interstitial expansion, as visualized by late gadolinium enhancement (LGE), may suggest a specific diagnosis [7]. As LGE patterns have been shown to be prognostic for VA in various CM, the guideline also acknowledges the role of CMR in risk stratification [15, 16]. A key message is the Class I recommendation for all individuals with a CM to undergo contrast-enhanced CMR during the initial assessment and during follow-up to detect disease progression. Of note, the interpretation of imaging results should always consider the clinical context, including personal and family history, clinical presentation, for example, arrhythmias and other more specific tests such as genetic testing if an inheritable CM is suspected.
A resting 12-lead electrocardiogram (ECG) is often an early diagnostic step. ECG abnormalities in patients with CM are common and can occur years before overt morphological or functional abnormalities (Fig. 1). Most ECG abnormalities are nonspecific. For example, epsilon waves may occur in ARVC but also in other CM such as cardiac sarcoidosis (Fig. 3; [17, 18]). Recently, an easily applicable algorithm including PR prolongation and the surface area of the maximum R’ wave in leads V1 through V3 has been demonstrated to distinguish cardiac sarcoidosis from ARVC. This QRS terminal activation in precordial leads V1 through V3 may reflect disease-specific scar patterns [19]. Unspecific ECG abnormalities should always be interpreted in conjunction with imaging findings, patient history, and clinical features and may provide important clues to the underlying diagnosis [17]. Some features, such as atrioventricular (AV) block, pseudo pre-excitation, low QRS voltages, or the distribution of repolarization changes may also be helpful (Fig. 1). Accordingly, the guideline recommends acquiring an ECG at the first visit from all patients with suspected or known CM and during regular follow-up. As arrhythmias are common in CM, ambulatory ECG monitoring at initial presentation and in regular intervals (e.g. every 1–2 years in stable patients [Class I]) is also advised for stroke and SCD risk stratification. On an individual basis a biopsy may be helpful [20, 21], but is only specifically mentioned for RCM (Class IIa).

A 49-year-old male with biopsy-proven cardiac sarcoidosis. a Atypical right bundle branch block (RBBB) with prominent second component of the QRS complex in the presence of atrioventricular block I. b Sustained hemodynamically tolerated ventricular tachycardia (CL 430 ms). c–f Electro-anatomical CARTO activation map and bipolar right ventricular voltage map with extensive fibrosis that correlated with the cardiac magnetic resonance imaging illustrated by late gadolinium enhancement
Atrial fibrillation in cardiomyopathies
AF is by far the most common sustained arrhythmia in patients with CM and associated with a worse prognosis, i.e., increased mortality and incidence of stroke [22]. Early-onset AF may be an early marker for an underlying syndromic or metabolic cardiomyopathy. A CM may on the other hand be an important predisposing factor for AF in the elderly [23]. The EURObservational Research Programme (EORP) CM registry [24, 25] demonstrated an AF prevalence of 28% at baseline (with an overall annual incidence of 3%) with more severe symptoms and worse prognosis for CM patients with AF. The incidence of stroke was increased threefold as compared to the general population. Short atrial runs may better predict AF in CM patients than in other patients but this needs to be evaluated, for example, with loop recorders [26]. Although the “Atrial Fibrillation Better Care (ABC)” approach [25, 27] has not been comprehensively assessed in patients with CM, two randomized trials (the RACE-III trial [28] and the mobile Atrial Fibrillation App Trial (mAFA-II trial) [29, 30]) provided some evidence in favor of an integrated approach in CM. Thus, the guideline recommends modification of risk factors such as obesity, reduced physical activity, as well as alcohol consumption, hypertension, diabetes, and chronic obstructive pulmonary disease in CM patients with recurrent AF (Class I). CM seem to have different thromboembolic risks. Several observational studies reported an increased risk in HCM, RCM, and cardiac amyloidosis. In an unselected transesophageal echocardiography population, 20% of AF patients with a left atrial appendage (LAA) thrombus had a CHA2DS2-VASc score < 2 [31] and there is evidence suggesting that the CHA2DS2-VASc score, which has not been validated for CM patients, may insufficiently assess thromboembolic risks in patients with HCM, RCM, or cardiac amyloidosis. Thus, the current guideline recommends oral anticoagulation (OAC) for all patients with HCM and cardiac amyloidosis and AF (Class I) and gives a Class IIa recommendation for RCM. In contrast, for AF patients with DCM, NDLVC, or ARVC, anticoagulation is recommended utilizing the CHA2DS2-VASc score for risk stratification (Class I). As in patients without CM, non-vitamin K-dependent OAC (NOAC) is preferred, except in patients with severe mitral stenosis and/or mechanical valve replacement.
Acknowledging the lack of consistent data for CM patients, rate control may be considered for all patients with AF, especially as AF-induced cardiac dysfunction may worsen the underlying CM [32]. As diastolic filling is reduced with increasing heart rates, AF can result in hemodynamic and clinical decompensation. This can be especially relevant for CM patients. In parallel to AF patients without CM, a lenient rate control (resting < 110 bpm) may be sufficient, but observational studies [33, 34] also suggest that CM patients with heart failure may benefit from lower heart rates. As there is no reliable data regarding the choice of pharmacological rate control in CM patients the guideline recommends beta-blockers as the preferred agent due to their established safety profile. Digoxin may be considered as an alternative; however, caution is advised due to potentially harmful effects in particular in combination with antiarrhythmic drugs (AAD) [35, 36] and inconsistent data regarding mortality in observational studies [37, 38]. In patients with left ventricular outflow tract obstruction (LVOTO), digoxin should be avoided (Class IIa). Non-dihydropyridine calcium channel blockers such as verapamil or diltiazem should only be used in CM patients with LVEF ≥ 40% though verapamil may be beneficial in combination with Class III AAD in reducing proarrhythmia [39]. AAD therapy is mostly limited to amiodarone and sotalol [40] in CM patients with significantly reduced left ventricular function (HFrEF) or significant left ventricular hypertrophy. For some CM such as ARVC [41], there is evidence that patients may benefit from flecainide [42]. In the presence of an implantable cardioverter (ICD), an individualized AAD strategy balancing risks and benefits of, for example, class III vs. class I AAD should be considered. In the case of insufficient rate control, AV-node ablation with concomitant pacemaker or preferably CRT is an alternative for selected patients (e.g., unsuitable for ablation or failed ablation) [43] and may promise symptomatic improvement in those aged > 75 years [44]. Perspectively, the potential benefit of conduction system pacing compared to CRT after AV-node ablation has to be identified [45].
Benefits of rhythm versus rate control in cardiomyopathies
More recently, the benefits of rhythm control over rate control have been extensively discussed for symptomatic and asymptomatic [46] patients with AF [47, 48]. In the EAST trial, attaining sinus rhythm seemed to be the key mediator for reduction in cardiovascular outcomes in patients with recently diagnosed AF and cardiovascular risk factors [49]. Patients with AF at the 12-month follow-up visit did not benefit from rhythm control during the remaining 4 years of follow-up. Whether these results are transferable to CM has not been investigated yet. Nevertheless, the current guideline prefers rhythm control with maintaining sinus rhythm at an early stage, especially in symptomatic CM patients with limited success of long-term AAD therapy and known relevant clinical and experimentally proven side effects [50,51,52], but also for asymptomatic patients without major risk factors for AF recurrence (Class IIa). AF ablation should be considered as first-line therapy to improve symptoms in selected CM patients (Class IIa). For patients with genetic CM (other than HCM) the guideline recognizes a lack of data whilst at the same time emphasizing the potential of proarrhythmia when using class I antiarrhythmic drugs [53, 54] in the presence of structural abnormalities in CM patients. Consequently, catheter ablation is recommended as a safe and superior alternative to AAD for rhythm control, alleviating symptoms, and improving quality of life, and in selected patients to improve survival and reduce heart failure hospitalizations (Class IIa). In addition, AF ablation is recommended for all symptomatic CM patients after failed AAD therapy or in case of tachycardia-induced LV dysfunction regardless of symptoms (Class I). When comparing AF ablation to AAD in patients with heart failure, several trials have demonstrated impressive benefits in favor of ablation [55]. For patients suffering from heart failure with preserved ejection fraction (HFpEF) some observational ablation studies have also suggested a benefit in AF burden and mortality [56].
The role of AF ablation in CM subtypes has been investigated in several registries, mostly in HCM patients and to a lesser extent for rarer diseases such as cardiac amyloidosis [57,58,59] or sarcoidosis [60]. In HCM (see below), rhythm control is achieved in about two thirds of patients; however, repeat procedures and/or continuation of AAD is often needed. CM patients have a higher risk of AF recurrences, particularly in the presence of atrial fibrosis/remodeling, for example, in patients with HCM, a laminopathy, or cardiac amyloidosis. In HCM patients with planned septal myectomy, surgical ablation and/or LAA occlusion/resection may be considered (Class IIb). Further studies will have to clarify who benefits most, which technology should be used (single tip, vs. balloon vs. variable loop catheters [3, 61]), and whether, for example, gender differences [62] regarding outcome of antiarrhythmic therapy are present in CM.
Management of ventricular arrhythmias and prevention of sudden cardiac death in cardiomyopathies
Data regarding management of VA in patients with specific genetic cardiomyopathies is scarce; however, the new ESC guideline illustrates some general concepts. Referencing the 2022 ESC Guidelines for the management of patients with VA and the prevention of SCD [2, 5, 6], the Task Force highlights the importance of identifying reversible and precipitating factors. Acute termination of sustained VA should be accomplished by cardioversion, AAD, or pacing, depending on hemodynamic tolerance, etiology, and patient profile. In a patient with electrical storm, mild-to-moderate or even deep sedation should be considered. If patients are not responding to AAD therapy, catheter ablation is recommended. In refractory cases, autonomic modulation/mechanical circulatory support can be considered. For prevention of VA recurrences, AAD (in scar-related VA mostly limited to beta blockers, amiodarone, or sotalol [40]), and catheter ablation (especially for sustained monomorphic VA, or polymorphic VA triggered by VEs of similar morphology) are indicated.
Shared-decision making that is evidence-based and takes individual preferences, circumstances, and values into account, guides ICD recommendations (Class I). An ICD is only reasonable when good quality survival of longer than 1 year is expected. Furthermore, counseling about inappropriate shocks, implant complications, and social, occupational, and driving implications is obligatory before implantation (Class I). For secondary prevention, ICD implantation is recommended irrespective of CM phenotype (Class I). For primary prevention (Table 1), a comprehensive SCD risk stratification is recommended at initial presentation and at 1‑ to 2‑year intervals, or whenever there is a change in clinical status (Class I). The use of validated risk scores (Table 2) to aid decision-making is recommended in HCM (Class I) and should be considered in DCM, NDLVC, and ARVC (Class IIa). It must always be evaluated whether a patient might profit from CRT (Class I). Given the expected need for CMR during follow-up, simpler ICD devices (e.g. single chamber or subcutaneous [63,64,65] coils in particular in children [66]) should be considered. Data on benefits of the wearable cardioverter defibrillator for primary prevention, for example, in myocarditis or peripartum CM, are so scarce that no recommendations are given.
Hypertrophic cardiomyopathy and selected phenocopies
The guideline provides a focused update of the 2014 ESC guidelines on diagnosis and management of HCM [67] considering advances in genetics, cardiac imaging, as well as novel therapies such as cardiac myosin inhibitors. The annual incidence for cardiovascular death in HCM is around 1–2%, with SCD, heart failure, and thromboembolic events in the presence of a high AF prevalence (up to 40%) being the main causes of death. Spontaneous ventricular fibrillation (VF) is often associated with fatal events; however, asystole, AV block, and pulseless electrical activity have also been documented. Of note, most HCM patients are asymptomatic and have a normal lifespan [1] but should be monitored on a regular basis (including ECG and Holter monitoring). A concise assessment of SCD risk is a key part of clinical management and a detailed discussion of clinical features associated with increased risk of SCD is provided (age, non-sustained VT, maximum LV wall thickness, family history, syncope, left atrial diameter, and LVOT diameter). There is lack of randomized data to support pharmacological therapy for prevention of SCD. One small study [68] suggested a lower incidence of SCD with amiodarone [40, 51] but other observational studies [69] found no such effect. Nevertheless, beta-blockers and/or amiodarone are recommended in patients with an ICD and recurrence of symptomatic VA, paroxysmal AF, or repeated shocks despite optimal treatment and ICD programming. Regarding risk stratification, the validated HCM risk-SCD calculator (https://qxmd.com/calculate/calculator_303/hcm-risk-scd) is recommended as the primary tool for estimating risk of sudden death at 5 years and guiding ICD implantation in primary prevention (Class I). It is recommended to assess the risk initially, and thereafter at 1‑ to 2‑year intervals or when a change in clinical status occurs (Class I). Based on risk score and further clinical risk factors (LVEF, LGE), implantation of an ICD may be considered with shared decision-making (Class IIa for high risk > 6%, Class IIb for intermediate risk between 4% and 6%). For the low-risk category (< 4%), the presence of extensive LGE (> 15%) may be considered in shared decision-making (Class IIb). For secondary prevention of SCD, ICD implantation is recommended after cardiac arrest due to VT/VF, or after sustained VA causing syncope or hemodynamic compromise. The role of subcutaneous ICD (S-ICD) in HCM with a potentially increased rate of inappropriate shocks is a matter of debate [70, 71].
Anderson–Fabry disease with inborn deficient or absent enzyme alpha-galactosidase A may be a clinically challenging phenocopy of HCM (Fig. 1). It is often diagnosed in patients with left ventricular hypertrophy and additional cardiac and extracardiac red flags [1]. Bradycardia, chronotropic incompetence, short PQ intervals, and first to third degree AV blocks have been described. AF as well as VA may occur in patients with late diagnosis (Fig. 4).

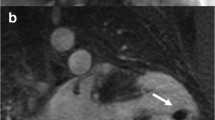
A 51-year-old male with genetically proven Anderson–Fabry disease and a history of aborted sudden cardiac death 4 years before presentation. After initial diagnosis of an assumed viral myocarditis, the patient presented 4 years later to the authors’ department with recurrent ventricular tachycardia (VT) (b). Late-stage Fabry disease with cardiac, neurological, and renal impairment was diagnosed. Cardiac magnetic resonance imaging (d,f) demonstrated apical and extensive posterolateral scarring that correlated to electroanatomical bipolar CARTO maps (c,e). The patient underwent endo- and epicardial VT ablation
Cardiac amyloidosis is another potential phenocopy of HCM in the form of an RCM that deserves special consideration in cardiac electrophysiology (Fig. 5). Electrical conduction disease with symptomatic bradycardia and advanced AV block or pseudo Q waves may be red flags for amyloidosis. Data on specific antiarrhythmic therapy including ablation for AF or VT and/or ICD implantation for primary prevention is too limited for evidence-based recommendations. The role of non-specific antiarrhythmic therapy such as transthyretin stabilization for AF and/or VT/VF prevention will hopefully be clarified in the next years.

Electrocardiogram (a) of a 74-year-old male with wild-type ATTR amyloidosis initially presenting with a sustained ventricular tachycardia (b). Note the prominent diffuse late gadolinium enhancement on cardiac magnetic resonance imaging (c,d)
Arrhythmogenic right ventricular cardiomyopathy (ARVC)
The term ARVC—originating from the pre-genetic and pre-CMR era—already indicates the interaction of arrhythmias with a cardiomyopathy. ARVC usually manifests at the second to fourth decade of life and is characterized by fibro-fatty replacement of myocardial fibers resulting in a fertile environment for arrhythmias. Though many patients with ARVC have left ventricular involvement or may even present with only LV disease (left dominant ARVC) the new guideline has preserved the term arrhythmogenic “right ventricular” CM. This may be debatable in view of concealed and subclinical as well as left-dominant disease forms. Some physicians nowadays prefer the more umbrella like term “arrhythmogenic CM.” However, the task force does not recommend its use as it lacks a morphological or functional definition consistent with the guidelines proposed classification scheme. Moreover, a significant number of conditions, which could be defined as ACM, are now classified as NDLVC.
The overall prevalence of ARVC is around 0.08%. Many patients initially present with ECG abnormalities and VA. Of note, cardiac sarcoidosis may be an important phenocopy ([18,19,20]; Fig. 3). In addition, in up to 20% of patients with acute myocarditis ARVC is diagnosed [1]. Characteristic ECG findings include T wave inversion in V1–V3 with terminal activation delay often combined with atypical right bundle branch block ([19, 72]; Fig. 6). Ventricular extrasystoles (VEs) and/or VT with left bundle branch block (LBBB) morphology (with a superior axis) are a hallmark and must be distinguished from idiopathic RVOT VEs/VT with LBBB and inferior axis [5, 73]. AF is also relatively common with a prevalence around 9–30%. Rhythm control is preferred in the case of symptoms and/or heart failure/LV dysfunction [1].

A 21-year-old female competitive athlete who presented with a syncope while playing handball. The electrocardiogram (a) was already suspicious for arrhythmogenic right ventricular cardiomyopathy (right precordial negative T waves and VEs with superior and inferior axis). Please also note the unspecific signs of inferolateral early repolarisation. During programmed electrical stimulation, a sustained ventricular tachycardia (VT) with left bundle branch block inferior axis was inducible. Electroanatomic bipolar (color range 0.5–1.5 mV) (b) and unipolar (color range 5–8 mV) maps illustrate the predominately epicardial low voltage areas. The VT was successfully ablated from the endocardial anterior right ventricular outflow tract (c)

Complete atrioventricular block (a) in a 40-year-old male with non-ischemic cardiomyopathy with mildly reduced left ventricular function but prominent intraseptal late gadolinium enhancement (b,c); the patient was found to have a likely pathogenic laminopathy (Lamin A/C) gene mutation. Marked P: sinus p waves and one P* atrial extrasystole in the ECG illustrate complete AV-dissociation in (a)
Data on antiarrhythmic therapy in asymptomatic ARVC is scarce but there is good evidence for beta-blocker therapy in symptomatic patients with VEs or VT (Class I). If beta-blockers fail to control arrhythmia-related symptoms flecainide or amiodarone are options (Class IIa). Patients with a sustained VT—even if hemodynamically tolerated—are candidates for an ICD (Class IIa). There is some evidence that ARVC patients may also benefit from S‑ICD [74], but due to lack of pacing most physicians prefer conventional ICDs. In general, pregnancy in ARVC has not been associated with negative-long term outcome, but previous VT in pregnant women represent a WHO risk class III indicating close follow-up at an expert center. Patients with high-risk features (i.e., arrhythmic syncope, non-sustained VT, RVEF < 40%, LVEF < 45%, sustained VT with programmed electrical stimulation [PES]) should also be offered an ICD (Class IIa) (see also arvcrisk.com; Table 2). Of note, the role of PES is not well defined in those who are asymptomatic illustrating some parallels to other fields in electrophysiology such as primary electrical diseases (e.g., Brugada syndrome [75,76,77]). In the case of incessant VT or frequent ICD interventions catheter ablation (with availability of epicardial approach) should be considered (Class IIa). In cardiac sarcoidosis, VT ablation may also reduce episodes and shocks, but the VT recurrence rate is relatively high [78, 79]. Based on disease acceleration with high-intensity exercise, the ESC guideline recommends against competitive sports in ARVC (Class III) including those who are genotype positive/phenotype negative (with less evidence: Class IIb).
Dilated cardiomyopathy and non-dilated left ventricular cardiomyopathy
The etiology of DCM is heterogenous and includes inherited and acquired forms. The guideline focuses mainly on genetic DCM. The diagnostic work-up of these patients and especially the results of CMR and genetic testing are relevant for risk stratification and management of VA. A recent retrospective study demonstrated a higher rate of major antiarrhythmic events in patients with genetic DCM variants compared to genotype-negative patients [80]. In certain genotypes, an increased arrhythmogenic risk was observed irrespective of LVEF [80]. The following genes are, for example, associated with increased risk: LMNA, PLN, FLNC, RBM20, EMD, TMEM43, DSP, DSG2, DSC2, and PKP2 [80,81,82,83,84]. In addition to these genetic variants, myocardial scaring determined by LGE on CMR has been established as a strong risk marker for all-cause mortality and VA, both in prospective and retrospective studies. Certain LGE distribution patterns, for example, a “ring-like” pattern, have also been found to be highly prognostic for VA [7, 85].
Regarding primary prevention of SCD the Task Force points out conflicting data from randomized trials, which have included patients with a LVEF < 35%, but overall, an only modest survival benefit in patients with ICD in DCM is observed, as demonstrated by a recent meta-analysis [86]. Nevertheless, patients with high-risk genotypes and additional risk factors (syncope, LGE) should be considered for ICD implantation, even if no LV dysfunction is evident (Class IIa). If no risk factors are present an ICD may even be considered in genotype-positive patients with a LVEF is > 35% (Class IIb). In contrast, in genotype-negative patients with a LVEF > 35%, an ICD may be only considered if risk factors (syncope, LGE) are present (Class IIb). In patients with LVEF ≤ 35% and symptomatic heart failure after > 3 months of optimal medical therapy, an ICD should be considered (Class IIa).
The management of patients with NDLVC regarding prevention of SCD is identical to patients with DCM, albeit with a lower evidence level, mirroring the lack of randomized trials in patients with absent or up to moderate LV dysfunction. Of note, the significance of resting and ambulatory ECG is highlighted for patients with NDLVC, as specific ECG features may suggest an underlying genetic cause (Fig. 1). Therefore, ambulatory ECG monitoring is recommended in patients with NDLVC annually or when there a change in clinical status (Class I). For specific genotypes, namely LMNA and PLN, validated risk calculators are recommended (Table 2). In patients with unexplained syncope, PES may provide useful information concerning the underlying cause: in view of lack of very conclusive data supporting the use of PES for risk stratification it experiences a renaissance [2,3,4]. In summary, the guideline leaves a lot of room for individualized and shared decision-making regarding ICD implantation, but there is no Class I recommendation for primary prevention.
Conclusions
Next to other areas of specialization, cardiac electrophysiology plays an integral role in the management of patients with CM. A multidisciplinary and expert approach to CM “has the patient and its family at its heart” [1]. Important innovations of the new guideline include a new phenotype-based classification system with emphasis on a multimodal imaging approach for diagnosis and risk stratification. Patient pathways guide clinicians from the initial presentation to diagnosis. Whilst providing general guidelines for CM management, the primary objective of the new guidelines is to ascertain the disease etiology for facilitating a disease-specific, individualized management.
References
Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C et al (2023) 2023 ESC guidelines for the management of cardiomyopathies. Eur Heart J. https://doi.org/10.1093/eurheartj/ehad194
Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA et al (2022) 2022 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 43:3997–4126
Eckardt L, Doldi F, Busch S, Duncker D, Estner H, Kuniss M et al (2023) 10-year follow-up of interventional electrophysiology: updated German survey during the COVID-19 pandemic. Clin Res Cardiol 112:784–794
Eckardt L, Frommeyer G, Sommer P, Steven D, Deneke T, Estner HL et al (2018) Updated survey on Interventional electrophysiology: 5‑year follow-up of infrastructure, procedures, and training positions in Germany. JACC Clin Electrophysiol 4:820–827
Könemann H, Ellermann C, Zeppenfeld K, Eckardt L (2023) Management of ventricular arrhythmias worldwide: comparison of the latest ESC, AHA/ACC/HRS, and CCS/CHRS guidelines. JACC Clin Electrophysiol 9:715–728
Könemann H, Dagres N, Merino JL, Sticherling C, Zeppenfeld K, Tfelt-Hansen J et al (2023) Spotlight on the 2022 ESC guideline management of ventricular arrhythmias and prevention of sudden cardiac death: 10 novel key aspects. EP Europace 25. https://doi.org/10.1093/europace/euad091
Bietenbeck M, Meier C, Korthals D, Theofanidou M, Stalling P, Dittmann S et al (2023) Possible causes and clinical relevance of a “ring-like” late gadolinium enhancement pattern. JACC Cardiovasc Imaging. https://doi.org/10.1016/j.jcmg.2023.08.004
Bruch C, Gotzmann M, Stypmann J, Wenzelburger F, Rothenburger M, Grude M et al (2005) Electrocardiography and doppler echocardiography for risk stratification in patients with chronic heart failure: incremental prognostic value of QRS duration and a restrictive mitral filling pattern. J Am Coll Cardiol 45:1072–1075
Alba AC, Gaztañaga J, Foroutan F, Thavendiranathan P, Merlo M, Alonso-Rodriguez D et al (2020) Prognostic value of late gadolinium enhancement for the prediction of cardiovascular outcomes in dilated cardiomyopathy: an international, multi-institutional study of the MINICOR group. Circ: Cardiovascular Imaging 13:e10105
Baggiano A, Boldrini M, Martinez-Naharro A, Kotecha T, Petrie A, Rezk T et al (2020) Noncontrast magnetic resonance for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging 13:69–80
Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA et al (2010) Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Eur Heart J 31:806–814
Halliday BP, Baksi AJ, Gulati A, Ali A, Newsome S, Izgi C et al (2019) Outcome in dilated cardiomyopathy related to the extent, location, and pattern of late gadolinium enhancement. JACC Cardiovasc Imaging 12:1645–1655
Holmström M, Kivistö S, Heliö T, Jurkko R, Kaartinen M, Antila M et al (2011) Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. J Cardiovasc Magn Reson 13:30
Sado DM, White SK, Piechnik SK, Banypersad SM, Treibel T, Captur G et al (2013) Identification and assessment of anderson-fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ: Cardiovascular Imaging 6:392–398
Barison A, Aimo A, Ortalda A, Todiere G, Grigoratos C, Passino C et al (2018) Late gadolinium enhancement as a predictor of functional recovery, need for defibrillator implantation and prognosis in non-ischemic dilated cardiomyopathy. Int J Cardiol 250:195–200
Di Marco A, Anguera I, Schmitt M, Klem I, Neilan TG, White JA et al (2017) Late gadolinium enhancement and the risk for ventricular arrhythmias or sudden death in dilated cardiomyopathy: systematic review and meta-analysis. JACC Heart Fail 5:28–38
Willy K, Dechering DG, Reinke F, Bögeholz N, Frommeyer G, Eckardt L (2021) The ECG in sarcoidosis - a marker of cardiac involvement? Current evidence and clinical implications. J Cardiol 77:154–159
Dechering DG, Kochhäuser S, Wasmer K, Zellerhoff S, Pott C, Köbe J et al (2013) Electrophysiological characteristics of ventricular tachyarrhythmias in cardiac sarcoidosis versus arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm 10:158–164
Hoogendoorn JC, Venlet J, Out YNJ, Man S, Kumar S, Sramko M et al (2021) The precordial R’ wave: a novel discriminator between cardiac sarcoidosis and arrhythmogenic right ventricular cardiomyopathy in patients presenting with ventricular tachycardia. Heart Rhythm 18:1539–1547
Dechering DG, Kochhäuser S, Zellerhoff S, Frommeyer G, Eckardt L (2016) Three-dimensional electroanatomic voltage mapping to guide biopsy sampling in unexplained cardiomyopathies: a proof-of-principle case series. Clin Res Cardiol 105:186–188
Zumhagen S, Spieker T, Rolinck J, Baba HA, Breithardt G, Böcker W et al (2009) Absence of pathognomonic or inflammatory patterns in cardiac biopsies from patients with brugada syndrome. Circ Arrhythmia Electrophysiol 2:16–23
Nielsen JC, Lin Y‑J, de Oliveira Figueiredo MJ, Sepehri Shamloo A, Alfie A, Boveda S et al (2020) European heart rhythm association (EHRA)/heart rhythm society (HRS)/Asia pacific heart rhythm society (APHRS)/Latin American heart rhythm society (LAHRS) expert consensus on risk assessment in cardiac arrhythmias: use the right tool for the right outcome, in the right population. EP Europace 22:1147–1148
Wasmer K, Eckardt L, Breithardt G (2017) Predisposing factors for atrial fibrillation in the elderly. J Geriatr Cardiol 14:179–184
Charron P, Elliott PM, Gimeno JR, Caforio ALP, Kaski JP, Tavazzi L et al (2018) The cardiomyopathy registry of the EURobservational research programme of the European society of cardiology: baseline data and contemporary management of adult patients with cardiomyopathies. Eur Heart J 39:1784–1793
Mizia-Stec K, Caforio ALP, Charron P, Gimeno JR, Elliott P, Kaski JP et al (2020) Atrial fibrillation, anticoagulation management and risk of stroke in the cardiomyopathy/Myocarditis registry of the EURObservational research programme of the European society of cardiology. Esc Heart Fail 7:3601–3609
Kochhäuser S, Dechering DG, Dittrich R, Reinke F, Ritter MA, Ramtin S et al (2014) Supraventricular premature beats and short atrial runs predict atrial fibrillation in continuously monitored patients with cryptogenic stroke. Stroke 45:884–886
Hindricks G, Potpara T, Dagres N, Arbelo E, Bax JJ, Blomström-Lundqvist C et al (2021) 2020 ESC guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European association for cardio-thoracic surgery (EACTS): the task force for the diagnosis and management of atrial fibrillation of the European society of cardiology (ESC) developed with the special contribution of the European heart rhythm association (EHRA) of the ESC. Eur Heart J 42:373–498
Rienstra M, Hobbelt AH, Alings M, Tijssen JGP, Smit MD, Brügemann J et al (2018) Targeted therapy of underlying conditions improves sinus rhythm maintenance in patients with persistent atrial fibrillation: results of the RACE 3 trial. Eur Heart J 39:2987–2996
Guo Y, Lane DA, Wang L, Zhang H, Wang H, Zhang W et al (2020) Mobile health technology to improve care for patients with atrial fibrillation. J Am Coll Cardiol 75:1523–1534
Guo Y, Guo J, Shi X, Yao Y, Sun Y, Xia Y et al (2020) Mobile health technology-supported atrial fibrillation screening and integrated care: a report from the mAFA-II trial long-term extension cohort. Eur J Intern Med 82:105–111
Wasmer K, Köbe J, Dechering D, Milberg P, Pott C, Vogler J et al (2013) CHADS(2) and CHA(2)DS (2)-VASc score of patients with atrial fibrillation or flutter and newly detected left atrial thrombus. Clin Res Cardiol 102:139–144
Willems S, Meyer C, de Bono J, Brandes A, Eckardt L, Elvan A et al (2019) Cabins, castles, and constant hearts: rhythm control therapy in patients with atrial fibrillation. Eur Heart J 40:3793–3799c
Sartipy U, Savarese G, Dahlström U, Fu M, Lund LH (2019) Association of heart rate with mortality in sinus rhythm and atrial fibrillation in heart failure with preserved ejection fraction. European J of Heart Fail 21:471–479
Hess PL, Sheng S, Matsouaka R, DeVore AD, Heidenreich PA, Yancy CW et al (2020) Strict versus lenient versus poor rate control among patients with atrial fibrillation and heart failure (from the get with the guidelines - heart failure program). Am J Cardiol 125:894–900
Frommeyer G, Puckhaber D, Ellermann C, Dechering DG, Kochhäuser S, Leitz P et al (2017) Interactions of digitalis and class-III antiarrhythmic drugs: amiodarone versus dronedarone. Int J Cardiol 228:74–79
Frommeyer G, Milberg P, Schulze Grotthoff J, Dechering DG, Kochhäuser S, Stypmann J et al (2015) Dronedarone and digitalis: individually reduced post-repolarization refractoriness enhances life-threatening arrhythmias. EP Europace 17:1300–1308
Bode N, Hochadel M, Andresen D, Zahn R, Spitzer SG, Brachmann J et al (2021) Cardiac glycosides are not associated with increased mortality or hospitalization rates in ICD and CRT-ICD patients after adjustment for baseline-characteristics at one-year follow-up: results from the German DEVICE registry. Int J Cardiol 338:109–114
Frommeyer G, Brachmann J, Ince H, Spitzer SG, Thomas D, Willems S et al (2019) Digitalis therapy is associated with higher comorbidities and poorer prognosis in patients undergoing ablation of atrial arrhythmias: data from the German ablation registry. Clin Res Cardiol 108:1083–1092
Milberg P, Reinsch N, Osada N, Wasmer K, Mönnig G, Stypmann J et al (2005) Verapamil prevents torsade de pointes by reduction of transmural dispersion of repolarization and suppression of early afterdepolarizations in an intact heart model of LQT3. Basic Res Cardiol 100:365–371
Milberg P, Ramtin S, Mönnig G, Osada N, Wasmer K, Breithardt G et al (2004) Comparison of the in vitro electrophysiologic and proarrhythmic effects of amiodarone and sotalol in a rabbit model of acute atrioventricular block. J Cardiovasc Pharmacol 44:278–286
Wichter T, Paul TM, Eckardt L, Gerdes P, Kirchhof P, Böcker D et al (2005) Arrhythmogenic right ventricular cardiomyopathy. Antiarrhythmic drugs, catheter ablation, or ICD? Herz 30:91–101
Rolland (2022) Safety and efficacy of flecainide associated with beta-blockers in arrhythmogenic right ventricular cardiomyopathy. Europace 24:278
Brignole M, Pentimalli F, Palmisano P, Landolina M, Quartieri F, Occhetta E et al (2021) AV junction ablation and cardiac resynchronization for patients with permanent atrial fibrillation and narrow QRS: the APAF-CRT mortality trial. Eur Heart J 42:4731–4739
Wasmer K, Hochadel M, Wieneke H, Spitzer SG, Brachmann J, Straube F et al (2019) Long-term symptom improvement and patient satisfaction after AV-node ablation vs. Pulmonary vein isolation for symptomatic atrial fibrillation: results from the German ablation registry. Clin Res Cardiol 108:395–401
Huang W, Wang S, Su L, Fu G, Su Y, Chen K et al (2022) His-bundle pacing vs biventricular pacing following atrioventricular nodal ablation in patients with atrial fibrillation and reduced ejection fraction: a multicenter, randomized, crossover study-the ALTERNATIVE-AF trial. Heart Rhythm 19:1948–1955
Willems S, Borof K, Brandes A, Breithardt G, Camm AJ, Crijns HJGM et al (2022) Systematic, early rhythm control strategy for atrial fibrillation in patients with or without symptoms: the EAST-AFNET 4 trial. Eur Heart J 43:1219–1230
Eckardt L, Wolfes J, Frommeyer G (2023) Benefits of early rhythm control of atrial fibrillation. Trends Cardiovasc Med. https://doi.org/10.1016/j.tcm.2023.04.001
Schnabel RB, Marinelli EA, Arbelo E, Boriani G, Boveda S, Buckley CM et al (2023) Early diagnosis and better rhythm management to improve outcomes in patients with atrial fibrillation: the 8th AFNET/EHRA consensus conference. EP Europace 25:6–27
Eckardt L, Sehner S, Suling A, Borof K, Breithardt G, Crijns H et al (2022) Attaining sinus rhythm mediates improved outcome with early rhythm control therapy of atrial fibrillation: the EAST-AFNET 4 trial. Eur Heart J 43:4127–4144
Eckardt L, Haverkamp W, Johna R, Böcker D, Deng MC, Breithardt G et al (2000) Arrhythmias in heart failure: current concepts of mechanisms and therapy. J Cardiovasc Electrophysiol 11:106–117
Frommeyer G, Milberg P, Witte P, Stypmann J, Koopmann M, Lücke M et al (2011) A new mechanism preventing proarrhythmia in chronic heart failure: rapid phase-III repolarization explains the low proarrhythmic potential of amiodarone in contrast to sotalol in a model of pacing-induced heart failure. European J of Heart Fail 13:1060–1069
Eckardt L, Haverkamp W, Mertens H, Johna R, Clague JR, Borggrefe M et al (1998) Drug-related torsades de pointes in the isolated rabbit heart: comparison of clofilium, d,l-sotalol, and erythromycin. J Cardiovasc Pharmacol 32:425–434
Frommeyer G, Eckardt L (2016) Drug-induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol 13:36–47
Schleberger R, Metzner A, Kuck K‑H, Andresen D, Willems S, Hoffmann E et al (2021) Antiarrhythmic drug therapy after catheter ablation for atrial fibrillation-insights from the German ablation registry. Pharmacol Res Perspect 9:e880
Casula M, Pignalosa L, Quilico F, Scajola LV, Rordorf R (2023) A comprehensive meta-analysis comparing radiofrequency ablation versus pharmacological therapy for the treatment of atrial fibrillation in patients with heart failure. Int J Cardiol 377:66–72
Gu G, Wu J, Gao X, Liu M, Jin C, Xu Y (2022) Catheter ablation of atrial fibrillation in patients with heart failure and preserved ejection fraction: a meta-analysis. Clin Cardiol 45:786–793
Briasoulis A, Kourek C, Papamichail A, Loritis K, Bampatsias D, Repasos E et al (2023) Arrhythmias in patients with cardiac amyloidosis: a comprehensive review on clinical management and devices. J Cardiovasc Dev Dis (10)
Brachmann J, Lewalter T, Kuck K‑H, Andresen D, Willems S, Spitzer SG et al (2017) Long-term symptom improvement and patient satisfaction following catheter ablation of supraventricular tachycardia: insights from the German ablation registry. Eur Heart J 38:1317–1326
Hoffmann E, Straube F, Wegscheider K, Kuniss M, Andresen D, Wu L‑Q et al (2019) Outcomes of cryoballoon or radiofrequency ablation in symptomatic paroxysmal or persistent atrial fibrillation. EP Europace 21:1313–1324
Willy K, Dechering DG, Wasmer K, Köbe J, Bögeholz N, Ellermann C et al (2019) Outcome of catheter ablation of supraventricular tachyarrhythmias in cardiac sarcoidosis. Clin Cardiol 42:1121–1125
Bittner A, Mönnig G, Zellerhoff S, Pott C, Köbe J, Dechering D et al (2011) Randomized study comparing duty-cycled bipolar and unipolar radiofrequency with point-by-point ablation in pulmonary vein isolation. Heart Rhythm 8:1383–1390
Zylla MM, Brachmann J, Lewalter T, Hoffmann E, Kuck K‑H, Andresen D et al (2016) Sex-related outcome of atrial fibrillation ablation: insights from the German ablation registry. Heart Rhythm 13:1837–1844
Lambiase PD, Theuns DA, Murgatroyd F, Barr C, Eckardt L, Neuzil P et al (2022) Subcutaneous implantable cardioverter-defibrillators: long-term results of the EFFORTLESS study. Eur Heart J 43:2037–2050
Holtstiege V, Meier C, Bietenbeck M, Chatzantonis G, Florian A, Köbe J et al (2020) Clinical experience regarding safety and diagnostic value of cardiovascular magnetic resonance in patients with a subcutaneous implanted cardioverter/defibrillator (S-ICD) at 1.5 T. J Cardiovasc Magn Reson 22:35
Bögeholz N, Willy K, Niehues P, Rath B, Dechering DG, Frommeyer G et al (2019) Spotlight on S‑ICD™ therapy: 10 years of clinical experience and innovation. EP Europace 21:1001–1012
Bettin M, Larbig R, Rath B, Fischer A, Frommeyer G, Reinke F et al (2017) Long-term experience with the subcutaneous implantable cardioverter-defibrillator in teenagers and young adults. JACC Clin Electrophysiol 3:1499–1506
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P et al (2014) 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European society of cardiology (ESC). Eur Heart J 35:2733–2779
McKenna WJ, Oakley CM, Krikler DM, Goodwin JF (1985) Improved survival with amiodarone in patients with hypertrophic cardiomyopathy and ventricular tachycardia. Br Heart J 53:412–416
Melacini P, Maron BJ, Bobbo F, Basso C, Tokajuk B, Zucchetto M et al (2007) Evidence that pharmacological strategies lack efficacy for the prevention of sudden death in hypertrophic cardiomyopathy. Heart 93:708–710
Köbe J, Reinke F, Meyer C, Shin D‑I, Martens E, Kääb S et al (2013) Implantation and follow-up of totally subcutaneous versus conventional implantable cardioverter-defibrillators: a multicenter case-control study. Heart Rhythm 10:29–36
Frommeyer G, Dechering DG, Zumhagen S, Löher A, Köbe J, Eckardt L et al (2016) Long-term follow-up of subcutaneous ICD systems in patients with hypertrophic cardiomyopathy: a single-center experience. Clin Res Cardiol 105:89–93
Kaya E, Fischer C, Eckardt L (2013) EKG-Befunde bei primär neurologischen Erkrankungen, Systemerkrankungen und primären Kardiomyopathien (ECG changes in primary neurological disorders, systemic diseases and primary cardioymopathies). Herzschrittmacherther Elektrophysiol 24:109–114
Ribbing M, Wasmer K, Mönnig G, Kirchhof P, LOH P, Breithardt G et al (2003) Endocardial mapping of right ventricular outflow tract tachycardia using noncontact activation mapping. J Cardiovasc Electrophysiol 14:602–608
Honarbakhsh S, Protonotarios A, Monkhouse C, Hunter RJ, Elliott PM, Lambiase PD (2023) Right ventricular function is a predictor for sustained ventricular tachycardia requiring anti-tachycardic pacing in arrhythmogenic ventricular cardiomyopathy: insight into transvenous vs. Subcutaneous implantable cardioverter defibrillator insertion. EP Europace (25)
Gaborit N, Wichter T, Varro A, Szuts V, Lamirault G, Eckardt L et al (2009) Transcriptional profiling of ion channel genes in brugada syndrome and other right ventricular arrhythmogenic diseases. Eur Heart J 30:487–496
Paul M, Gerss J, Schulze-Bahr E, Wichter T, Vahlhaus C, Wilde AAM et al (2007) Role of programmed ventricular stimulation in patients with brugada syndrome: a meta-analysis of worldwide published data. Eur Heart J 28:2126–2133
Eckardt L, Kirchhof P, Schulze-Bahr E, Rolf S, Ribbing M, Loh P et al (2002) Electrophysiologic investigation in brugada syndrome; yield of programmed ventricular stimulation at two ventricular sites with up to three premature beats. Eur Heart J 23:1394–1401
Papageorgiou N, Providência R, Bronis K, Dechering DG, Srinivasan N, Eckardt L et al (2018) Catheter ablation for ventricular tachycardia in patients with cardiac sarcoidosis: a systematic review. EP Europace 20:682–691
Siontis KC, Santangeli P, Muser D, Marchlinski FE, Zeppenfeld K, Hoogendoorn JC et al (2022) Outcomes associated with catheter ablation of ventricular tachycardia in patients with cardiac sarcoidosis. JAMA Cardiol 7:175–183
Escobar-Lopez L, Ochoa JP, Mirelis JG, Espinosa MÁ, Navarro M, Gallego-Delgado M et al (2021) Association of genetic variants with outcomes in patients with nonischemic dilated cardiomyopathy. J Am Coll Cardiol 78:1682–1699
Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal J‑M, Androulakis AFA et al (2016) Long-term arrhythmic and nonarrhythmic outcomes of Lamin A/C mutation carriers. J Am Coll Cardiol 68:2299–2307
Verstraelen TE, van Lint FHM, Bosman LP, de Brouwer R, Proost VM, Abeln BGS et al (2021) Prediction of ventricular arrhythmia in phospholamban p.Arg14del mutation carriers-reaching the frontiers of individual risk prediction. Eur Heart J 42:2842–2850
Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC et al (2020) Desmoplakin Cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 141:1872–1884
Gigli M, Stolfo D, Graw SL, Merlo M, Gregorio C, Nee Chen S et al (2021) Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation 144:1600–1611
Chen W, Qian W, Zhang X, Li D, Qian Z, Xu H et al (2021) Ring-like late gadolinium enhancement for predicting ventricular tachyarrhythmias in non-ischaemic dilated cardiomyopathy. European Heart Journal - Cardiovascular Imaging 22:1130–1138
Alba AC, Foroutan F, Duero Posada J, Battioni L, Schofield T, Alhussein M et al (2018) Implantable cardiac defibrillator and mortality in non-ischaemic cardiomyopathy: an updated meta-analysis. Heart 104:230–236
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
D. Korthals and L. Eckardt declare that they have no competing interests.
For this article no studies with human participants or animals were performed by any of the authors. All studies mentioned were in accordance with the ethical standards indicated in each case.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.

Scan QR code & read article online
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Korthals, D., Eckardt, L. The new European Society of Cardiology guideline for the management of cardiomyopathies: key messages for cardiac electrophysiologists. Herzschr Elektrophys 34, 311–323 (2023). https://doi.org/10.1007/s00399-023-00975-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00399-023-00975-y