
Abstract
In the context of myocardial infarction, the burst of superoxide generated by reverse electron transport (RET) at complex I in mitochondria is a crucial trigger for damage during ischaemia/reperfusion (I/R) injury. Here we outline the necessary conditions for superoxide production by RET at complex I and how it can occur during reperfusion. In addition, we explore various pathways that are implicated in generating the conditions for RET to occur and suggest potential therapeutic strategies to target RET, aiming to achieve cardioprotection.
Similar content being viewed by others


Introduction
Despite extensive research, therapies to prevent ischaemia/reperfusion (I/R) injury in myocardial infarction (MI) remain elusive [65,66,67]. However, one thing is clear: mitochondria play a critical role in the damaging events of I/R injury [21, 42, 43, 52, 177]. Cardiomyocytes rely on mitochondria to oxidise various fuel sources, maintaining the essential ATP/ADP ratio and consuming oxygen in the process, due to their high energetic demands [60, 119]. As the organelles responsible for consuming over 90% of the oxygen intake [156], it is logical that mitochondria play a central role in ischaemic pathologies where oxygen supply to tissues is rapidly depleted [67, 80]. Moreover, mitochondria are not merely ATP producers; they also serve as key metabolic and signalling hubs that underpin (patho)physiology [129], playing a multifaceted and complex role in cardiac I/R injury [42, 101, 120].
Apart from their involvement in the tricarboxylic acid (TCA) cycle as providers of electron transport chain (ETC) cofactors, mitochondrial metabolites intricately interact with the rest of the cell, shaping its phenotype and response to stimuli [33, 55, 129]. This precise interplay becomes particularly evident during ischaemia and reperfusion, defining the response to the shift from homeostasis and ultimately the phenomenon of I/R injury [63, 66, 101]. In this overview, we will explore the mitochondria-centric mechanism that triggers damage in I/R injury by generating the proximal reactive oxygen species (ROS) superoxide by reverse electron transport (RET) and potential therapeutic opportunities in this domain.
RET in I/R injury pathophysiology
Thermodynamics of RET in I/R injury
For a long time, it has been hypothesised that a surge of reactive oxygen species (ROS) originating from mitochondria plays a crucial role in I/R injury. Initially, ROS was believed to be a non-specific oxidative event triggered by the reintroduction of oxygen into the ischaemic region [56, 64, 178]. However, more recent research has led to the proposal of a specific mechanistic pathway leading to the production of superoxide [40, 41]. Now, RET at complex I is thought to be the predominant mechanism of the mitochondrial superoxide burst upon reperfusion [40, 41, 121, 173]. This notion is a result of a large body of work detailing the metabolic changes occurring during I/R and their contribution to driving the production of superoxide [40, 41, 113, 141, 173].
As complex I sits close to thermodynamic equilibrium, it is considered to be a reversible catalytic machine, with the direction of electron flow dependent on the protonmotive force (Δp) and relative Coenzyme Q (CoQ) and NADH pool reduction states [3, 121, 176]. This leads to complex I being able to produce superoxide in two catalytic states: forward and reverse electron transport [97, 121, 137]. Superoxide production during forward electron transport (FET) occurs in a situation where the flavin mononucleotide (FMN) group in complex I is reduced. The reduction of the FMN is set by the NADH/NAD+ ratio, therefore perturbations in complex I function such as respiratory chain inhibition (e.g. Q-site inhibitor rotenone) or dysfunction lead to an increased NADH/NAD+ ratio, driving superoxide production by FET. The second catalytic state of complex I that can produce superoxide is RET [121, 137, 141, 169]. RET occurs when electrons are forced back from the CoQ pool onto FMN and thus able to reduce NAD+ to NADH [121, 137, 169].
Thermodynamically, for forward electron transport, the reduction potential difference of the NAD+/NADH and CoQ/CoQH2 redox couples (ΔEh) for 2 electrons to transport across complex I must be large enough to pump 4 protons against the protonmotive force (Δp) across the mitochondrial inner membrane [29, 121]. Therefore, for 2 electrons passing through complex I, 4 protons would be pumped so 2ΔEh > 4Δp (Fig. 1A). For RET, the reverse must be true, in that the reduction potential difference between the NAD+/NADH and CoQ/CoQH2 pools must be less than the energy required to pump 4 protons across the inner membrane [32, 121, 169]. These conditions would be met in instances of a high CoQ pool reduction state and a large Δp, meaning that 4Δp > 2ΔEh, leading to RET through complex I and onto FMN, with either subsequent reduction of NAD+ to NADH, or of oxygen to generate superoxide (Fig. 1B) [121, 169]. Furthermore, RET generates the most superoxide out of all the mitochondrial superoxide production mechanisms [121].

Schematic of conditions required for RET and superoxide production. A Complex I operating forward electron transport, oxidising NADH to generate the protonmotive force (Δp) which is used for ATP synthesis. The reduction potential difference between the NAD+/NADH and the CoQ/CoQH2 redox couples across complex I (ΔEh) has to be larger than that required to pump protons across the IMM against the Δp. As 4 protons are pumped per 2 electrons through complex I, for forward electron transport 2ΔEh > 4Δp. B In conditions of a large Δp, such as no ATP synthesis, so that 2ΔEh < 4Δp, electrons can be forced backwards from the CoQ pool and onto the complex I FMN which can transfer an electron onto oxygen to produce superoxide. IMS intermembrane space, IMM inner mitochondrial membrane, SDH succinate dehydrogenase, FMN flavin mononucleotide, Δp protonmotive force, cyt c cytochrome c, O2●− superoxide, DIC mitochondrial dicarboxylate carrier
For RET to be relevant in I/R injury, we must understand whether the conditions upon early reperfusion are sufficient to drive superoxide production via this mechanism. The low Km of complex IV for oxygen means that upon reperfusion, the oxygen present will rapidly restore electron flow through the ETC [156]. The restoration in ETC activity generates a large Δp as complexes III and IV pump protons across the inner mitochondrial membrane (IMM) [40, 41, 91]. Under normal conditions, the Δp would be utilised by ATP synthase, shuttling the protons back across the IMM and into the matrix to power the generation and release of ATP from ATP synthase, as well as export of ATP to the cytosol by the adenine nucleotide translocator (ANT) [94, 144]. However, during the conditions of early reperfusion, ATP synthesis is limited due to the lack of availability of adenine nucleotides for phosphorylation [40, 41]. This is due to the extensive degradation of the adenine nucleotide pool during ischaemia [40, 106]. Limiting ATP synthesis on reperfusion minimises the utilisation of the Δp across the IMM [153]. Therefore, the metabolic changes during ischaemia seem to be able to satisfy the condition of 4Δp > 2ΔEh for RET to occur (Fig. 1B). On reperfusion, electrons will be transferred from the CoQ pool, through the ETC and onto oxygen. The conditions for RET to occur requires a source of electrons to maintain a highly reduced CoQ pool in early reperfusion [121]. Therefore a substrate that is highly accumulated in ischaemia, rapidly oxidised within mitochondria and feeds electrons directly into the CoQ pool could drive RET at complex I in early reperfusion.
Succinate at the heart of RET in cardiac I/R injury
A critical finding tying together the idea of ordered RET-derived superoxide production initiating I/R injury is the TCA cycle metabolite succinate. Succinate accumulation in oxygen-compromised environments was well documented in the comparative physiology field [69, 70], however, the link to I/R injury was not made until the precise metabolomic profiling of ischaemic tissues [40]. Succinate was significantly accumulated in numerous ischaemic mouse tissues, including the heart [40]. Furthermore, succinate was one of only three metabolites that accumulated in all the tissue types tested and the only mitochondrial metabolite [40]. The other two metabolites were hypoxanthine and xanthine – both adenine nucleotide degradation products [20, 40, 85, 106]. The many-fold accumulation of succinate in the in vivo ischaemic mouse heart has also been recapitulated in rat, rabbit, pig and human hearts [40, 88, 89, 106], thus succinate accumulation is a conserved hallmark of ischaemia.
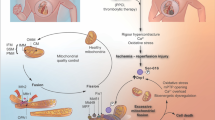
The mechanism of succinate accumulation in ischaemia remains uncertain with evidence from varying models pointing to either succinate dehydrogenase (SDH) reversal or canonical TCA cycle activity [38, 40, 151, 174]. SDH is a key node of oxidative metabolism due to its dual role as an enzyme within both the TCA cycle and the ETC. The data from an ex vivo perfused heart model have suggested that ischaemic succinate accumulation occurs predominantly by canonical TCA cycle activity [174]. Here, it was shown that glutaminolysis drives the anaplerotic entry of carbons into the TCA cycle via α-ketoglutarate and that dimethyl α-ketoglutarate could further boost the levels of ischaemic succinate [174]. However, in vivo studies support succinate accumulation occurring due to a reversal in SDH activity, reducing fumarate to succinate (Fig. 2A) [40, 151]. In ischaemia, the CoQ pool becomes highly reduced due to the lack of the terminal electron acceptor oxygen [59, 141]. Thus electrons from cofactors, such as NADH via complex I, reduce the CoQ but can no longer be transferred onto oxygen. Instead, the electrons from the CoQ pool can be used by SDH to reduce fumarate to succinate, with fumarate, therefore, acting as a terminal electron acceptor and succinate accumulating [40, 151]. As no mammalian mitochondrial fumarate transporter has been identified [76] it has been postulated that fumarate transport across the IMM in cardiac mitochondria arises from malate via a partial reversal of the TCA cycle [40, 41]. As malate can be transported freely across the IMM by the mitochondrial dicarboxylate carrier (DIC), this enables numerous cytosolic pathways to converge at malate and then enter mitochondria to enable succinate accumulation (Fig. 2A) [53]. The degradation of the adenine nucleotide pool leads to the production of fumarate via the purine nucleotide cycle (PNC) by the action of adenylosuccinate lyase (ASL) in the cytosol [159]. This cytosolic fumarate can be converted to malate by cytosolic fumarate hydratase (FH) [1, 7]. In addition, aspartate may be an important anaplerotic route driving ischaemic succinate accumulation. Aspartate can feed into the cytosolic production of malate by participating in the PNC as a substrate for adenylosuccinate synthetase [40, 159], or aspartate may be transaminated to oxaloacetate and subsequently metabolised to malate in the cytosol of the ischaemic heart tissue [40, 44].

Succinate at the heart of I/R injury A Pathways involved in ischaemic succinate accumulation. In ischaemic tissue, succinate accumulation occurs primarily by SDH reversal. This process is driven by the reduced CoQ pool, fed by NADH oxidation by complex I, enabling electrons from the CoQ pool to reduce fumarate to succinate. There are multiple avenues for substrates to lead to succinate accumulation. The degradation of AMP in the purine nucleotide cycle leads to the production of fumarate in the cytosol, which is subsequently converted to malate. The transamination of aspartate to oxaloacetate and subsequent reduction to malate provides another pathway to produce cytosolic malate. This malate can be transported into mitochondria by the DIC in exchange for succinate, also leading to a cytosolic pool of succinate accumulating. The mitochondrial malate can be subsequently dehydrated to fumarate which can feed the production of succinate. Ischaemic succinate accumulation may also occur by the canonical TCA cycle via glutaminolysis. B Succinate as a driver of RET on reperfusion. Upon reperfusion, the reintroduction of oxygen restarts the electron transport chain and proton pumping by complexes III and IV. As ATP synthesis is limited due to the degraded adenine nucleotide pool, a large Δp is generated. The CoQ pool remains reduced by the oxidation of the pool of succinate, sustained by the transport of cytosolic succinate into mitochondria by the DIC. Succinate oxidation together with the large Δp drives RET, with electrons from the CoQ pool going on the complex I FMN and onto oxygen, generating superoxide. A proportion of the cytosolic pool of succinate is effluxed from cardiomyocytes via MCT1. IMS intermembrane space, IMM inner mitochondrial membrane, SDH succinate dehydrogenase, FMN flavin mononucleotide, Δp protonmotive force, cyt c cytochrome c, O2●− superoxide, DIC mitochondrial dicarboxylate carrier, IMP inosine monophosphate
As malate may be produced in the cytosol of the ischaemic cardiomyocytes, an important step is its transport into mitochondria. Both malate and succinate are substrates for the DIC, thus the cytosolic malate can enter mitochondria in exchange for succinate [53]. This has the dual effect of succinate being diluted in the cytosol preventing the fumarate/succinate ratio from affecting SDH reversal (or maintaining canonical TCA cycle activity [102]), as well as maintaining a supply of malate to sustain succinate production (Fig. 2A) [40]. With the cytosol acting as an electron sink, this enables succinate to accumulate to 2–5 mM as quantified in ischaemic mouse heart tissue [131]. Depending on the conditions and time from the initiation of ischaemia, it may be likely that both canonical TCA cycle and SDH reversal are important in achieving ischaemic succinate accumulation. Nevertheless, regardless of the intricacies of the mechanism, it is clear that succinate is profoundly elevated in ischaemic tissue.
After a short period of reperfusion (~ 1 to 3 min), succinate levels return to those at normoxia [40, 131, 174]. Approximately half of the accumulated succinate effluxes from cardiomyocytes into the circulation via the monocarboxylate transporter 1 (MCT1), while the other half is oxidised by SDH in mitochondria (Fig. 2B) [6, 131, 174]. This rapid oxidation of succinate by SDH provides a source of electrons to maintain a highly reduced CoQ pool during early reperfusion [40, 141]. Together with the large Δp, succinate oxidation maintains a reduced CoQ pool driving RET at complex I and the subsequent superoxide production in early reperfusion (Fig. 2B) [40, 114, 141]. The conditions of early reperfusion are akin to isolated heart mitochondria incubated with succinate in low/non-phosphorylating conditions, where superoxide is maximally produced by RET [121]. Within the reperfused tissue, the cytosolic store of succinate can be transported back into mitochondria via the DIC, sustaining succinate oxidation and returning the succinate levels back to those at normoxia, while driving RET and initiating the damage in I/R injury (Fig. 2B) [41, 53, 121]. With succinate oxidation driving RET and a subsequent burst of superoxide, this initiates the downstream cascade of cell death and injury [40, 80, 95]. The burst of superoxide on reperfusion together with calcium dyshomeostasis and restoration of pH leads to the opening of the mitochondrial permeability transition pore (mPTP) [10, 16, 123]. mPTP opening leads to mitochondrial swelling and rupture activating a series of downstream events resulting in cell death. A number of different mechanisms of cell death have been implicated with I/R, with an initial wave of necrosis and apoptosis in the acute phase of reperfusion (< 24 h) [66]. Later stages of cell death (> 24 h) are now associated with ferroptosis, in an iron-dependent programmed cell death mediated by lipid peroxides [31].
Therefore, the finding of succinate accumulation and oxidation in I/R was instrumental in our current understanding of the ordered production of superoxide upon reperfusion by RET which drives cellular damage and injury.
Targeting RET therapeutically
As superoxide production via complex I RET is a key initiating event in the pathology of I/R injury, the conditions required to drive RET also provide an opportunity for therapeutic intervention and may help explain mechanistically the basis of cardioprotection of several agents. Below we outline the key factors required for RET-derived superoxide and the current evidence towards pharmacological approaches targeting these to prevent I/R injury.
Succinate accumulation
The accumulation of succinate during ischaemia is the key to providing both driving forces for superoxide production by RET upon reperfusion: Δp and ΔEh. Therefore, targeting the accumulation of succinate during ischaemia is a potential intervention in preventing I/R injury. Indeed, preventing succinate accumulation during ischaemia was a pivotal approach that helped define the importance of succinate metabolism in I/R injury [40]. Succinate accumulation is now seen as a hallmark of ischaemia. Considering the role it plays in driving RET upon reperfusion, interventions that prevent its accumulation may play a role in combatting I/R injury.
Preventing succinate accumulation was first found to be a therapeutic option using dimethyl malonate (DMM) [40]. DMM is an ester prodrug of the potent, competitive SDH inhibitor malonate [93, 165], but can freely diffuse across biological membranes and can be hydrolysed by cellular esterases to release malonate in its active form. As SDH reversal leads to succinate accumulation, inhibiting SDH may prevent succinate accumulation during ischaemia (Fig. 3A). When mice were treated with DMM prior to left anterior descending coronary artery (LAD) ligation, succinate accumulation was prevented in the ischaemic tissue [40]. Furthermore, DMM-treated mice also had smaller infarct sizes than control animals [40]. This showed in vivo, not only that SDH reversal contributed to succinate accumulation but also that preventing succinate accumulation during ischaemia may be a promising therapeutic option. In addition, DMM pre-treatment has been used in the isolated Langendorff-perfused heart model with healthy or diabetic rats [78, 158]. Here, DMM led to modest cardioprotection, however, as the levels of succinate and malonate were not measured, it is difficult to ascertain whether SDH was inhibited or whether the diabetic heart affects the metabolism of DMM. However, these results suggest that targeting succinate metabolism may also be of use in treating I/R injury in patients with comorbidities [78, 158]. As well as in MI, DMM has shown utility in preventing cardiac succinate accumulation in organ transplantation [106], haemorrhagic shock [154] and in combination with hypothermic reperfusion [89].

Targeting succinate metabolism to prevent RET. A Inhibiting routes to succinate accumulation. Preventing ischaemic succinate accumulation has been shown experimentally by inhibiting SDH reversal with malonate, complex I with rotenone, adenylosuccinate lyase with AICAR and aspartate transaminase with AOA. We hypothesise that inhibiting fumarate hydratase with FHIN-1 or DIC with butylmalonate may also prevent succinate accumulation. Inhibitors in red have experimental evidence of preventing ischaemic succinate accumulation, whereas inhibitors in pink are hypothesised to prevent ischaemic accumulation. B Modulating succinate oxidation and transport on reperfusion. Inhibiting SDH with either malonate or AA5 experimentally have shown to be able to prevent RET by slowing succinate oxidation. Malonate selectively enters ischaemic cardiomyocytes on reperfusion due to the low pH protonating malonate into its monocarboxylate form, allowing it to be transported by MCT1. Malonate is subsequently transported into mitochondria by the DIC to selectively inhibit SDH in the at-risk tissue. Blocking succinate efflux via MCT1 with AR-C155858 has been shown to increase superoxide production on reperfusion by RET, due to increasing the proportion of the accumulated succinate oxidised. It is also hypothesised that AR-C141990 and AZD3965 would lead to similar effects in vivo. Targeting the DIC with butylmalonate may also prevent RET by preventing the cytosolic succinate pool from being oxidised by SDH in mitochondria. Inhibitors in red have experimental evidence of modulating RET, whereas inhibitors in pink are hypothesised to affect RET by modulating succinate transport. IMS intermembrane space, IMM inner mitochondrial membrane, OMM outer mitochondrial membrane, SDH succinate dehydrogenase, FMN flavin mononucleotide, Δp protonmotive force, cyt c cytochrome c, O2●− superoxide, DIC mitochondrial dicarboxylate carrier, IMP inosine monophosphate, MDH malate dehydrogenase, FH fumarate hydratase, ASL adenylosuccinate lyase, MCT1 monocarboxylate transporter 1
The immune-relevant metabolite itaconate competitively inhibits SDH [98, 116]. The ester prodrug of itaconate, dimethyl itaconate (DMI) was found to be cardioprotective, and its hydrolysis product was presumed to act via the same mechanism as DMM by preventing succinate accumulation [98]. However, it was subsequently found that DMI is not hydrolysed to release itaconate [13, 49, 116]. Rather, the conjugated α-β unsaturation acts as a Michael acceptor and it reacts with free or protein thiols [13, 49, 116] and thus may act similarly to the cardioprotective compound dimethyl fumarate by activating the downstream Nrf2 antioxidant response [7, 92]. Therefore, a key experiment when assessing any potential cardioprotective compound is to measure the levels of the active compound and succinate to assess the impact. It also remains unknown whether Nrf2 activators themselves affect ischaemic succinate accumulation.
As aspartate can feed into at least two different pathways to produce malate within the cytosol of ischaemic tissue, this presents a novel approach which could be targeted therapeutically. Aminooxyacetic acid (AOA) has been used as a pharmacological inhibitor of aspartate aminotransferase (Fig. 3A) [152]. When AOA was infused in vivo, it was able to reduce the levels of accumulated succinate in ischaemic heart tissue [40]. A similar reduction in ischaemic succinate was seen using AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) as an inhibitor of adenylosuccinate lyase (ASL) (Fig. 3A), thus preventing a route for aspartate conversion into cytosolic malate [40]. Both compounds have also been shown to be cardioprotective when infused prior to ischaemia in Langendorff heart models [46, 152]. Although their complete mechanism of action may be complex, their ability to prevent succinate accumulation and thereby prevent RET may have been the reason. A major limitation of both AOA and AICAR is their lack of specificity, with multiple other targets and pathway interactions [147, 164]. Owing to the lack of selectivity, these two compounds may be less useful pharmacological agents to prevent I/R injury for translation, however, there is the potential that other compounds with transient, selective inhibition of either aspartate transamination or adenylosuccinate lyase could be developed.
As fumarate is considered an important intermediate both in the cytosol and mitochondria for ischaemic succinate accumulation [40, 41], targeting fumarate metabolism may be another option to prevent this. As fumarate hydratase (FH) is responsible for the formation of malate in the cytosol to enable its entry into mitochondria, as well as the subsequent production of fumarate from malate in the mitochondrial matrix, FH inhibitors may serve as a strategy to prevent succinate accumulation. FH knockout hearts are protected against I/R injury, which was attributed to the accumulated fumarate leading to antioxidant upregulation via Nrf2 stabilisation [1, 7]. It may well be that FH KO mouse hearts are protected as the lack of FH activity renders the ischaemic heart tissue to be unable to accumulate succinate, however this is currently unknown. FH can be inhibited by the potent, non-competitive drug FHIN-1, which has shown target engagement in vitro and in vivo [37]. With FH localised to both the cytosol and mitochondrial matrix, the effects of FHIN-1 likely represent inhibiting both enzymes, thus may have some mechanistic utility in understanding ischaemic succinate accumulation (Fig. 3A). However, as FH inhibition may be detrimental to cardiac and immune cell function [7, 73], it remains to be seen whether a potent, competitive inhibitor of FH could be developed to transiently inhibit FH to prevent ischaemic succinate accumulation.
Insights from different models may also help bring about new therapeutic targets to prevent ischaemic succinate accumulation. For example, it was recently found that the cardioprotected cyclophilin D (CyD) knockout mouse hearts do not accumulate succinate during ischaemia [132]. While the chronic loss of CyD is responsible for the lack of ischaemic succinate accumulation [132], it may also be of some merit to explore whether chronic cyclosporine A treatment could similarly prevent succinate accumulation, or whether patients taking cyclosporine A chronically for other conditions have smaller infarcts/improved outcomes after MI. Furthermore, comparing the mammalian heart with other species may provide insights into ischaemic succinate accumulation. Anoxia-tolerant animals provide an interesting contrast to mammals, due to their ability to readily experience long periods of anoxia and reoxygenation, without detrimental effects [18]. The succinate levels in the anoxic hearts of red-eared slider turtles (Trachemys scripta) are orders of magnitude lower than the ischaemic mouse heart, thus succinate does not accumulate to a level capable of driving RET on reoxygenation [26, 27]. Whether the reduced succinate accumulation is solely due to the colder temperatures or reduced metabolic rate and whether the turtle hearts are similarly protected against ischaemia and not just anoxia remains unknown. Consequently, comprehensive profiling of the pathways involved in preventing succinate accumulation in ischaemic/anoxic tissues may be a useful approach to generate novel therapeutic targets.
While preventing succinate accumulation may be an effective method of preventing I/R injury, its clinical relevance, at least in the context of MI, is debatable. To be effective, a compound capable of preventing succinate accumulation would need to be infused prior to the ischaemic event, which in acute MI is likely to be highly unpredictable. Without this knowledge, the therapeutic window of a compound designed to prevent ischaemic succinate accumulation would be missed. Therefore, compounds preventing succinate accumulation may be more applicable when the period of ischaemia is known, such as elective surgery or organ transplantation when the patient or organ could be pre-treated before the ischaemic event [106]. Furthermore, interventions such as cooling reduce the metabolic rate of the tissue, lessening succinate accumulation during ischaemia [106]; however, how this may be achieved in the setting of a MI may be technically challenging.
Succinate oxidation
The action of SDH oxidising succinate is also responsible for the rapid return to normoxic succinate levels upon reperfusion [40, 131, 174]. The electrons from succinate help maintain the reduced CoQ pool and drive RET at complex I, leading to the production of the proximal ROS superoxide and the initiation of the damaging cascade in I/R injury. Therefore, approaches to prevent succinate oxidation during reperfusion may attenuate the downstream superoxide production and signalling cascade, preventing I/R injury. Furthermore, targeting succinate oxidation therapeutically represents a highly clinically relevant point of intervention, as a pharmacological agent could be simply administered at the point of removing the vessel occlusion and creating the opportunistic therapeutic window.
The use of the competitive SDH inhibitor malonate was described above in the context of preventing succinate accumulation with its esterified form DMM. DMM was effective at preventing succinate accumulation, however, when DMM was infused at the point of reperfusion, it was not found to be cardioprotective in a murine MI model [134]. The lack of cardioprotection was because hydrolysis of DMM was too slow to achieve sufficient malonate to perturb succinate oxidation by SDH. The rapidly hydrolysable malonate ester prodrug diacetoxymethyl malonate (MAM) [15, 47, 134] could release malonate far more rapidly into cardiomyocytes than DMM and when administered at reperfusion, sufficient malonate was released to reduce succinate oxidation and elicit protection against cardiac I/R injury in vivo [15, 134]. Therefore, competitive inhibition of SDH is a suitable strategy to inhibit RET in a clinically relevant therapeutic window (Fig. 3B), however, an important point is that the drug must rapidly reach its target site in the active form to be effective.
Intriguingly, it was also shown that malonate as the disodium salt (disodium malonate; DSM) was protective against I/R injury in an ex vivo mouse model and in vivo pig MI model [162, 163]. Malonate could enter cardiomyocytes and inhibit SDH on reperfusion leading to reduced superoxide production and smaller infarct size. Curiously, in normoxic mouse tissue infused with malonate, succinate levels only doubled from baseline and were ~ 7 times lower than those achieved during ischaemia, suggesting little SDH inhibition in normoxic tissue [162]. As malonate carries two negative charges at physiological pH, it suggests that malonate uptake into normoxic cardiomyocytes is inefficient, however, can still lead to SDH inhibition and reduced succinate oxidation in I/R [130, 162, 163]. The paradox of malonate’s low cellular permeability, yet robust cardioprotection was recently discovered to be due to malonate’s preferential uptake into ischaemic tissue [130, 146]. In the ischaemic environment, the pH is significantly more acidic, which facilitates the protonation of one of the carboxylate groups of malonate, generating monocarboxylate malonate [130], enabling it to be a substrate for the plasma membrane transporter monocarboxylate transporter 1 (MCT1) (Fig. 3B) [130]. Therefore, malonate can selectively enter the ischaemic tissue upon reperfusion, sparing the healthy tissue where the pH remains unaltered [130, 162]. Once within the cytosol, malonate is rapidly transported into the mitochondria by the DIC, subsequently inhibiting succinate oxidation, superoxide formation and injury [130, 162, 163]. Furthermore, MCT1-dependent malonate uptake could be harnessed to improve the uptake of malonate into the heart tissue by developing a low pH formulation [130]. The low pH malonate formulation increases the proportion of monocarboxylate malonate and leads to lower doses of malonate required for cardioprotection (Fig. 3B) [130]. Another approach which directly targets malonate to mitochondria is via the use of the well-characterised triphenylphosphonium mitochondria-targeting group [143, 150]. Although this approach has been shown to be able to deliver malonate in vivo, whether the levels would be sufficient to prevent succinate oxidation and therefore I/R injury is unknown [133].
The TCA cycle intermediate oxaloacetate is an even more potent competitive inhibitor of SDH than malonate and thus may be an interesting therapeutic candidate for I/R injury [166]. Interestingly, oxaloacetate can also be oxidised by hydrogen peroxide and converted to malonate but whether this process occurs in vivo is unknown [105]. Oxaloacetate has a very high affinity for SDH, with SDH in isolated systems often initially inactive due to endogenous oxaloacetate being bound [135]. However, the understanding of oxaloacetate in biology and its therapeutic use is limited. This may be due to the rapid, spontaneous decarboxylation of oxaloacetate meaning it has a short biological half-life (~ 10 min) [168], as well as being problematic for measurement by the techniques of traditional liquid chromatography-mass spectrometry metabolomics [61]. On top of this, the transport of oxaloacetate into mammalian mitochondria from the cytosol is slow [126]. Therefore, despite oxaloacetate being a very potent competitive SDH inhibitor, its therapeutic utility may be negligible until there are robust methods to accurately measure oxaloacetate in biological samples.
Targeting SDH using the potent, selective, and non-competitive inhibitor atpenin A5 (AA5) has shown cardioprotection (Fig. 3B) [114, 167]. In the Langendorff-perfused heart, when AA5 was infused at reperfusion, it blunted the burst of superoxide upon reperfusion, leading to reduced infarct size, consistent with AA5 preventing RET by blocking succinate oxidation [114]. Although AA5 may be a viable approach in the isolated heart, like other non-competitive inhibitors of SDH, it is unlikely to be a useful approach due to it chronically inhibiting SDH in vivo.
An additional advantage to the use of malonate over other SDH inhibitors is its favourable pharmacokinetics. Malonate has a short plasma half-life (t1/2 < 40 min, unpublished data), low toxicity and via acidic formulation lower doses can be used to achieve cardioprotection. Malonate is rapidly renally excreted and thereby can limit any off-target effects [145]. Overall, targeting succinate oxidation looks to be a promising approach to targeting RET for cardioprotection.
Succinate transport
As succinate oxidation is an essential driver of RET, altering the supply of succinate independently of SDH inhibition may provide novel therapeutic targets for cardioprotection (Fig. 3). The current model for succinate accumulation suggests that fumarate acts as a terminal electron acceptor within the mitochondria, being reduced into succinate which can be transported into the cytosol to act as an electron sink [41]. As the succinate concentration would be dramatically diluted in the cytosol, this may facilitate succinate accumulating to high levels in ischaemic heart tissue [40, 131]. Upon reperfusion, there are multiple fates for succinate. To drive RET, succinate is rapidly transported into mitochondria, to enable its SDH-mediated oxidation [40, 131]. However, a proportion of succinate can also leave cardiomyocytes, enter the circulation and thus not contribute to fuelling RET on reperfusion [6, 131, 174]. Therefore, altering the compartmentalisation of succinate may present an attractive option to change the dynamics of the accumulated succinate pool towards RET.
Succinate efflux from the ischaemic heart during reperfusion has been shown to occur in a variety of in vitro and in vivo IR/MI models, including ST-elevated MI patients during primary percutaneous coronary intervention [88, 131, 174]. Approximately 50% of the accumulated succinate pool can efflux from the ischaemic heart tissue [131, 174], thus representing a significant mechanism to rewire the fate of succinate destined for oxidation by SDH. Succinate efflux is driven by the lowered pH of ischaemic tissue, which increases the proportion of the protonated succinate monocarboxylate form [6, 131, 139]. Monocarboxylate succinate can act as a substrate for the monocarboxylate transporter 1 (MCT1; SLC16A1), which is highly expressed on the plasma membrane of cardiomyocytes [6, 130, 131]. This provides an outlay for succinate to leave the cardiomyocytes, reducing the amount oxidised by SDH. Despite this efflux of succinate, considerable succinate remains within the cardiomyocytes and is sufficient to drive RET on reperfusion [131]. When MCT1 was selectively inhibited in the Langendorff heart, preventing the release of succinate from the heart on reperfusion, there was an increase in the amount of superoxide produced and damage to the cardiac tissue [114]. This suggests that by blocking succinate efflux from the heart, a greater proportion of the accumulated pool is oxidised by SDH, driving increased RET, superoxide and damage upon reperfusion (Fig. 3B). This result differs from when an MCT1 inhibitor was used in an acute murine MI model, where the inhibitor was found to elicit cardioprotection [131]. This difference may be due to several reasons, including the use of different inhibitors and non-cardiac/off-target effects and the added complexity of the situation in vivo. Nevertheless, both results indicate that targeting cardiomyocyte succinate transport can affect cardioprotection and efforts inhibiting or enhancing this transport may be of use. The impact of other MCT isoforms, genetic variants of MCT1 or MCT1 post-translational modifications on cardioprotection is currently unknown, as well as their expression levels in the heart with comorbidities, which may affect susceptibility to damage.
Succinate compartmentalisation between the cytosol and mitochondria may also play an important role in preventing RET and I/R injury [41]. As succinate from the cytosol fuels RET on reperfusion by re-entering mitochondria, targeting this transport process may be an alternative strategy to disrupt ischaemic succinate accumulation and oxidation on reperfusion (Fig. 3). Succinate transport across the IMM principally occurs via the mitochondrial dicarboxylate carrier (DIC; SLC25A10) [53]. The primarily characterised inhibitor of the DIC is butylmalonate [81], however, its potential use in I/R injury is currently unknown. Butylmalonate is a competitive inhibitor of the DIC, but its low potency means high concentrations are required to achieve adequate DIC inhibition [81, 142]. Furthermore, in the I/R injury environment, butylmalonate must compete with the millimolar levels of succinate within cardiomyocytes [59, 131], acting within the cytosol as it cannot enter mitochondria, therefore its utility may be limited until more potent and selective alternatives are developed.
Modulating succinate transport may therefore be an important target in preventing RET by rerouting succinate away from SDH. However, the consequences of succinate leaving cardiomyocytes and entering the circulation are not fully understood. The succinate receptor (GPR91; SUCNR1) is highly expressed on the surface of inflammatory cells such as macrophages, thus succinate may play a role in inflammation in the later phase of I/R injury [82, 104, 131, 160]. Furthermore, succinate-driven RET is also thought to be a key driver for the production of pro-inflammatory cytokines in macrophages [115]. Therefore, the interplay of interfering with succinate transport may be an interesting double-edged sword on one hand preventing RET in cardiomyocytes but increasing the potential negative effects of extra-cardiomyocyte succinate.
Targeting mitochondrial complex I
As the burst of superoxide upon reperfusion is thought to originate from RET at complex I, complex I remains an important potential target in preventing I/R injury (Figs. 2 and 4) [39, 40]. Additionally, complex I can adopt a catalytically active or deactive state which is important in I/R injury (Fig. 4A) [8, 28]. In conditions where substrate turnover is limited, such as ischaemia, complex I can undergo what is called an active/deactive transition which has been historically defined by the exposure of a solvent-accessible cysteine residue Cys39 on the ND3 subunit of complex I (Fig. 4A) [2]. Alkylation of exposed Cys39 in the deactive state renders complex I catalytically inactive, even when the conditions favouring substrate oxidation return [2, 8, 28]. In this deactive state, complex I cannot perform RET thus transiently keeping complex I in its deactive state during early reperfusion may be a highly targeted method of cardioprotection (Fig. 4A) [28, 45]. This is exemplified by the ND6-P25L mouse where ND6-P25L complex I cannot perform RET but oxidises NADH in the forward catalytic state the same as wildtype [103, 108, 173]. ND6-P25L complex I collapses rapidly into the deactive state due to the mutation creating a disordered Q-site [173]. While forward electron transport can efficiently revert ND6-P25L complex I to the active state, thus catalysing NADH oxidation efficiently, RET conditions cannot consequently complex I remains in its deactive state and RET does not occur [173]. Consistent with RET being a key driver of I/R injury, ND6-P25L mouse hearts are profoundly protected against cardiac I/R injury in vivo, despite all the drivers of RET being present [173].

Targeting complex I to inhibit RET. A Modulating complex I active/deactive transition. During ischaemia, complex I transitions to a deactive conformation, leading to ND3 cys39 becoming exposed (Cys39-SH). Upon reperfusion, complex I rapidly reactivates, producing a burst of superoxide by RET. Cys39 can be transiently modified by MitoSNO, locking complex I in its deactive state and preventing superoxide production by RET. The modification will be removed from Cys39 by the glutathione or thioredoxin systems, with complex I slowly returning back to its active state. It is also hypothesised that other cysteine modifying agents such as H2S, H2O2 and NaNO2 may act by modifying Cys39 to prevent RET. B The inhibitor of complex I rotenone has been shown to inhibit superoxide production by preventing RET and can lead to a protective effect. Furthermore, it is hypothesised that other complex I inhibitors such as piericidin, amobarbital and metformin may provide similar cardioprotective effects by preventing RET on reperfusion. IMS intermembrane space, IMM inner mitochondrial membrane, SDH succinate dehydrogenase, FMN flavin mononucleotide, Δp protonmotive force, cyt c cytochrome c, O2●− superoxide
Compounds capable of transiently modifying Cys39 of ND3 in complex I are attractive strategies to prevent RET and I/R injury. Several compounds that have shown cardioprotection have the potential to modify cysteines, thus there is the possibility that these act by transiently modifying Cys39 on ND3 of complex I to prevent RET (Fig. 4A). An example of this is the mitochondria-targeted S-nitrosothiol MitoSNO which has shown protection in vivo against acute cardiac I/R injury [39, 136] and post-MI heart failure [110]. Mechanistically, MitoSNO has been shown to be able to S-nitrosate the ND3 Cys39 on complex I when administered at reperfusion, locking complex I in its deactive state, preventing RET and affording protection (Fig. 4A) [39]. The modification of Cys39 is transient, with the glutathione and thioredoxin systems capable of reducing the cysteine back to its unmodified form, enabling the complex to switch back to its active form (Fig. 4A) [39, 136]. Similar mechanisms of cysteine modification and RET prevention may be part of the mechanism of protection from several agents that have been previously shown to be cardioprotective including hydrogen sulphide [50], hydrogen peroxide [57], sodium nitrite [148] and other S-nitrosothiols (Fig. 4A) [122]. Furthermore, the active/deactive transition may be relevant in the cardioprotection gained by preconditioning, where multiple rounds of ischaemia and reperfusion sequentially increase the proportion of transiently deactive complex I [90].
The recent finding that complex I ND3 Cys39 can be exposed during active catalysis but labelling has no impact on catalysis raises further questions surrounding molecular characterisation of the active/deactive transition [28]. This suggests that strategies employed to selectively modify deactive complex I may also hit active complex I but not affect its catalytic activity. The subtleties of this remain unknown, however, the significance of active complex I modified ND3 Cys39 in cardioprotection may have implications for RET, cardioprotection and more chronic effects of complex I on pathology. Furthermore, as shown with the recently developed mitochondria-targeted hydrogen sulphide donor MitoPerSulf, it may also be that in part, like hydrogen sulphide, MitoSNO may also transiently inhibit complex IV on reperfusion, reducing respiration and thereby reducing RET-derived superoxide at complex I [111].
As RET requires the flow of electrons from the CoQ pool backwards through complex I, it suggests that strategies to inhibit complex I may be cardioprotective (Fig. 4B). The use of complex I inhibitors, such as the potent and selective inhibitor rotenone, helped distinguish RET as an important mechanism in I/R injury [40, 100]. Under conditions of forward electron transport, rotenone leads to enhanced superoxide production by preventing electrons from NADH reducing the CoQ pool, rather the FMN in complex I will be fully reduced which subsequently reacts with oxygen [121]. However, paradoxically, complex I inhibition prevents superoxide production by RET [121, 137, 169]. Rotenone has been shown to inhibit RET and reduce superoxide production upon reperfusion and therefore may contribute to the mitoprotective effects during ischaemia seen in numerous studies (Fig. 4B) [35, 39, 40, 100, 101], though there is little evidence of rotenone providing cardioprotection after reperfusion ex vivo or in vivo. The reversible, short-acting barbiturate amobarbital can transiently inhibit complex I, thought to be by binding to the same site as rotenone, leading to a significant reduction in infarct size and reduced ROS production on reperfusion [4, 34, 36, 100]. As the amobarbital was added prior to ischaemia, whether this protective effect is by inhibiting RET on reperfusion, or alternative effects during ischaemia is unknown. Furthermore, complex I inhibition has also been shown to prevent succinate accumulation in isolated mitochondria under pseudo-ischaemic conditions (Fig. 3A) [174]. As electrons from NADH feed into the CoQ pool via complex I during ischaemia and can be used to reduce fumarate to succinate, inhibiting complex I with rotenone may prevent this by attenuating succinate accumulation. Therefore, rotenone may play a dual role in cardioprotection, however, the prevention of succinate accumulation by complex I inhibition in tissues and in vivo is unclear.
A limitation of many of the complex I inhibitors is their dose-limiting toxicities [77, 172, 175]. As complex I inhibitors often bind non-reversibly to the Q-site of complex I, their hydrophobicity increases membrane permeability and potency, enhancing off-target effects [172, 175]. As such, the translatability of such compounds has recently been questioned after unfavourable human trials [17, 77, 172]. Biguanides, such as metformin, can inhibit complex I [24, 25] and intriguingly prefer binding complex I in its deactive state when tested in vitro [107]. Metformin has shown protection against acute cardiac I/R injury in MI when high doses were administered at reperfusion [14, 125]. The mechanism of protection by metformin is likely to be highly complex due to its utility in many pathways, however, if metformin can inhibit complex I, inhibiting RET may be part of this cardioprotective mechanism. The current evidence for metformin inhibiting complex I in vivo is limited [68, 157]. As metformin is a hydrophilic, positively charged compound and requires high concentrations to achieve substantial complex I inhibition [24, 25], there are many barriers limiting its effectiveness [54]. While the organic cation transporter 1 (OCT1) that transports metformin is present on the surface of cardiomyocytes [58], how and the rate at which metformin enters mitochondria is unknown [54]. Therefore, despite metformin representing a hydrophilic, highly tolerated and clinically approved drug, many unknowns of the action of metformin on mitochondria exist.
Overall, complex I may be an important druggable target to prevent RET for cardioprotection. As complex I is important for cardiac mitochondrial function, interventions must be transient to enable inhibition of RET on reperfusion, yet subsequently allow for normal respiration. Targeting RET selectively, such as transiently modifying ND3 Cys39 in deactive complex I is an attractive prospect, with endogenous mechanisms to reverse the modification and relieve complex I inhibition. However, it remains to be seen whether these compounds have detrimental off-target cysteine modifications and their translatability in large animal models and humans.
Manipulating the mitochondrial membrane potential
A large Δp across the mitochondrial inner membrane is important for superoxide production via RET. As mitochondrial membrane potential (Δψ) is a large component of Δp (Δp = Δψ—61ΔpH [124]), methods which can reduce mitochondrial membrane potential during early reperfusion may be protective against I/R injury (Fig. 5). By interfering with the mitochondrial membrane potential and short-circuiting the ETC, by providing a route for protons to re-enter the mitochondrial matrix, this leads to oxidation of the CoQ pool, thus nullifying another driver of RET.

Manipulating mitochondrial membrane potential to prevent RET. Uncoupling the mitochondrial membrane potential with uncouplers FCCP and 2,4-DNP can prevent superoxide production by RET in I/R injury. By short-circuiting the IMM, protons flow from the IMS to the matrix and therefore reduce the Δψ. As Δψ is a large proportion of Δp, (Δp = Δψ – 61ΔpH; given that Δp ~ 180 mV and ΔpH ~ 0.5 U) small changes in Δψ will significantly affect Δp and thereby the driving force for RET to occur. It is also hypothesised that BAM15 may be able to similarly prevent RET from occurring due to its selective uncoupling of the inner mitochondrial membrane. IMS intermembrane space, IMM inner mitochondrial membrane, SDH succinate dehydrogenase, FMN flavin mononucleotide, Δp protonmotive force, Δψ mitochondrial membrane potential, cyt c cytochrome c, O2●− superoxide
The importance of mitochondrial membrane potential in I/R injury is highlighted by studies showing cardioprotection dependent on uncoupling protein 1 (UCP1) [71, 74]. UCP2 and UCP3 have been implicated in protection against cardiac I/R injury, however, their mechanism of action is independent of mitochondrial membrane potential uncoupling [30]. Several previous studies have shown the cardioprotective effects of pharmacological uncouplers, however, their mechanisms of action were determined to be due to impacting mitochondrial membrane potential-dependent ion channel function, rather than electron transport and the CoQ pool directly [22, 23, 30]. FCCP was shown to be cardioprotective and mechanistically initially was thought to afford protection like preconditioning by affecting the mitochondrial KATP channel [30]. Low doses of FCCP infused into isolated hearts or treatment of ventricular cardiomyocytes were cardioprotective, independent of effects on KATP channel activation [23]. However, these effects were also independent of membrane potential changes detected by TMRM, though the dynamic range of TMRM may not have been sufficient to detect the membrane potential changes [22]. Furthermore, when isolated rat hearts were reperfused with 2,4-dinitrophenol (DNP), this led to a presumed block in mitochondrial calcium entry and cardioprotective effect [51]. As RET is highly sensitive to small membrane potential changes, it is likely that even these concentrations of uncoupler RET would be affected and may be responsible for the cardioprotective effects seen (Fig. 5) [117].
A key limitation of the ‘classical’ mitochondrial uncouplers is their lack of discrimination for mitochondrial membranes leading to uncoupling of the plasma membrane as well. Impacting the plasma membrane proton gradient may have a multitude of effects, including affecting lactate transport by MCT1 which may provide a secondary mechanism of action of these uncouplers in cardioprotection. Furthermore, as pH plays a critical role in opening of the mPTP, alteration of mitochondrial and cytosolic proton flux may play a key part in the protective mechanism independently of the effects on the respiratory chain. The recently developed mitochondria-membrane selective uncoupler BAM15 has shown potent uncoupling activity, without changes in plasma membrane ion movement [83]. The mechanism of the selectivity of BAM15 for the mitochondrial inner membrane is unknown, however, it has been postulated that the lower mitochondrial pH or difference in lipid composition may be involved [83]. The BAM15 has shown a multitude of beneficial effects in various disease models including renal I/R injury [83], sepsis [161] and diet-induced obesity [5], thus it may have utility against cardiac I/R injury.
While a limitation of the use of uncouplers has been their questionable safety profile, with DNP leading to several fatalities [127], the toxicity of these compounds may be somewhat mitigated in their use against cardiac I/R injury due to their single use. Also, liver-targeted and controlled-release formulations of DNP have significantly reduced toxicity [127, 128]. Low doses of selective uncouplers at reperfusion may transiently depolarise mitochondria and prevent RET on reperfusion without long-term undue toxicity.
Adenine nucleotide pool degradation
It was previously thought that ATP is rapidly depleted during ischaemia, leading to a large pool of ADP readily available for ATP synthesis upon reperfusion [11, 84]. More recently, it has become apparent that during ischaemia adenylate kinase catalyses the conversion of 2 ADP to ATP and AMP, thus maintaining a source of ATP in non-oxidative conditions [40, 79, 106]. This leads to elevated AMP, which is consequently degraded, with large elevations in xanthine and hypoxanthine as remnants from their adenine nucleotide past (Fig. 6) [40, 106]. The depletion of the ADP pool is an important aspect of the ability of mitochondria to produce superoxide via RET. A depleted ADP pool in ischaemic tissue reduces the ADP available for phosphorylation upon reperfusion, thereby preventing the Δp from being used for ATP synthesis [121]. High mitochondrial Δp is a prerequisite for RET to occur, thus using the Δp for ATP synthesis reduces its magnitude across the IMM and prevents RET. This has been shown experimentally in isolated heart mitochondria respiring on succinate, where the addition of ADP reduces the levels of RET-derived H2O2 [113]. Therefore, preventing the depletion of the adenine nucleotide pools to sustain ADP levels may be another strategy for preventing RET.

Adenine nucleotide pool degradation targeting to prevent RET. Preventing adenine nucleotide pool degradation, may prevent RET by a dual mechanism. Firstly, as shown with AICAR inhibition of ASL [40] (or hypothesised by FH inhibition by FHIN-1 [155] or ASS1 inhibition by MDLA [75]), targeting this pathway may prevent succinate accumulation during ischaemia, thus prevent succinate oxidation driving RET on reperfusion. Otherwise, it may be hypothesised that inhibitors such as Ap5A [96] which inhibits ADK may prevent ADP breakdown to AMP, thus maintaining ADP levels to allow ATP synthesis using the Δp and thus preventing RET. Other hypothesised targets and inhibitors of the adenine nucleotide degradation pathway include: AMP deamination to IMP (AMPD1 inhibition by Cpd3 [75]), IMP hydrolysis to inosine (nucleotidase inhibition by IBTI [149]), hydrolysis of inosine to hypoxanthine (PNP inhibition by 8-aminoguanine [19] or forodesine [12]) or hypoxanthine oxidation to xanthine and subsequently to uric acid (XO inhibited by allopurinol [85]). These interventions may be hypothesised to maintain the adenine nucleotide pool intermediates and expedite resynthesis of the adenine nucleotide pool. ASL adenylosuccinate lyase, ASS1 adenylosuccinate synthase 1, MDLA α-methyl-DL-aspartic acid, ADK adenylate kinase, IMP inosine monophosphate, AMPD1 AMP deaminase 1, PNP purine nucleoside phosphorylase, XO xanthine oxidase
Several reports have suggested the cardioprotective effects of kynurenic acid, however, the mechanism of protection has been poorly understood [109, 140, 170]. Recently, it has been proposed that a key part of the protection of kynurenic acid is through salvaging the ATP pool, preventing ATP hydrolysis by reverse action of ATP synthase, during ischaemia [170]. Here, kynurenic acid is thought to act as a ligand of GPR35 on the plasma membrane [112], with the receptor subsequently internalised and trafficked to the mitochondrial outer membrane [170]. The internalised GPR35 is postulated to be able to inhibit mitochondrial adenylate cyclase, prevent protein kinase A phosphorylation of ATP synthase inhibitory factor subunit 1 (ATPIF1) and thereby promote ATP synthase dimerisation, preventing ATP hydrolysis [170]. The details of how GPR35 enacts its effects at the level of the mitochondrial matrix, despite binding at the outer mitochondrial membrane remain incomplete. Further investigation into whether activation of GRP35 prevents degradation of the adenine nucleotide pool, and not just ATP, is warranted as this aspect remains unclear. If degradation of the adenine nucleotide pool is prevented, this may lead to increased ADP levels in the ischaemic tissue, which may provide a rapid means to reduce the Δp via its phosphorylation by ATP synthase and prevent RET in early reperfusion. Despite this, the actions of kynurenic acid on the ATP pool suggest that strategies to prevent ATP hydrolysis and subsequent adenine nucleotide pool degradation may be effective at preventing cardiac I/R injury.
Many studies have interfered with the adenine nucleotide degradation pathway genetically and have shown their impact on cardioprotection [20, 62, 72, 85, 138]. However, historically these often have used conventional knockout of the gene from conception in the mice, which may lead to the upregulation of compensatory pathways to maintain an appropriate ATP/ADP ratio and thus complicate the interpretation and relevance. Several drugs targeting enzymes within the adenine nucleotide degradation pathway show protection against cardiac I/R injury [40, 41, 85, 138]. Their cardioprotective effects have mostly been attributed to their prevention of oxidative stress, such as with allopurinol inhibiting xanthine oxidase superoxide production [85], however, an alternative mechanism could be via their interference with adenine nucleotide breakdown [20, 85]. Preventing the complete breakdown of adenine nucleotides during ischaemia may facilitate their salvage and synthesis into adenine nucleotides upon reperfusion. By more rapidly re-synthesising the adenine nucleotide pools, this may again lead to utilising the Δp and preventing RET. Figure 6 highlights pharmacological inhibitors of the adenine nucleotide degradation pathway, which may be hypothesised to affect RET. Despite the potential to prevent the complete breakdown of the nucleotide pool, whether this impacts RET directly remains unknown. Also, the cardioprotection by compounds targeting certain enzymes of the adenine nucleotide degradation pathway is poorly characterised, thus further work may be required in this area to understand its relevance. Another caveat to targeting adenine nucleotide degradation is the requirement for treatment prior to the onset of ischaemia, therefore even if interventions may be able to lead to conditions able to prevent RET, they are less likely to be suitable for acute MI.
Furthermore, targeting adenine nucleotide pool degradation may also impact the accumulation of succinate, considering it is thought to be an anaplerotic source of substrate for SDH reversal to produce succinate (Fig. 6) [40, 41]. The rerouting and regeneration of AMP in the PNC leads to the production of fumarate via ASL, as detailed above (see Sect. "Succinate accumulation" and Fig. 3A). This fumarate is converted to malate by fumarate hydratase, which can be transported into mitochondria and act as a precursor to produce succinate during ischaemia. Therefore, affecting the pool of adenine nucleotides may subsequently affect the flux through the PNC and subsequent production of succinate, via the PNC-derived fumarate.
The degradation of adenine nucleotide pools is a hallmark of ischaemia and may impact both the accumulation of succinate and the conditions required to drive RET. Little direct evidence exists as to whether interventions at the nucleotide pool level alter succinate accumulation or can prevent RET, thus future insights into the usefulness of targeting this pathway may be warranted.
Conclusions and future perspectives
Overall, RET-derived superoxide upon reperfusion is an important initiator of the damage in I/R injury. The extreme conditions of ischaemia remodel cardiac metabolism and lead to the environment satisfying the thermodynamic requirements for RET to occur on reperfusion. Therefore, targeting the pathways which enable the conditions for RET to occur may be an effective strategy to minimise cardiac I/R injury (Table 1). Furthermore, due to the far-reaching changes in metabolism in ischaemia which help support RET upon reperfusion, it may be that several drugs historically found to be cardioprotective may have acted by affecting succinate accumulation or preventing RET-derived superoxide production. Currently, targeting succinate oxidation with malonate seems to have the greatest experimental evidence, with protection in small and large animal models and ischaemia-selective properties. Malonate has also shown effectiveness in resuscitation [171], ischaemic stroke [118] and cardiac regeneration [9] and thus may help break the cycle of failures in preventing cardiac I/R injury. Despite this, history tells us that many further studies are required for successful translation from the bench to the clinic in this field [21, 65, 99, 146]. Currently, there is no data surrounding the use of malonate in human myocardial tissue and whether the same mechanisms from animal models are applicable [48, 86, 87]. Furthermore, there is a lack of evidence of systemic malonate administration and cardioprotection in large animal models [86]. Also, how malonate may affect the normal physiology of conscious large animals needs to be addressed [99]. Aside from malonate, many questions remain for all these therapeutic approaches, in particular: is targeting RET effective in conjunction with comorbidities, are there any gender differences in the protection, are these interventions still effective in aged animals and how does inhibiting RET translate to long-term outcomes? Even though many questions remain open, the orthogonal approaches targeting RET and leading to cardioprotection are promising that RET is a worthwhile target to prevent cardiac I/R injury.
Data availability
There is no data associated with this manuscript.
References
Adam J, Yang M, Bauerschmidt C, Kitagawa M, O’Flaherty L, Maheswaran P, Özkan G, Sahgal N, Baban D, Kato K, Saito K, Iino K, Igarashi K, Stratford M, Pugh C, Tennant DA, Ludwig C, Davies B, Ratcliffe PJ, El-Bahrawy M, Ashrafian H, Soga T, Pollard PJ (2013) A role for cytosolic fumarate hydratase in urea cycle metabolism and renal neoplasia. Cell Rep 3:1440–1448. https://doi.org/10.1016/j.celrep.2013.04.006
Agip AA, Blaza JN, Bridges HR, Viscomi C, Rawson S, Muench SP, Hirst J (2018) Mitochondria in two biochemically defined states. Nat Struct Mol Biol 25:1–12. https://doi.org/10.1038/s41594-018-0073-1
Agip A-NA, Blaza JN, Fedor JG, Hirst J (2019) Mammalian respiratory complex I through the lens of cryo-EM. Annu Rev Biophys 48:165–184. https://doi.org/10.1146/annurev-biophys-052118-115704
Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AKS (2008) Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc Res 77:406–415. https://doi.org/10.1016/j.cardiores.2007.08.008
Alexopoulos SJ, Chen S-Y, Brandon AE, Salamoun JM, Byrne FL, Garcia CJ, Beretta M, Olzomer EM, Shah DP, Philp AM, Hargett SR, Lawrence RT, Lee B, Sligar J, Carrive P, Tucker SP, Philp A, Lackner C, Turner N, Cooney GJ, Santos WL, Hoehn KL (2020) Mitochondrial uncoupler BAM15 reverses diet-induced obesity and insulin resistance in mice. Nat Commun 11:2397. https://doi.org/10.1038/s41467-020-16298-2
Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, Halestrap AP (2017) The role of succinate and ROS in reperfusion injury—A critical appraisal. J Mol Cell Cardiol 110:1–4
Ashrafian H, Czibik G, Bellahcene M, Aksentijević D, Smith AC, Mitchell SJ, Dodd MS, Kirwan J, Byrne JJ, Ludwig C, Isackson H, Yavari A, Støttrup NB, Contractor H, Cahill TJ, Sahgal N, Ball DR, Birkler RID, Hargreaves I, Tennant DA, Land J, Lygate CA, Johannsen M, Kharbanda RK, Neubauer S, Redwood C, de Cabo R, Ahmet I, Talan M, Günther UL, Robinson AJ, Viant MR, Pollard PJ, Tyler DJ, Watkins H (2012) Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab 15:361–371. https://doi.org/10.1016/j.cmet.2012.01.017
Babot M, Birch A, Labarbuta P, Galkin A (2014) Characterisation of the active/de-active transition of mitochondrial complex I. Biochimica et Biophysica Acta (BBA) 1837:1083–1092. https://doi.org/10.1016/j.bbabio.2014.02.018
Bae J, Salamon RJ, Brandt EB, Paltzer WG, Zhang Z, Britt EC, Hacker TA, Fan J, Mahmoud AI (2021) Malonate promotes adult cardiomyocyte proliferation and heart regeneration. Circulation 143:1973–1986. https://doi.org/10.1161/CIRCULATIONAHA.120.049952
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662. https://doi.org/10.1038/nature03434
Bak MI, Ingwall JS (1994) Acidosis during ischemia promotes adenosine triphosphate resynthesis in postischemic rat heart. In vivo regulation of 5’-nucleotidase. J Clin Investig 93:40–49. https://doi.org/10.1172/JCI116974
Balakrishnan K, Nimmanapalli R, Ravandi F, Keating MJ, Gandhi V (2006) Forodesine, an inhibitor of purine nucleoside phosphorylase, induces apoptosis in chronic lymphocytic leukemia cells. Blood 108:2392–2398. https://doi.org/10.1182/blood-2006-03-007468
Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang L-H, Duncan D, Bregman H, Keskin A, Santeford A, Apte RS, Sehgal R, Johnson B, Amarasinghe GK, Soares MP, Satoh T, Akira S, Hai T, de Guzman SC, Auclair K, Roddy TP, Biller SA, Jovanovic M, Klechevsky E, Stewart KM, Randolph GJ, Artyomov MN (2018) Electrophilic properties of itaconate and derivatives regulate the IκBζ–ATF3 inflammatory axis. Nature 556:501–504. https://doi.org/10.1038/s41586-018-0052-z
Bates L, Krause-Hauch M, Wang H, Fatmi MK, Li Z, Chen Q, Ren D, Li J, Lesnefsky EJ (2023) Acute, high dose metformin therapy at reperfusion decreases infarct size in the high-risk aging heart. Aging Dis. https://doi.org/10.14336/AD.2023.0205
Beach TE, Prag HA, Pala L, Logan A, Huang MM, Gruszczyk AV, Martin JL, Mahbubani K, Hamed MO, Hosgood SA, Nicholson ML, James AM, Hartley RC, Murphy MP, Saeb-Parsy K (2020) Targeting succinate dehydrogenase with malonate ester prodrugs decreases renal ischemia reperfusion injury. Redox Biol 36:101640. https://doi.org/10.1016/j.redox.2020.101640
Bernardi P, Di Lisa F (2015) The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78:100–106. https://doi.org/10.1016/j.yjmcc.2014.09.023
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306. https://doi.org/10.1038/81834
Bickler PE, Buck LT (2007) Hypoxia tolerance in reptiles, amphibians, and fishes: life with variable oxygen availability. Annu Rev Physiol 69:145–170. https://doi.org/10.1146/annurev.physiol.69.031905.162529
Birder LA, Wolf-Johnston A, Wein AJ, Cheng F, Grove-Sullivan M, Kanai AJ, Watson AM, Stoltz D, Watkins SC, Robertson AM, Newman D, Dmochowski RR, Jackson EK (2020) Purine nucleoside phosphorylase inhibition ameliorates age-associated lower urinary tract dysfunctions. JCI Insight. https://doi.org/10.1172/jci.insight.140109
Borkowski T, Lipinski M, Kaminski R, Krzyminska-Stasiuk E, Langowska M, Raczak G, Slominska EM, Smolenski RT (2008) Modulation of AMP deaminase in rat hearts subjected to ischemia and reperfusion by purine riboside. Nucleosides Nucleotides Nucleic Acids 27:876–880. https://doi.org/10.1080/15257770802146551
Bøtker HE, Cabrera-Fuentes HA, Ruiz-Meana M, Heusch G, Ovize M (2020) Translational issues for mitoprotective agents as adjunct to reperfusion therapy in patients with ST-segment elevation myocardial infarction. J Cell Mol Med 24:2717–2729. https://doi.org/10.1111/jcmm.14953
Brennan J, Berry R, Baghai M, Duchen M, Shattock M (2006) FCCP is cardioprotective at concentrations that cause mitochondrial oxidation without detectable depolarisation. Cardiovasc Res 72:322–330. https://doi.org/10.1016/j.cardiores.2006.08.006
Brennan J, Southworth R, Medina R, Davidson S, Duchen M, Shattock M (2006) Mitochondrial uncoupling, with low concentration FCCP, induces ROS-dependent cardioprotection independent of KATP channel activation. Cardiovasc Res 72:313–321. https://doi.org/10.1016/j.cardiores.2006.07.019
Bridges HR, Blaza JN, Yin Z, Chung I, Pollak MN (1979) Hirst J (2023) Structural basis of mammalian respiratory complex I inhibition by medicinal biguanides. Science 379:351–357. https://doi.org/10.1126/science.ade3332
Bridges HR, Jones AJY, Pollak MN, Hirst J (2014) Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J 462:475–487. https://doi.org/10.1042/BJ20140620
Bundgaard A, Gruszczyk AV, Prag HA, Williams C, McIntyre A, Ruhr IM, James AM, Galli GLJ, Murphy MP, Fago A (2023) Low production of mitochondrial reactive oxygen species after anoxia and reoxygenation in turtle hearts. J Exp Biol. https://doi.org/10.1242/jeb.245516
Bundgaard A, James AM, Gruszczyk AV, Martin J, Murphy MP, Fago A (2019) Metabolic adaptations during extreme anoxia in the turtle heart and their implications for ischemia−reperfusion injury. Sci Rep 9:2850. https://doi.org/10.1038/s41598-019-39836-5
Burger N, James AM, Mulvey JF, Hoogewijs K, Ding S, Fearnley IM, Loureiro-López M, Norman AAI, Arndt S, Mottahedin A, Sauchanka O, Hartley RC, Krieg T, Murphy MP (2022) ND3 Cys39 in complex I is exposed during mitochondrial respiration. Cell Chem Biol 29:636-649.e14. https://doi.org/10.1016/j.chembiol.2021.10.010
Cadenas E, Boveris A, Ragan CI, Stoppani AOM (1977) Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys 180:248–257. https://doi.org/10.1016/0003-9861(77)90035-2
Cadenas S (2018) Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim Biophys Acta (BBA) - Bioenerg 1859:940–950. https://doi.org/10.1016/j.bbabio.2018.05.019
Cai W, Liu L, Shi X, Liu Y, Wang J, Fang X, Chen Z, Ai D, Zhu Y, Zhang X (2023) Alox15/15-HpETE aggravates myocardial ischemia−reperfusion injury by promoting cardiomyocyte ferroptosis. Circulation 147:1444–1460. https://doi.org/10.1161/CIRCULATIONAHA.122.060257
Chance B, Hollunger G (1961) The interaction of energy and electron transfer reactions in mitochondria. I. General properties and nature of the products of succinate-linked reduction of pyridine nucleotide. J Biol Chem 236:1534–1543
Chandel NS (2021) Signaling and metabolism. Cold Spring Harb Perspect Biol 13:a040600. https://doi.org/10.1101/cshperspect.a040600
Chen Q (2005) Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J Pharmacol Exp Ther 316:200–207. https://doi.org/10.1124/jpet.105.091702
Chen Q, Camara AKS, Stowe DF, Hoppel CL, Lesnefsky EJ (2007) Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol 292:C137–C147. https://doi.org/10.1152/ajpcell.00270.2006
Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ (2006) Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J Pharmacol Exp Ther 319:1405–1412. https://doi.org/10.1124/jpet.106.110262
Cheng J, Liu Y, Yan J, Zhao L, Zhou Y, Shen X, Chen Y, Chen Y, Meng X, Zhang X, Jiang P (2022) Fumarate suppresses B-cell activation and function through direct inactivation of LYN. Nat Chem Biol 18:954–962. https://doi.org/10.1038/s41589-022-01052-0
Chinopoulos C (2019) Succinate in ischemia: where does it come from? Int J Biochem Cell Biol 115:105580. https://doi.org/10.1016/j.biocel.2019.105580
Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, Ding S, James AM, Cochemé HM, Reinhold J, Lilley KS, Partridge L, Fearnley IM, Robinson AJ, Hartley RC, Smith RAJ, Krieg T, Brookes PS, Murphy MP (2013) Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat Med 19:753–759. https://doi.org/10.1038/nm.3212
Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. https://doi.org/10.1038/nature13909
Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP (2016) A unifying mechanism for mitochondrial superoxide production during ischemia−reperfusion injury. Cell Metab 23:254–263. https://doi.org/10.1016/j.cmet.2015.12.009
Davidson SM, Adameová A, Barile L, Cabrera-Fuentes HA, Lazou A, Pagliaro P, Stensløkken K-O, Garcia-Dorado D (2020) Mitochondrial and mitochondrial-independent pathways of myocardial cell death during ischaemia and reperfusion injury. J Cell Mol Med 24:3795–3806. https://doi.org/10.1111/jcmm.15127
Davidson SM, Ferdinandy P, Andreadou I, Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM, Hausenloy DJ, Garcia-Dorado D (2019) Multitarget strategies to reduce myocardial ischemia/reperfusion injury. J Am Coll Cardiol 73:89–99. https://doi.org/10.1016/j.jacc.2018.09.086
Drake KJ, Sidorov VY, Mcguinness OP, Wasserman DH, Wikswo JP (2012) Amino acids as metabolic substrates during cardiac ischemia. Exp Biol Med 237:1369–1378. https://doi.org/10.1258/ebm.2012.012025
Dröse S, Stepanova A, Galkin A (2016) Ischemic A/D transition of mitochondrial complex I and its role in ROS generation. Biochim Biophys Acta (BBA) - Bioenerg 1857:946–957. https://doi.org/10.1016/j.bbabio.2015.12.013
Du J, Li H, Song J, Wang T, Dong Y, Zhan A, Li Y, Liang G (2022) AMPK activation alleviates myocardial ischemia−reperfusion injury by regulating Drp1-mediated mitochondrial dynamics. Front Pharmacol. https://doi.org/10.3389/fphar.2022.862204
Ehinger JK, Piel S, Ford R, Karlsson M, Sjövall F, Frostner EÅ, Morota S, Taylor RW, Turnbull DM, Cornell C, Moss SJ, Metzsch C, Hansson MJ, Fliri H, Elmér E (2016) Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat Commun 7:12317. https://doi.org/10.1038/ncomms12317
Eickelmann C, Lieder HR, Shehada S-E, Thielmann M, Heusch G, Kleinbongard P (2023) Mitochondrial respiration analysis in permeabilized porcine left ventricular and human right atrial specimens with ischemia−reperfusion. Am J Physiol-Heart Circ Physiol 325:H125–H135. https://doi.org/10.1152/ajpheart.00172.2023
ElAzzouny M, Tom CTMB, Evans CR, Olson LL, Tanga MJ, Gallagher KA, Martin BR, Burant CF (2017) Dimethyl itaconate is not metabolized into itaconate intracellularly. J Biol Chem 292:4766–4769. https://doi.org/10.1074/jbc.C117.775270
Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow C-W, Lefer DJ (2007) Hydrogen sulfide attenuates myocardial ischemia−reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci 104:15560–15565. https://doi.org/10.1073/pnas.0705891104
Elz JS, Nayler WG (1988) Calcium gain during postischemic reperfusion. The effect of 2,4-dinitrophenol. Am J Pathol 131:137–145
Ferdinandy P, Andreadou I, Baxter GF, Bøtker HE, Davidson SM, Dobrev D, Gersh BJ, Heusch G, Lecour S, Ruiz-Meana M, Zuurbier CJ, Hausenloy DJ, Schulz R (2023) Interaction of cardiovascular nonmodifiable risk factors, comorbidities and comedications with ischemia/reperfusion injury and cardioprotection by pharmacological treatments and ischemic conditioning. Pharmacol Rev 75:159–216. https://doi.org/10.1124/pharmrev.121.000348
Fiermonte G, Palmieri L, Dolce V, Lasorsa FM, Palmieri F, Runswick MJ, Walker JE (1998) The sequence, bacterial expression, and functional reconstitution of the rat mitochondrial dicarboxylate transporter cloned via distant homologs in yeast and Caenorhabditis elegans. J Biol Chem 273:24754–24759. https://doi.org/10.1074/jbc.273.38.24754
Fontaine E (2018) Metformin-induced mitochondrial complex I inhibition: facts, uncertainties, and consequences. Front Endocrinol (Lausanne). https://doi.org/10.3389/fendo.2018.00753
Frezza C (2017) Mitochondrial metabolites: undercover signalling molecules. Interface Focus 7:20160100. https://doi.org/10.1098/rsfs.2016.0100
Garlick PB, Davies MJ, Hearse DJ, Slater TF (1987) Direct detection of free radicals in the reperfused rat heart using electron spin resonance spectroscopy. Circ Res 61:757–760. https://doi.org/10.1161/01.RES.61.5.757
Gorenkova N, Robinson E, Grieve DJ, Galkin A (2013) Conformational change of mitochondrial complex I increases ROS sensitivity during ischemia. Antioxid Redox Signal 19:1459–1468. https://doi.org/10.1089/ars.2012.4698
Grube M, Ameling S, Noutsias M, Köck K, Triebel I, Bonitz K, Meissner K, Jedlitschky G, Herda LR, Reinthaler M, Rohde M, Hoffmann W, Kühl U, Schultheiss H-P, Völker U, Felix SB, Klingel K, Kandolf R, Kroemer HK (2011) Selective regulation of cardiac organic cation transporter novel type 2 (OCTN2) in dilated cardiomyopathy. Am J Pathol 178:2547–2559. https://doi.org/10.1016/j.ajpath.2011.02.020
Gruszczyk AV, Casey AM, James AM, Prag HA, Burger N, Bates GR, Hall AR, Allen FM, Krieg T, Saeb-Parsy K, Murphy MP (2022) Mitochondrial metabolism and bioenergetic function in an anoxic isolated adult mouse cardiomyocyte model of in vivo cardiac ischemia−reperfusion injury. Redox Biol 54:102368. https://doi.org/10.1016/j.redox.2022.102368
Hahn VS, Petucci C, Kim M-S, Bedi KC, Wang H, Mishra S, Koleini N, Yoo EJ, Margulies KB, Arany Z, Kelly DP, Kass DA, Sharma K (2023) Myocardial metabolomics of human heart failure with preserved ejection fraction. Circulation 147:1147–1161. https://doi.org/10.1161/CIRCULATIONAHA.122.061846
Han J, Gagnon S, Eckle T, Borchers CH (2013) Metabolomic analysis of key central carbon metabolism carboxylic acids as their 3-nitrophenylhydrazones by UPLC/ESI-MS. Electrophoresis 34:2891–2900. https://doi.org/10.1002/elps.201200601
Harrison GJ, Willis RJ, Headrick JP (1998) Extracellular adenosine levels and cellular energy metabolism in ischemically preconditioned rat heart. Cardiovasc Res 40:74–87. https://doi.org/10.1016/S0008-6363(98)00123-0
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia−reperfusion injury: a neglected therapeutic target. J Clin Investig 123:92–100. https://doi.org/10.1172/JCI62874
Hess M, Manson N (1984) Molecular oxygen: friend and foe*The role of the oxygen free radical system in the calcium paradox, the oxygen paradox and ischemia/reperfusion injury. J Mol Cell Cardiol 16:969–985. https://doi.org/10.1016/S0022-2828(84)80011-5
Heusch G (2017) Critical Issues for the translation of cardioprotection. Circ Res 120:1477–1486. https://doi.org/10.1161/CIRCRESAHA.117.310820
Heusch G (2020) Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Heusch G, Gersh BJ (2016) The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J. https://doi.org/10.1093/eurheartj/ehw224
Heusch G, Skyschally A, Kleinbongard P (2018) Translation, translation, translation. Circ Res 123:931–933. https://doi.org/10.1161/CIRCRESAHA.118.313947
Hochachka PW, Mustafa T (1972) Invertebrate facultative anaerobiosis. Science (1979) 178:1056–1060. https://doi.org/10.1126/science.178.4065.1056
Hochachka PW, Storey KB (1975) Metabolic consequences of diving in animals and man. Science 187:613–621. https://doi.org/10.1126/science.163485
Hoerter J, Gonzalez-Barroso M-M, Couplan E, Mateo P, Gelly C, Cassard-Doulcier A-M, Diolez P, Bouillaud F (2004) Mitochondrial uncoupling protein 1 expressed in the heart of transgenic mice protects against ischemic-reperfusion damage. Circulation 110:528–533. https://doi.org/10.1161/01.CIR.0000137824.30476.0E
Hohl CM (1999) AMP deaminase in piglet cardiac myocytes: effect on nucleotide metabolism during ischemia. Am J Physiol-Heart and Circ Physiol 276:H1502–H1510. https://doi.org/10.1152/ajpheart.1999.276.5.H1502
Hooftman A, Peace CG, Ryan DG, Day EA, Yang M, McGettrick AF, Yin M, Montano EN, Huo L, Toller-Kawahisa JE, Zecchini V, Ryan TAJ, Bolado-Carrancio A, Casey AM, Prag HA, Costa ASH, De Los SG, Ishimori M, Wallace DJ, Venuturupalli S, Nikitopoulou E, Frizzell N, Johansson C, Von Kriegsheim A, Murphy MP, Jefferies C, Frezza C, O’Neill LAJ (2023) Macrophage fumarate hydratase restrains mtRNA-mediated interferon production. Nature 615:490–498. https://doi.org/10.1038/s41586-023-05720-6
Hou D, Fu H, Zheng Y, Lu D, Ma Y, Yin Y, Zhang L, Bao D (2022) Uncoupling protein 1 knockout aggravates isoproterenol-induced acute myocardial ischemia via AMPK/mTOR/PPARα pathways in rats. Transgenic Res 31:107–118. https://doi.org/10.1007/s11248-021-00289-0
Huang Z, Wang T-S, Zhao Y-C, Zuo R-J, Deng W-B, Chi Y-J, Yang Z-M (2014) Cyclic adenosine monophosphate-induced argininosuccinate synthase 1 expression is essential during mouse decidualization. Mol Cell Endocrinol 388:20–31. https://doi.org/10.1016/j.mce.2014.02.005
Iacobazzi V, Infantino V, Castegna A, Menga A, Palmieri EM, Convertini P, Palmieri F (2017) Mitochondrial carriers in inflammation induced by bacterial endotoxin and cytokines. Biol Chem 398:303–317. https://doi.org/10.1515/hsz-2016-0260
Janku F, LoRusso P, Mansfield AS, Nanda R, Spira A, Wang T, Melhem-Bertrandt A, Sugg J, Ball HA (2021) First-in-human evaluation of the novel mitochondrial complex I inhibitor ASP4132 for treatment of cancer. Invest New Drugs 39:1348–1356. https://doi.org/10.1007/s10637-021-01112-7
Jespersen NR, Hjortbak MV, Lassen TR, Støttrup NB, Johnsen J, Tonnesen PT, Larsen S, Kimose H-H, Bøtker HE (2020) Cardioprotective effect of succinate dehydrogenase inhibition in rat hearts and human myocardium with and without diabetes mellitus. Sci Rep 10:10344. https://doi.org/10.1038/s41598-020-67247-4
Johnson TA, Jinnah HA, Kamatani N (2019) Shortage of cellular ATP as a cause of diseases and strategies to enhance ATP. Front Pharmacol. https://doi.org/10.3389/fphar.2019.00098
Kalogeris T, Baines CP, Krenz M, Korthuis RJ (2012) Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 298:229–317. https://doi.org/10.1016/B978-0-12-394309-5.00006-7
Kaplan RS, Pedersen PL (1985) Isolation and reconstitution of the n-butylmalonate-sensitive dicarboxylate transporter from rat liver mitochondria. J Biol Chem 260:10293–10298. https://doi.org/10.1016/S0021-9258(17)39246-3
Keiran N, Ceperuelo-Mallafré V, Calvo E, Hernández-Alvarez MI, Ejarque M, Núñez-Roa C, Horrillo D, Maymó-Masip E, Rodríguez MM, Fradera R, de la Rosa JV, Jorba R, Megia A, Zorzano A, Medina-Gómez G, Serena C, Castrillo A, Vendrell J, Fernández-Veledo S (2019) SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol 20:581–592. https://doi.org/10.1038/s41590-019-0372-7
Kenwood BM, Weaver JL, Bajwa A, Poon IK, Byrne FL, Murrow BA, Calderone JA, Huang L, Divakaruni AS, Tomsig JL, Okabe K, Lo RH, Cameron Coleman G, Columbus L, Yan Z, Saucerman JJ, Smith JS, Holmes JW, Lynch KR, Ravichandran KS, Uchiyama S, Santos WL, Rogers GW, Okusa MD, Bayliss DA, Hoehn KL (2014) Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab 3:114–123. https://doi.org/10.1016/j.molmet.2013.11.005
Kermode H, Williams AJ, Sitsapesan R (1998) The interactions of ATP, ADP, and inorganic phosphate with the sheep cardiac ryanodine receptor. Biophys J 74:1296–1304. https://doi.org/10.1016/S0006-3495(98)77843-9
Kinugasa Y, Ogino K, Furuse Y, Shiomi T, Tsutsui H, Yamamoto T, Igawa O, Hisatome I, Shigemasa C (2003) Allopurinol improves cardiac dysfunction after ischemia−reperfusion via reduction of oxidative stress in isolated perfused rat hearts. Circ J 67:781–787. https://doi.org/10.1253/circj.67.781
Kleinbongard P, Gedik N, Kirca M, Stoian L, Frey U, Zandi A, Thielmann M, Jakob H, Peters J, Kamler M, Heusch G (2018) Mitochondrial and contractile function of human right atrial tissue in response to remote ischemic conditioning. J Am Heart Assoc. https://doi.org/10.1161/JAHA.118.009540
Kleinbongard P, Kuthan P, Eickelmann C, Jakobs P, Altschmied J, Haendeler J, Ruhparwar A, Thielmann M, Heusch G (2022) Triiodothyronine improves contractile recovery of human atrial trabeculae after hypoxia/reoxygenation. Int J Cardiol 363:159–162. https://doi.org/10.1016/j.ijcard.2022.06.050
Kohlhauer M, Dawkins S, Costa ASH, Lee R, Young T, Pell VR, Choudhury RP, Banning AP, Kharbanda RK, Saeb-Parsy K, Murphy MP, Frezza C, Krieg T, Channon KM (2018) Metabolomic profiling in acute ST-segment–elevation myocardial infarction identifies succinate as an early marker of human ischemia−reperfusion injury. J Am Heart Assoc. https://doi.org/10.1161/JAHA.117.007546
Kohlhauer M, Pell VR, Burger N, Spiroski AM, Gruszczyk A, Mulvey JF, Mottahedin A, Costa ASH, Frezza C, Ghaleh B, Murphy MP, Tissier R, Krieg T (2019) Protection against cardiac ischemia−reperfusion injury by hypothermia and by inhibition of succinate accumulation and oxidation is additive. Basic Res Cardiol 114:18. https://doi.org/10.1007/s00395-019-0727-0
Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C (2011) Simultaneous measurement of protein oxidation and S -Nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res 108:418–426. https://doi.org/10.1161/CIRCRESAHA.110.232173
Korge P, Ping P, Weiss JN (2008) Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation. Circ Res 103:873–880. https://doi.org/10.1161/CIRCRESAHA.108.180869
Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, Calabresi PA, Snyder SH (2018) Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science (1979) 360:449–453. https://doi.org/10.1126/science.aan4665
Kotlyar AB, Vinogradov AD (1984) Interaction of the membrane-bound succinate dehydrogenase with substrate and competitive inhibitors. Biochim Biophys Acta (BBA)/Protein Struct Mol 784:24–34. https://doi.org/10.1016/0167-4838(84)90168-7
Kühlbrandt W (2019) Structure and mechanisms of F-type ATP synthases. Annu Rev Biochem 88:515–549. https://doi.org/10.1146/annurev-biochem-013118-110903
Kula-Alwar D, Prag HA, Krieg T (2019) Targeting succinate metabolism in ischemia/reperfusion injury. Circulation 140:1968–1970. https://doi.org/10.1161/CIRCULATIONAHA.119.042791
Kurebayashi N, Kodama T, Ogawa Y (1980) P1, P5-Di(Adenosine-5′)Pentaphosphate(Ap5A) as an inhibitor of adenylate kinase in studies of fragmented sarcoplasmic reticulum from bullfrog skeletal muscle1. J Biochem 88:871–876. https://doi.org/10.1093/oxfordjournals.jbchem.a133041
Kussmaul L, Hirst J (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA 103:7607–7612. https://doi.org/10.1073/pnas.0510977103
Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC-C, Griss T, Weinheimer CJ, Khader S, Randolph GJ, Pearce EJ, Jones RG, Diwan A, Diamond MS, Artyomov MN (2016) Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab 24:158–166. https://doi.org/10.1016/j.cmet.2016.06.004
Lecour S, Andreadou I, Bøtker HE, Davidson SM, Heusch G, Ruiz-Meana M, Schulz R, Zuurbier CJ, Ferdinandy P, Hausenloy DJ, Adamovski P, Andreadou I, Batirel S, Barteková M, Bertrand L, Beauloye C, Biedermann D, Borutaite V, Bøtker HE, Chlopicki S, Dambrova M, Davidson S, Devaux Y, Di Lisa F, Djuric D, Erlinge D, Falcao-Pires I, Ferdinandy P, Galatou E, Garcia-Sosa A, Girao H, Giricz Z, Gyongyosi M, Hausenloy DJ, Healy D, Heusch G, Jakovljevic V, Jovanic J, Kararigas G, Kerkal R, Kolar F, Kwak B, Leszek P, Liepinsh E, Lonborg J, Longnus S, Marinovic J, Muntean DM, Nezic L, Ovize M, Pagliaro P, Da Costa Gomes CP, Pernow J, Persidis A, Pischke SE, Podesser B, Potočnjak I, Prunier F, Ravingerova T, Ruiz-Meana M, Serban A, Slagsvold K, Schulz R, van Royen N, Turan B, Vendelin M, Walsh S, Zidar N, Zuurbier C, Yellon D (2021) IMproving preclinical assessment of cardioprotective therapies (IMPACT) criteria: guidelines of the EU-CARDIOPROTECTION COST action. Basic Res Cardiol 116:52. https://doi.org/10.1007/s00395-021-00893-5
Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL (2004) Blockade of electron transport during ischemia protects cardiac mitochondria. J Biol Chem 279:47961–47967. https://doi.org/10.1074/jbc.M409720200
Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL (2017) Mitochondrial dysfunction and myocardial ischemia−reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol 57:535–565. https://doi.org/10.1146/annurev-pharmtox-010715-103335
Li X, Wu F, Beard DA (2013) Identification of the kinetic mechanism of succinyl-CoA synthetase. Biosci Rep 33(1):e00014
Lin CS, Sharpley MS, Fan W, Waymire KG, Sadun AA, Carelli V, Ross-Cisneros FN, Baciu P, Sung E, McManus MJ, Pan BX, Gil DW, MacGregor GR, Wallace DC (2012) Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc Natl Acad Sci 109:20065–20070. https://doi.org/10.1073/pnas.1217113109
Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S, Valeaux S, Gommermann N, Rubic-Schneider T, Junt T, Carballido JM (2016) GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med. https://doi.org/10.1084/jem.20160061
Long LH, Halliwell B (2012) Biochemical and biophysical research communications the effects of oxaloacetate on hydrogen peroxide generation from ascorbate and epigallocatechin gallate in cell culture media: potential for altering cell metabolism. Biochem Biophys Res Commun 417:446–450. https://doi.org/10.1016/j.bbrc.2011.11.136
Martin JL, Costa ASH, Gruszczyk AV, Beach TE, Allen FM, Prag HA, Hinchy EC, Mahbubani K, Hamed M, Tronci L, Nikitopoulou E, James AM, Krieg T, Robinson AJ, Huang MM, Caldwell ST, Logan A, Pala L, Hartley RC, Frezza C, Saeb-Parsy K, Murphy MP (2019) Succinate accumulation drives ischaemia-reperfusion injury during organ transplantation. Nat Metab 1:966–974. https://doi.org/10.1038/s42255-019-0115-y
Matsuzaki S, Humphries KM (2015) Selective inhibition of deactivated mitochondrial complex I by biguanides. Biochemistry 54:2011–2021. https://doi.org/10.1021/bi501473h
McManus MJ, Picard M, Chen H-W, De Haas HJ, Potluri P, Leipzig J, Towheed A, Angelin A, Sengupta P, Morrow RM, Kauffman BA, Vermulst M, Narula J, Wallace DC (2019) Mitochondrial DNA variation dictates expressivity and progression of nuclear DNA mutations causing cardiomyopathy. Cell Metab 29:78-90.e5. https://doi.org/10.1016/j.cmet.2018.08.002
Melhem NJ, Chajadine M, Gomez I, Howangyin K-Y, Bouvet M, Knosp C, Sun Y, Rouanet M, Laurans L, Cazorla O, Lemitre M, Vilar J, Mallat Z, Tedgui A, Ait-Oufella H, Hulot J-S, Callebert J, Launay J-M, Fauconnier J, Silvestre J-S, Taleb S (2021) Endothelial cell indoleamine 2, 3-dioxygenase 1 alters cardiac function after myocardial infarction through kynurenine. Circulation 143:566–580. https://doi.org/10.1161/CIRCULATIONAHA.120.050301
Methner C, Chouchani ET, Buonincontri G, Pell VR, Sawiak SJ, Murphy MP, Krieg T (2014) Mitochondria selective S -nitrosation by mitochondria-targeted S -nitrosothiol protects against post-infarct heart failure in mouse hearts. Eur J Heart Fail 16:712–717. https://doi.org/10.1002/ejhf.100
Miljkovic JLJ, Burger N, Gawel JM, Mulvey JF, Norman AAI, Nishimura T, Tsujihata Y, Logan A, Sauchanka O, Caldwell ST, Morris JL, Prime TA, Warrington S, Prudent J, Bates GR, Aksentijević D, Prag HA, James AM, Krieg T, Hartley RC, Murphy MP (2022) Rapid and selective generation of H2S within mitochondria protects against cardiac ischemia−reperfusion injury. Redox Biol 55:102429. https://doi.org/10.1016/j.redox.2022.102429
Milligan G (2011) Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol Sci 32:317–325. https://doi.org/10.1016/j.tips.2011.02.002
Milliken AS, Kulkarni CA, Brookes PS (2020) Acid enhancement of ROS generation by complex-I reverse electron transport is balanced by acid inhibition of complex-II: Relevance for tissue reperfusion injury. Redox Biol 37:101733. https://doi.org/10.1016/j.redox.2020.101733
Milliken AS, Nadtochiy SM, Brookes PS (2022) Inhibiting succinate release worsens cardiac reperfusion injury by enhancing mitochondrial reactive oxygen species generation. J Am Heart Assoc 11:e026135. https://doi.org/10.1161/JAHA.122.026135
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167:457-470.e13. https://doi.org/10.1016/j.cell.2016.08.064
Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sévin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, Dinkova-Kostova AT, Hartley RC, Murphy MP, O’Neill LA (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556:113–117. https://doi.org/10.1038/nature25986
Miwa S, Brand MD (2003) Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem Soc Trans 31:1300–1301. https://doi.org/10.1042/bst0311300
Mottahedin A, Prag HA, Dannhorn A, Mair R, Schmidt C, Yang M, Sorby-Adams A, Lee JJ, Burger N, Kulaveerasingam D, Huang MM, Pluchino S, Peruzzotti-Jametti L, Goodwin R, Frezza C, Murphy MP, Krieg T (2023) Targeting succinate metabolism to decrease brain injury upon mechanical thrombectomy treatment of ischemic stroke. Redox Biol 59:102600. https://doi.org/10.1016/j.redox.2023.102600
Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, Rabinowitz JD, Frankel DS, Arany Z (2020) Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science (1979) 370:364–368. https://doi.org/10.1126/science.abc8861
Murphy E, Steenbergen C (2008) Mechanisms underlying acute protection from cardiac ischemia−reperfusion injury. Physiol Rev 88:581–609. https://doi.org/10.1152/physrev.00024.2007
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13. https://doi.org/10.1042/BJ20081386
Nadtochiy SM, Burwell LS, Ingraham CA, Spencer CM, Friedman AE, Pinkert CA, Brookes PS (2009) In vivo cardioprotection by S-nitroso-2-mercaptopropionyl glycine. J Mol Cell Cardiol 46:960–968. https://doi.org/10.1016/j.yjmcc.2009.01.012
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–658. https://doi.org/10.1038/nature03317
Nicholls DG, Ferguson SJ (2013) Bioenergetics, 4th edn. Academic Press, London
Palee S, Higgins L, Leech T, Chattipakorn SC, Chattipakorn N (2020) Acute metformin treatment provides cardioprotection via improved mitochondrial function in cardiac ischemia/reperfusion injury. Biomed Pharmacother 130:110604. https://doi.org/10.1016/j.biopha.2020.110604
Passarella S, Palmieri F, Quagliariello E (1977) The transport of oxaloacetate in isolated mitochondria. Arch Biochem Biophys 180:160–168. https://doi.org/10.1016/0003-9861(77)90020-0
Perry RJ, Kim T, Zhang X-M, Lee H-Y, Pesta D, Popov VB, Zhang D, Rahimi Y, Jurczak MJ, Cline GW, Spiegel DA, Shulman GI (2013) Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 18:740–748. https://doi.org/10.1016/j.cmet.2013.10.004
Perry RJ, Zhang D, Zhang X-M, Boyer JL, Shulman GI (2015) Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science (1979) 347:1253–1256. https://doi.org/10.1126/science.aaa0672
Picard M, Shirihai OS (2022) Mitochondrial signal transduction. Cell Metab 34:1620–1653. https://doi.org/10.1016/j.cmet.2022.10.008
Prag HA, Aksentijevic D, Dannhorn A, Giles AV, Mulvey JF, Sauchanka O, Du L, Bates G, Reinhold J, Kula-Alwar D, Xu Z, Pellerin L, Goodwin RJA, Murphy MP, Krieg T (2022) Ischemia-selective cardioprotection by malonate for ischemia/reperfusion injury. Circ Res 131:528–541. https://doi.org/10.1161/CIRCRESAHA.121.320717
Prag HA, Gruszczyk AV, Huang MM, Beach TE, Young T, Tronci L, Nikitopoulou E, Mulvey JF, Ascione R, Hadjihambi A, Shattock MJ, Pellerin L, Saeb-Parsy K, Frezza C, James AM, Krieg T, Murphy MP, Aksentijević D (2021) Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc Res 117:1188–1201. https://doi.org/10.1093/cvr/cvaa148
Prag HA, Kula-Alwar D, Bernardi P, Di Lisa F, Murphy MP, Krieg T (2022) Cyclophilin D knockout mice do not accumulate succinate during cardiac ischemia. J Mol Cell Cardiol 173:73–74. https://doi.org/10.1016/j.yjmcc.2022.09.006
Prag HA, Kula-Alwar D, Pala L, Caldwell ST, Beach TE, James AM, Saeb-Parsy K, Krieg T, Hartley RC, Murphy MP (2020) Selective delivery of dicarboxylates to mitochondria by conjugation to a lipophilic cation via a cleavable linker. Mol Pharm 17:3526–3540. https://doi.org/10.1021/acs.molpharmaceut.0c00533
Prag HA, Pala L, Kula-Alwar D, Mulvey JF, Luping D, Beach TE, Booty LM, Hall AR, Logan A, Sauchanka V, Caldwell ST, Robb EL, James AM, Xu Z, Saeb-Parsy K, Hartley RC, Murphy MP, Krieg T (2022) Ester prodrugs of malonate with enhanced intracellular delivery protect against cardiac ischemia−reperfusion injury in vivo. Cardiovasc Drugs Ther 36:1–13. https://doi.org/10.1007/s10557-020-07033-6
Priegnitz A, Brzhevskaya ON, Wojtczak L (1973) Tight binding of oxaloacetate to succinate dehydrogenase. Biochem Biophys Res Commun 51:1034–1041
Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, Vitturi DA, Patel RP, Hiley CR, Abakumova I, Requejo R, Chouchani ET, Hurd TR, Garvey JF, Taylor CT, Brookes PS, Smith RAJ, Murphy MP (2009) A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia−reperfusion injury. Proc Natl Acad Sci USA 106:10764–10769. https://doi.org/10.1073/pnas.0903250106
Pryde KR, Hirst J (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J Biol Chem 286:18056–18065. https://doi.org/10.1074/jbc.M110.186841
Pucar D, Bast P, Gumina RJ, Lim L, Drahl C, Juranic N, Macura S, Janssen E, Wieringa B, Terzic A, Dzeja PP (2002) Adenylate kinase AK1 knockout heart: energetics and functional performance under ischemia−reperfusion. Am J Physiol-Heart Circ Physiol 283:H776–H782. https://doi.org/10.1152/ajpheart.00116.2002
Reddy A, Bozi LHM, Yaghi OK, Mills EL, Xiao H, Nicholson HE, Paschini M, Paulo JA, Garrity R, Laznik-Bogoslavski D, Ferreira JCB, Carl CS, Sjøberg KA, Wojtaszewski JFP, Jeppesen JF, Kiens B, Gygi SP, Richter EA, Mathis D, Chouchani ET (2020) pH-Gated succinate secretion regulates muscle remodeling in response to exercise. Cell 183:62-75.e17. https://doi.org/10.1016/j.cell.2020.08.039
Ristagno G, Fries M, Brunelli L, Fumagalli F, Bagnati R, Russo I, Staszewsky L, Masson S, Li Volti G, Zappalà A, Derwall M, Brücken A, Pastorelli R, Latini R (2013) Early kynurenine pathway activation following cardiac arrest in rats, pigs, and humans. Resuscitation 84:1604–1610. https://doi.org/10.1016/j.resuscitation.2013.06.002
Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, Murphy MP (2018) Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 293:9869–9879. https://doi.org/10.1074/jbc.RA118.003647
Robinson BH, Williams GR (1970) The sensitivity of dicarboxylate anion exchange reactions to transport inhibitors in rat-liver mitochondria. Biochim Biophys Acta (BBA) - Bioenerg 216:63–70. https://doi.org/10.1016/0005-2728(70)90159-3
Ross MF, Kelso GF, Blaikie FH, James AM, Cochemé HM, Filipovska A, Da Ros T, Hurd TR, Smith RAJ, Murphy MP (2005) Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochem Mosc 70:222–230. https://doi.org/10.1007/s10541-005-0104-5
Ruprecht JJ, Kunji ERS (2020) The SLC25 mitochondrial carrier family: structure and mechanism. Trends Biochem Sci 45:244–258. https://doi.org/10.1016/j.tibs.2019.11.001
Samarzija I, Molnar V, Frömter E (1981) THE stoichiometry OF Na+ coupled anion absorption across the brushborder membrane of rat renal proximal tubule. Kidney and body fluids. Elsevier, pp 419–423
Schulz R, Heusch G (2022) Targeted mito- and cardioprotection by malonate. Circ Res 131:542–544. https://doi.org/10.1161/CIRCRESAHA.122.321582
Sherry AD, Zhao P, Wiethoff AJ, Jeffrey FMH, Malloy CR (1998) Effects of aminooxyacetate on glutamate compartmentation and TCA cycle kinetics in rat hearts. Am J Physiol-Heart Circ Physiol 274:H591–H599. https://doi.org/10.1152/ajpheart.1998.274.2.H591
Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, Burwell L, Wang X, MacArthur PH, Shoja A, Raghavachari N, Calvert JW, Brookes PS, Lefer DJ, Gladwin MT (2007) Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J Exp Med 204:2089–2102. https://doi.org/10.1084/jem.20070198
Skladanowski AC, Sala GB, Newby AC (1989) Inhibition of IMP-specific cytosolic 5′-nucleotidase and adenosine formation in rat polymorphonuclear leucocytes by 5′-deoxy-5′-isobutylthio derivatives of adenosine and inosine. Biochem J 262:203–208. https://doi.org/10.1042/bj2620203
Smith RA, Hartley RC, Murphy MP (2011) Mitochondria-targeted small molecule therapeutics and probes. Antioxid Redox Signal. https://doi.org/10.1089/ars.2011.3969
Spinelli JB, Rosen PC, Sprenger H-G, Puszynska AM, Mann JL, Roessler JM, Cangelosi AL, Henne A, Condon KJ, Zhang T, Kunchok T, Lewis CA, Chandel NS, Sabatini DM (2021) Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science (1979) 374:1227–1237. https://doi.org/10.1126/science.abi7495
Støttrup NB, Løfgren B, Birkler RD, Nielsen JM, Wang L, Caldarone CA, Kristiansen SB, Contractor H, Johannsen M, Bøtker HE, Nielsen TT (2010) Inhibition of the malate–aspartate shuttle by pre-ischaemic aminooxyacetate loading of the heart induces cardioprotection. Cardiovasc Res 88:257–266. https://doi.org/10.1093/cvr/cvq205
St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277:44784–44790. https://doi.org/10.1074/jbc.M207217200
Taghavi S, Abdullah S, Toraih E, Packer J, Drury RH, Aras OAZ, Kosowski EM, Cotton-Betteridge A, Karim M, Bitonti N, Shaheen F, Duchesne J, Jackson-Weaver O (2022) Dimethyl malonate slows succinate accumulation and preserves cardiac function in a swine model of hemorrhagic shock. J Trauma Acute Care Surg 93:13–20. https://doi.org/10.1097/TA.0000000000003593
Takeuchi T, Schumacker PT, Kozmin SA (2015) Identification of fumarate hydratase inhibitors with nutrient-dependent cytotoxicity. J Am Chem Soc 137:564–567. https://doi.org/10.1021/ja5101257
Taylor CT (2008) Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J 409:19–26. https://doi.org/10.1042/BJ20071249
Techiryan G, Weil BR, Palka BA, Canty JM (2018) Effect of intracoronary metformin on myocardial infarct size in swine. Circ Res 123:986–995. https://doi.org/10.1161/CIRCRESAHA.118.313341
Tonnesen PT, Hjortbak MV, Lassen TR, Seefeldt JM, Bøtker HE, Jespersen NR (2021) Myocardial salvage by succinate dehydrogenase inhibition in ischemia–reperfusion injury depends on diabetes stage in rats. Mol Cell Biochem 476:2675–2684. https://doi.org/10.1007/s11010-021-04108-2
Tornheim K, Lowenstein JM (1972) The purine nucleotide cycle. The production of ammonia from aspartate by extracts of rat skeletal muscle. J Biol Chem 247:162–169
Trauelsen M, Hiron TK, Lin D, Petersen JE, Breton B, Husted AS, Hjorth SA, Inoue A, Frimurer TM, Bouvier M, O’Callaghan CA, Schwartz TW (2021) Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated Gq signaling. Cell Rep 35:109246. https://doi.org/10.1016/j.celrep.2021.109246
Tsuji N, Tsuji T, Yamashita T, Hayase N, Hu X, Yuen PST, Star RA (2023) BAM15 treats mouse sepsis and kidney injury, linking mortality, mitochondrial DNA, tubule damage, and neutrophils. J Clin Investig. https://doi.org/10.1172/JCI152401
Valls-Lacalle L, Barba I, Miró-Casas E, Alburquerque-Béjar JJ, Ruiz-Meana M, Fuertes-Agudo M, Rodríguez-Sinovas A, García-Dorado D (2016) Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res 109:374–384. https://doi.org/10.1093/cvr/cvv279
Valls-Lacalle L, Barba I, Miró-Casas E, Ruiz-Meana M, Rodríguez-Sinovas A, García-Dorado D (2018) Selective inhibition of succinate dehydrogenase in reperfused myocardium with intracoronary malonate reduces infarct size. Sci Rep 8:2442. https://doi.org/10.1038/s41598-018-20866-4
Višnjić D, Lalić H, Dembitz V, Tomić B, Smoljo T (2021) AICAr, a widely used AMPK activator with important AMPK-independent effects: a systematic review. Cells 10:1095. https://doi.org/10.3390/cells10051095
Wang H, Huwaimel B, Verma K, Miller J, Germain TM, Kinarivala N, Pappas D, Brookes PS, Trippier PC (2017) Synthesis and antineoplastic evaluation of mitochondrial complex II (succinate dehydrogenase) inhibitors derived from atpenin A5. ChemMedChem 12:1033–1044. https://doi.org/10.1002/cmdc.201700196
Wojtczak L, Wojtczak AB, Ernster L (1969) The inhibition of succinate dehydrogenase by oxaloacetate. Biochim Biophys Acta BBA Enzymol 191:10–21. https://doi.org/10.1016/0005-2744(69)90310-6
Wojtovich AP, Brookes PS (2009) The complex II inhibitor atpenin A5 protects against cardiac ischemia−reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol 104:121–129. https://doi.org/10.1007/s00395-009-0001-y
Wolfenden R, Lewis CA, Yuan Y (2011) Kinetic challenges facing oxalate, malonate, acetoacetate, and oxaloacetate decarboxylases. J Am Chem Soc 133:5683–5685. https://doi.org/10.1021/ja111457h
Wright JJ, Biner O, Chung I, Burger N, Bridges HR, Hirst J (2022) Reverse electron transfer by respiratory complex I catalyzed in a modular proteoliposome system. J Am Chem Soc 144:6791–6801. https://doi.org/10.1021/jacs.2c00274
Wyant GA, Yu W, Doulamis IIP, Nomoto RS, Saeed MY, Duignan T, McCully JD, Kaelin WG (2022) Mitochondrial remodeling and ischemic protection by G protein–coupled receptor 35 agonists. Science 377:621–629. https://doi.org/10.1126/science.abm1638
Xu J, Pan H, Xie X, Zhang J, Wang Y, Yang G (2018) Inhibiting succinate dehydrogenase by dimethyl malonate alleviates brain damage in a rat model of cardiac arrest. Neuroscience 393:24–32. https://doi.org/10.1016/j.neuroscience.2018.09.041
Yap TA, Daver N, Mahendra M, Zhang J, Kamiya-Matsuoka C, Meric-Bernstam F, Kantarjian HM, Ravandi F, Collins ME, Di FME, Dumbrava EE, Fu S, Gao S, Gay JP, Gera S, Han J, Hong DS, Jabbour EJ, Ju Z, Karp DD, Lodi A, Molina JR, Baran N, Naing A, Ohanian M, Pant S, Pemmaraju N, Bose P, Piha-Paul SA, Rodon J, Salguero C, Sasaki K, Singh AK, Subbiah V, Tsimberidou AM, Xu QA, Yilmaz M, Zhang Q, Li Y, Bristow CA, Bhattacharjee MB, Tiziani S, Heffernan TP, Vellano CP, Jones P, Heijnen CJ, Kavelaars A, Marszalek JR, Konopleva M (2023) Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: phase I trials. Nat Med 29:115–126. https://doi.org/10.1038/s41591-022-02103-8
Yin Z, Burger N, Kula-Alwar D, Aksentijević D, Bridges HR, Prag HA, Grba DN, Viscomi C, James AM, Mottahedin A, Krieg T, Murphy MP, Hirst J (2021) Structural basis for a complex I mutation that blocks pathological ROS production. Nat Commun 12:707. https://doi.org/10.1038/s41467-021-20942-w
Zhang J, Wang YT, Miller JH, Day MM, Munger JC, Brookes PS (2018) Accumulation of succinate in cardiac ischemia primarily occurs via canonical krebs cycle activity. Cell Rep 23:2617–2628. https://doi.org/10.1016/j.celrep.2018.04.104
Zhang X, Dang CV (2023) Time to hit pause on mitochondria-targeting cancer therapies. Nat Med 29:29–30. https://doi.org/10.1038/s41591-022-02129-y
Zu Y, Shannon RJ, Hirst J (2003) Reversible, electrochemical interconversion of NADH and NAD + by the catalytic (Iλ) subcomplex of mitochondrial NADH: ubiquinone oxidoreductase (complex I). J Am Chem Soc 125:6020–6021. https://doi.org/10.1021/ja0343961
Zuurbier CJ, Bertrand L, Beauloye CR, Andreadou I, Ruiz-Meana M, Jespersen NR, Kula-Alwar D, Prag HA, Eric Botker H, Dambrova M, Montessuit C, Kaambre T, Liepinsh E, Brookes PS, Krieg T (2020) Cardiac metabolism as a driver and therapeutic target of myocardial infarction. J Cell Mol Med 24:5937–5954. https://doi.org/10.1111/jcmm.15180
Zweier JL, Flaherty JT, Weisfeldt ML (1987) Direct measurement of free radical generation following reperfusion of ischemic myocardium. Proc Natl Acad Sci 84:1404–1407. https://doi.org/10.1073/pnas.84.5.1404
Funding
This work was supported by the British Heart Foundation (PG/20/10025 to T. Krieg); the Medical Research Council (MC_UU_00015/3 to M.P. Murphy) and the Wellcome Trust (220257/Z/20/Z to M.P. Murphy).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
HAP, MPM and TK have pending patents on targeting succinate metabolism in ischaemia/reperfusion injury. MPM and TK are directors of Camoxis Therapeutics Ltd.
Additional information
This article is part of the topical collection “Mitochondria at the heart of cardioprotection”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prag, H.A., Murphy, M.P. & Krieg, T. Preventing mitochondrial reverse electron transport as a strategy for cardioprotection. Basic Res Cardiol 118, 34 (2023). https://doi.org/10.1007/s00395-023-01002-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-023-01002-4