
Abstract
Objective
The aim of this study was to provide a full characterization of a cohort of 11 pediatric patients diagnosed with PTEN hamartoma tumor syndrome (PHTS).
Patients and methods
Eleven patients with genetic diagnostic of PHTS were recruited between February 2019 and April 2023. Clinical, imaging, demographic, and genetic data were retrospectively collected from their hospital medical history.
Results
Regarding clinical manifestations, macrocephaly was the leading sign, present in all patients. Frontal bossing was the most frequent dysmorphism. Neurological issues were present in most patients. Dental malformations were described for the first time, being present in 27% of the patients. Brain MRI showed anomalies in 57% of the patients. No tumoral lesions were present at the time of the study. Regarding genetics, 72% of the alterations were in the tensin-type C2 domain of PTEN protein. We identified four PTEN genetic alterations for the first time.
Conclusions
PTEN mutations appear with a wide variety of clinical signs and symptoms, sometimes associated with phenotypes which do not fit classical clinical diagnostic criteria for PHTS. We recommend carrying out a genetic study to establish an early diagnosis in children with significant macrocephaly. This facilitates personalized monitoring and enables anticipation of potential PHTS-related complications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
PTEN hamartoma tumor syndrome (PHTS) is a disease with a broad spectrum of signs and symptoms. First described in 1993, it includes the classical Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome (BRRS), Lhermitte-Duclos disease (LDD), and Proteus and Proteus-like syndrome. The phenotypic spectrum of PHTS has been evolving and expanding paralleling the increasing accessibility of genetic diagnosis. There are currently many other alterations related to PTEN pathogenic variants such as neurodevelopmental disorders, segmentary overgrowth, autistic spectrum disorder (ASD), or macrocephaly. In the pediatric population, classic forms with mucocutaneous manifestations, hamartomatous lesions, and malignancies are less common than development delay, brain magnetic resonance imaging (MRI) alterations, and growth disorders such as macrocephaly, overweight, or limb asymmetries [1]. The etiology of PHTS lies with the PTEN (phosphatase and tensin homolog deleted on chromosome 10) gene. The protein encoded by this gene is a dual phosphatase with both lipid and protein activity. It is ubiquitously expressed in different cells. As regards the lipid activity, it dephosphorylates phosphatidylinositol-3, 4, 5-phosphate (PIP3), a mediator in the MAPK pathway. This action induces cell cycle arrest in G1 and apoptosis, regulating cell growth. It is known as a tumor suppressor, mutated in different cancer types. Moreover, it has been associated with insulin regulation pathways and mitochondrial metabolism [2]. Pathogenic variants of this gene have been reported to cause PHTS. The exact prevalence of PHTS is unknown due to the high variability in the manifestations and the difficulties to carry out massive genetic testing. The prevalence of CS was estimated in 1:250,000 in the Dutch population, although the global real prevalence of PHTS is likely to be higher [3].
Different types of genetic variants have been reported that affect the PTEN gene without a clear genotype-phenotype correlation. An apparent lack of missense mutations has been described in LDD [3]. However, missense variant 5′ to or within the phosphate core motif has been associated with involvement of five or more organs, although none of these genotype-phenotype correlations has been confirmed in large case series [4]. PTEN pathogenic variants include missense and nonsense nucleotide variants, deletions, insertions, and splicing mutations, all of them with autosomal dominant inheritance [5]. Frequency of de novo mutations is estimated to be between 10.7 and 47.6% [6]. Mosaicism for PTEN has also been described in PHTS.
The penetrance of PHTS is near 100% by the fourth decade in patients with a pathogenic variant in PTEN [2]. The clinical manifestations can vary between different individuals: macrocephaly and neurodevelopmental disorders are more frequent in childhood while classical symptoms (intestinal polyps, malignancies) are more frequent in patients diagnosed in adulthood without a clear evolution from one spectrum to the other.
The literature regarding PHTS in children is limited. The largest series of cases published in Pubmed includes just sixteen cases, so we aim to describe a cohort of eleven children with a pathogenic variant of PTEN, diagnosed in the “Reference Unit of Rare Diseases Advanced Diagnosis” (DiERCyL) to provide more data to the literature detailing the clinical, genetic, and other issues related to this condition.
Patients and methods
This study includes patients diagnosed with PHTS between February 2019 and April 2023 in DiERCyL. Demographic, clinical, and genetic data were recorded retrospectively from their hospital medical history.
Genetic studies
The genetic studies were carried out in the Laboratory of Molecular Genetics and Pharmacogenetics at University Hospital of Salamanca. Genomic DNA was extracted from peripheral blood leukocytes by magnetics beads or silica-membrane-based nucleic acid purification. Whole exome sequencing was performed on the NextSeq 500 platform (Illumina; San Diego, CA). For this purpose, DNA libraries were prepared using TruSeq technology (Illumina; San Diego, CA) and captured using xGen Exome technology (Integrated DNA Technologies, IDT; Coralville, IA). A bioinformatic study of the DNA sequences obtained was performed by comparison with the reference genome version GRCh37/hg19. The genetic variants detected were confirmed by Sanger sequencing. The assessment of the pathogenicity of the variants was determined according to the American College of Medical Genetics and Genomics (ACMG) guidelines, using the ClinVar database and Varsome scores. Genetic studies were amplified to parents and siblings, when possible (six out of eleven index cases), although they do not present clinical features of PHTS.
Signs and symptoms
According to clinical signs, macrocephaly was defined as percentile > 97 and standard deviation (SD) was calculated according to the anthropometric standards published for the Spanish population [7]. Height and weight percentiles and SD were calculated according to Spanish growth studies from 2010 [8]. In case it was required, a clinical examination by a pediatric neurologist was performed in order to evaluate ASD and development delay (DD). MRI was performed in all but four patients. Other complementary tests and oncologic follow-up were individualized according to the clinical spectrum and the individual risk.
Results
Clinical and demographic data
As summarized in Table 1, we have studied 11 patients, 7 males and 4 females, diagnosed with PHTS. The mean age was 6.87 ± 3.02 (1.91–11.5) years. In our cohort, we have two siblings with the same mutation.
The age at diagnosis ranged from 1 year and 11 months to 11 years and 6 months. All genetic studies except one were requested by a pediatric neurologist, while the other one was requested by a pediatric endocrinologist. The most prevalent diagnoses prior to the genetic study were macrocephaly (11/11), developmental delay (5/11), and overgrowth (3/11). The mean head circumference SD score was +4.3 ranging from +2.6 to +5. According to their growth, three of our patients were diagnosed with some kind of overgrowth. One of them presented overweight (+3 SD), another one overheight (+2.35 SD), and the last one was diagnosed with overheight with no data available about her height.
In general, our patients did not present facial dysmorphism, and most of the anomalies were secondary to macrocephaly. Facial phenotype was not described in two of the patients. In the rest of them, frontal bossing (5/9), prognathism (3/9), and dental anomalies such as dental agenesis or dental malocclusion (3/9) were the most frequent phenotypic manifestations. No facial asymmetries were reported, although two of the patients showed slight asymmetry of the lower limbs.
Cutaneous signs were observed in three of the patients. Patient 1 had cafe-au-lait macules and a hamartoma on the thumb. Patient 2 also showed cafe-au-lait macules besides follicular keratosis and a keloid scar. Patient 3 had had a cervical lipoma removed. No other dermatological findings were described. Three of our patients showed joint laxity without presenting joint pain, dislocations, Marfan phenotype, or other manifestations related to collagen disturbance such as varicose veins, hernias, or uterine/anal prolapse. Four of the patients presented tonsillar hypertrophy, one of them requiring surgical removal due to recurrent tonsillitis and sleep apnea/hypopnea syndrome.
Seven of our patients (64%) suffered some kind of developmental delay, and five of them (71%) presented clinical improvement in their symptomatology. Our cohort showed a wide range of neuropsychiatric involvement. Six patients showed both speech and motor delay and two patients showed only motor delay. ASD was reported in three patients (27%), the rest showed normal social behavior. Specific intelligence quotient tests were not performed.
Currently, none of our patients has developed any malignancies, although they are still young and oncologic screening is being conducted. Only patient 5 has thyroid involvement, with multinodular goiter, and he is waiting for surgical removal due to mild dysphagia and aesthetic reasons. A special case is patient 4, with a diagnosis of intention tremor and macrocephaly without any other neurological symptoms. This patient’s phenotype included frontal bossing, prognathism, dental agenesis, short limbs and fingers, low-set ears, short philtrum, low nasal bridge, and hypertelorism. She had a cervical lipoma removed prior to diagnosis. As far as we know, none of these signs has been described in PHTS. Brain MRI showed an enlargement of perivascular spaces and a 2-mm tonsillar descent. A de novo nonsense mutation in PTEN gene was detected during the genetic study.
Imaging studies
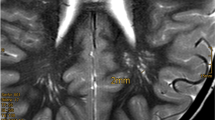
A brain MRI was performed in seven of our patients following symptomatic criteria. Three of the patients (43%) showed no abnormalities in MRI. Two patients presented enlarged perivascular spaces, one of them with a 2-mm tonsillar descent. Another patient was diagnosed with diffuse leukoencephalopathy and patient 8 showed cerebellar asymmetry and cortical dysplasia compatible with LDD.
Genetic studies
Genetic analysis was performed to all the patients, and Sanger sequencing or confirmed nine heterozygous pathogenic mutations, three of them not described before up to our knowledge. The most common mutation was nonsense point mutation, identified in six patients (55%). Two of the patients presented missense point mutation and the other two frameshift insertion. In patient 10, a deletion involving the region of the PTEN gene was found by CGH-arrays and confirmed by quantitative PCR.
Table 2 contains the genetic data of our patients. We were not able to establish a clear genotype-phenotype correlation because of the sample size, although the two missense mutations were in a hotspot region according to VarSome. This region includes a phosphatase domain that contains the CX5R signature motif for phosphatases [2]. One of our patients presented a deletion involving part of the PTEN gene. The rest of our patients’ mutations affect the tensin-type C2 domain and all of them are point nonsense mutations except for one frameshift insertion (patient 6). The mutation detected in patient 3 has been reported to generate a truncated protein affecting the functionality of a C-terminal C2 domain with different phenotype expressions [9,10,11]. Mutation in patient 8 affects a putative tyrosine phosphatase domain affecting the tertiary structure of the protein [12, 13]. The molecular effect of the rest of the mutations detected has not been described for the moment. Figure 1 shows the PTEN structure and the location of the detected mutations.

PTEN structure and location of the mutations detected
Segregation studies were performed in six of our patients, including both parents in four of them. The mutation found in the siblings (patient 9 and patient 11) was also present in their mother, suggesting maternal inheritance, although for the moment she does not present any symptoms or oncological history. Moreover, a de novo mutation was confirmed in two patients. Finally, in patients 1 and 2, just the father or the mother were studied, and no PTEN mutations were found.
Discussion
PHTS involves a broad clinical spectrum, where the oncologic risk is the main point under consideration. There is not much literature on this syndrome in the pediatric population. Through this retrospective study, we try to offer new data according to clinical, genetic, and neuroimaging findings in our PHTS cohort.
The molecular approach to certain pathologies has made it possible to know the underlying cause behind many of them. On some occasions, a known disease or syndrome has been subclassified into several different pathologies according to their molecular substrate. In other cases, such as PHTS, a variety of different syndromes (e.g., CS, BRRS, PS…) have shown a common etiology, in this case, alterations in the PTEN gene affecting its protein functionality. PHTS has been widely studied in adults due to its tendency to develop different kinds of tumors. However, it has barely been described in the pediatric population. We aim to offer new data in pediatric patients and to correlate our patients with the existing literature. In our study, we have identified four new PTEN alterations that need to be considered when studying PHTS.
There are no differences in prevalence associated to sex described in the literature. Several series of cases with PHTS show male preponderance while others do not show any sex predisposition [5, 14,15,16]. In our cohort of 11 patients, seven of them are male. As developmental disorder is more common in males and it was the leading sign in most of our patients, this might be associated with our findings [17].
As shown in the “Results” section, we were not able to establish a clear genotype-phenotype correlation according to previous studies, including a review of the clinical literature [18]. However, it has been hypothesized after considering some facts. For example, there are no data about missense variants detected in LDD disease in the literature, all of them are caused by a nonsense or frameshift variant which is in line with the clinical and genetic data of patient 8 [3]. It has been suggested that pathogenic variants 5′ to or within the phosphatase core are associated with higher severity and involvement of five or more organs; however, these studies are based on an adult population, and no conclusions can be established in our cohort, although a narrower oncologic screening may be implemented in these patients [1]. There is one study which hypothesizes about a correlation between the phenotype and post-transcriptional PTEN expression and its splice variants. However, we cannot add new data because none of our mutations was in the promoter region and no transcriptional studies were conducted [9]. A more recent study using an artificial humanized yeast model and evaluating lipid phosphatase activity in different PTEN mutations showed a higher phosphatase activity in ASD-associated variants [19]. This fact, albeit interesting, is difficult to evaluate in real patients, but it opens a window for future research and better understanding of the PHTS.
Macrocephaly is a major sign in PHTS. All our patients presented macrocephaly, as it has been reported in several other cohorts [5, 20]. Seventy-eight percent of our sample (7/9) had a head circumference SD score over + 4, greater than a French cohort and similar to an Italian cohort, which are the studies closest to the Spanish population [5, 20].
According to a PTEN review in childhood, the most common facial characteristics described were frontal bossing, depression of nasal bridge, horizontal eyebrows, and dolichocephaly [9]. Frontal bossing was present in approximately half of our patients, which could be secondary to the macrocephaly. Other common features described in the literature were less frequent in our patients. Only two patients presented depression of the nasal bridge and one patient showed acrocephaly, although dolichocephaly has been reported in other series [21]. Prognathism was described in three of our patients while we have just found one case in the literature reporting this sign [22]. It is also remarkable that teeth alterations such as agenesis and malocclusion were present in three of the patients. This fact has not been described up to our knowledge and could be secondary to gingival disturbances which are more common, but it is also possible that PTEN mutations imply dental issues because PTEN plays an important role in osteogenesis and proliferation in dental pulp cells [18, 23].
Neuropsychiatric involvement has also been described in the literature, with a wide spectrum of signs including motor and speech delay, ASD, and intellectual disability which tends to improve, although some alterations persist through adulthood [5, 18]. Seven of our patients (64%) presented some grade of developmental delay, which was only motor in two of them and motor and language-related in the rest. This prevalence is lower than the prevalence reported in other case series and comparable to findings from an Italian study [5, 21]. Four of the affected patients showed an improvement in successive controls and in two of the patients who did not present neurological improvement this might be due to a short follow-up period. Developmental impairment during childhood with a subsequent normal cognitive outcome in adulthood has been previously reported [24].
ASD is another of the main neuropsychiatric issues related to PTEN alterations. Three of our patients (27%) had some symptoms of the autistic spectrum. These data are similar to those of other studies suggesting a strong association between PTEN and ASD [5]. The underlying cause could be an alteration in neuronal growth, survival, and migration via the PI3K/AKT pathway as well as a synergy between PTEN mutations and other alterations in autism susceptibility genes [25].
There are several manifestations described in brain MRI in PHTS. However, it is difficult to assess accurately the prevalence of these alterations, especially in the pediatric population since MRI is not a diagnostic test exempt from certain risk. In general terms, it was just conducted in those patients who showed neuropsychiatric signs. In our series, brain MRI was performed in seven patients, and three were completely normal (43%) which is a greater percentage than reported in other studies, especially considering that, in the Italian cohort, MRI was performed in all patients, including those without neurological involvement [5, 15]. Enlarged perivascular spaces were found in 29% of our patients, which was less frequent than the 94% prevalence previously described [15]. In this study, 36% of their patients presented white matter abnormalities, defined as changes in signal intensity. This condition was detected in only one of our patients. One of our patients presented a tonsillar descent of 2 mm, which represents a lower prevalence than the 33–37% reported by other studies involving adults and children, respectively [5, 26]. LDD is difficult to assess in children due to the lack of the typical “tiger-stripe” in T2. One of our patients presented cortical cerebellar dysplasia compatible with LDD. Despite being a pathognomonic criterion for CS in adults, this finding has not been reported in several case series in children’s populations with PHTS and the relation between LDD and PHTS in pediatric populations remains unclear according to PHTS reviews in children, although in some serial of cases of pediatric patients with LDD, some of them meet CS clinical criteria [18].
Vascular features such as hemangiomas, cavernomas, or arteriovenous malformations, which are a major sign in several of the classic syndromes related to PTEN alterations (CS, BRRS…), have not been reported in our cohort, although they may appear later in the natural evolution of the disease [18].
PTEN alterations have been frequently associated with skin features, which were a CS criterion even before the discovery of the PTEN gene. Hamartomatous growths of the same or different tissues (e.g., trichilemmomas, fibromas, lipomas) can be detected in almost every part of the body, although they tend to appear on the face and surrounding orifices [5, 18]. Acral keratoses and tongue alterations are some of the most prevalent skin alterations in PHTS [18]. The presence of several of these features, especially in a macrocephalic child, is very suggestive of PHTS [5]. In our cohort, one patient presented a hamartoma on the thumb and another patient presented a cervical lipoma. The prevalence is lower than what has been reported in other studies, but this may be due to the age-related penetrance described in skin lesions [5, 18]. Two of our patients presented cafe-au-lait macules, which is not a typical manifestation of PHTS, and its prevalence has not been studied for the moment, although there are several case reports which include this alteration [27−29]. One of these patients also presented keloid scarring. The PTEN gene may play an important role in keloid scarring as suggested in a case control study that demonstrates underexpression of PTEN in keloid samples compared to normal controls [30]. Penile freckling is another classic hallmark of PHTS, especially in the pediatric population. It has been reported in childhood from the first age of life and was absent in our cohort [18].
One of the main concerns of PHTS is the oncological risk. PTEN mutations have been associated with thyroid, breast, endometrium, and kidney tumor, among others that are less frequent [1]. Defining the exact risk of developing malignancies is difficult. Studies involving CS, BRRS, or PS patients prior to molecular diagnosis had an important recruitment bias. In our cohort, many of the new diagnosed children were just studied because of macrocephaly and developmental problems. There is a lack of longitudinal studies in this group of patients to accurately estimate oncological risk. Although some malignant tumors have been reported in children, such as thyroid and renal cell carcinoma, granulosa cell tumor of the ovary, or colonic ganglioneuroma, none of our patients has presented any malignancy for the moment [31]. This lack of evidence in the literature makes it difficult to establish the best clinical management of these children. A 2019 review of PHTS in children proposes several management considerations for these patients, considering that in spite of the lack of data about the prevalence of oncologic complications in children, there have been some cases reported [18, 31]. Achieving an early diagnosis through a PTEN genetic study in pediatric patients with macrocephaly would allow an early diagnosis of possible malignancies by performing an accurate follow-up of these patients.
Conclusions
This study shows the wide variety of clinical signs and symptoms associated with PTEN mutations, which sometimes express phenotypes which do not meet any of the classic diagnostic criteria for CS.
All except one of our patients were referred by a pediatric neurologist, and macrocephaly and neurodevelopmental issues were the main reason to initiate genetic studies. We highly recommend looking for PTEN mutations in children with pronounced macrocephaly, especially if they present other symptoms such as neurodevelopmental disorders, ASD, certain facial dysmorphisms, or thyroid nodules.
In PHTS, as in other heterogenous syndromes, it is important to describe as many clinical manifestations as possible in order to get a better knowledge of the disorder and help other clinicians to reach an early diagnosis. Being able to diagnose PHTS during childhood makes it possible to keep a closer follow-up in the patients and detect certain complications associated earlier, improving the treatment and, subsequently, the prognosis and the quality of life. This is especially relevant in oncologic issues, in which an individualized screening for malignancies may be useful to detect tumors in earlier stages. Besides, as it is a hereditary syndrome, genetic counselling could be possible with an early diagnosis.
Molecular diagnosis may be difficult to carry out in many centers. Whole exome sequencing usually takes too much time and resources to get accurately to a diagnosis. That is why being able to direct the molecular study according to the clinic features will be very helpful to save both money and time.
Data Availability
Sequencing raw data is stored by the Reference Unit of Rare Diseases Advanced Diagnosis of Castile and Leon (DiERCyL). The clinical data mentioned in the manuscript was obtained from the hospital medical history of the patients.
References
Yehia L, Eng C (2001) PTEN hamartoma tumor syndrome. In: Adam MP et al (eds) GeneReviews®. University of Washington, Seattle
Chen CY, Chen J, He L, Stiles BL (2018) PTEN: tumor suppressor and metabolic regulator. Front Endocrinol 9:338. https://doi.org/10.3389/fendo.2018.00338
Nelen MR, Kremer H, Konings IB, Schoute F, van Essen AJ, Koch R, Woods CG, Fryns JP, Hamel B, Hoefsloot LH, Peeters EA, Padberg GW (1999) Novel PTEN mutations in patients with Cowden disease: absence of clear genotype-phenotype correlations. Eur J Hum Gen 7(3):267–273. https://doi.org/10.1038/sj.ejhg.5200289
Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, Liaw D, Caron S, Duboué B, Lin AY, Richardson AL, Bonnetblanc JM, Bressieux JM, Cabarrot-Moreau A, Chompret A, Demange L, Eeles RA, Yahanda AM, Fearon ER, Fricker JP, Gorlin RJ, Hodgson SV, Huson S, Lacombe D, Eng C et al (1998) Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet 7(3):507–515. https://doi.org/10.1093/hmg/7.3.507
Ciaccio C, Saletti V, D’Arrigo S, Esposito S, Alfei E, Moroni I, Tonduti D, Chiapparini L, Pantaleoni C, Milani D (2019) Clinical spectrum of PTEN mutation in pediatric patients. A bicenter experience. Eur J Med Gene 62(12):103596. https://doi.org/10.1016/j.ejmg.2018.12.001
Mester J, Eng C (2012) Estimate of de novo mutation frequency in probands with PTEN hamartoma tumor syndrome. Genet Med 14(9):819–822. https://doi.org/10.1038/gim.2012.51
Carrascosa A, Fernández JM, Fernández C, Ferrández A, López-Siguero JP, Sánchez E, Sobradillo B, Yeste D (2008) Spanish growth studies 2008. New anthropometric standards Endocrinol Nutr 55(10):484–506. https://doi.org/10.1016/S1575-0922(08)75845-5
Sánchez González E, Carrascosa Lezcano A, Fernández García JM, Ferrández Longás A, López de Lara D, López-Siguero JP (2011) Estudios españoles de crecimiento: situación actual, utilidad y recomendaciones de uso [Spanish growth studies: the current situation, their effectiveness and recommendations for their use]. An Pediatr (Barc) 74(3). https://doi.org/10.1016/j.anpedi.2010.10.005
Sarquis MS, Agrawal S, Shen L, Pilarski R, Zhou XP, Eng C (2006) Distinct expression profiles for PTEN transcript and its splice variants in Cowden syndrome and Bannayan-Riley-Ruvalcaba syndrome. J Hum Genet 79(1):23–30. https://doi.org/10.1086/504392
Wanner M, Celebi JT, Peacocke M (2001) Identification of a PTEN mutation in a family with Cowden syndrome and Bannayan-Zonana syndrome. J Am Acad Dermatol 44(2):183–187. https://doi.org/10.1067/mjd.2001.110390
Zhou X, Hampel H, Thiele H, Gorlin RJ, Hennekam RC, Parisi M, Winter RM, Eng C (2001) Association of germline mutation in the PTEN tumour suppressor gene and Proteus and Proteus-like syndromes. Lancet 358(9277):210–211. https://doi.org/10.1016/s0140-6736(01)05412-5
Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R (1997) Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16(1):64–67. https://doi.org/10.1038/ng0597-64
Tate G, Suzuki T, Endo Y, Mitsuya T (2008) A novel mutation of the PTEN gene in a Japanese patient with Cowden syndrome and bilateral breast cancer. Cancer Genet Cytogenet 184(1):67–71. https://doi.org/10.1016/j.cancergencyto.2008.03.013
Magaña M, Landeta-Sa AP, López-Flores Y (2022) Cowden disease: a review. The Am J Dermatopathol 44(10):705–717. https://doi.org/10.1097/DAD.0000000000002234
Plamper M, Born M, Gohlke B, Schreiner F, Schulte S, Splittstößer V, Woelfle J (2020) Cerebral MRI and clinical findings in children with PTEN hamartoma tumor syndrome: can cerebral MRI scan help to establish an earlier diagnosis of PHTS in children? Cells 9(7):1668. https://doi.org/10.3390/cells9071668
Plamper M, Schreiner F, Gohlke B, Kionke J, Korsch E, Kirkpatrick J, Born M, Aretz S, Woelfle J (2018) Thyroid disease in children and adolescents with PTEN hamartoma tumor syndrome (PHTS). Eur J Pediatr 177(3):429–435. https://doi.org/10.1007/s00431-017-3067-9
Chilosi AM, Brovedani P, Cipriani P, Casalini C (2023) Sex differences in early language delay and in developmental language disorder. J Neurosci Res 101(5):654–667. https://doi.org/10.1002/jnr.24976
Macken WL, Tischkowitz M, Lachlan KL (2019) PTEN hamartoma tumor syndrome in childhood: a review of the clinical literature. Am J Med Genet Part C, Seminars in medical genetics 181(4):591–610. https://doi.org/10.1002/ajmg.c.31743
Mighell TL, Evans-Dutson S, O’Roak BJ (2018) A saturation mutagenesis approach to understanding PTEN lipid phosphatase activity and genotype-phenotype relationships. Am J Hum Genet 102(5):943–955. https://doi.org/10.1016/j.ajhg.2018.03.018
Martin H, Bessis D, Bourrat E, Mazereeuw-Hautier J, Morice-Picard F, Balguerie X, Chiaverini C, Groupe de Recherche de la Société Française de Dermatologie Pédiatrique (2020) Cutaneous lipomas and macrocephaly as early signs of PTEN hamartoma tumor syndrome. Pediatr Dermatol 37(5):839–843. https://doi.org/10.1111/pde.14265
Kato K, Mizuno S, Inaba M, Fukumura S, Kurahashi N, Maruyama K, Ieda D, Ohashi K, Hori I, Negishi Y, Hattori A, Saitoh S (2018) Distinctive facies, macrocephaly, and developmental delay are signs of a PTEN mutation in childhood. Brain Dev 40(8):678–684
Patraquim C, Fernandes V, Martins S, Antunes A, Marques O, Carvalho JL, Correia-Pinto J, Meireles C, Ferreira AM (2017) A pediatric case of Cowden syndrome with Graves’ disease. Case Rep Pediatr 2017:2750523. https://doi.org/10.1155/2017/2750523
Nowwarote N, Osathanon T, Fournier BPJ, Theerapanon T, Yodsanga S, Kamolratanakul P, Porntaveetus T, Shotelersuk V (2023) PTEN regulates proliferation and osteogenesis of dental pulp cells and adipogenesis of human adipose-derived stem cells. Oral Dis 29(2):735–746. https://doi.org/10.1111/odi.14030
Parisi MA, Dinulos MB, Leppig KA, Sybert VP, Eng C, Hudgins L (2001) The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J Med Genet 38(1):52–58. https://doi.org/10.1136/jmg.38.1.52
Zhou J, Parada LF (2012) PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol 22(5):873–879. https://doi.org/10.1016/j.conb.2012.05.004
Stein MT, Elias ER, Saenz M, Pickler L, Reynolds A (2010) Autistic spectrum disorder in a 9-year-old girl with macrocephaly. J Dev Behav Pediatr 31(7):632–634. https://doi.org/10.1097/DBP.0b013e3181ef422a
Suzuki H, Hosokawa K, Ono M, Kojima Y, Kanda M, Shibasaki H (2017) Brain Nerve 69(12):1442–1446. https://doi.org/10.11477/mf.1416200932
Yotsumoto Y, Harada A, Tsugawa J, Ikura Y, Utsunomiya H, Miyatake S, Matsumoto N, Kanemura Y, Hashimoto-Tamaoki T (2020) Infantile macrocephaly and multiple subcutaneous lipomas diagnosed with PTEN hamartoma tumor syndrome: a case report. Mol Clin Oncol 12(4):329–335. https://doi.org/10.3892/mco.2020.1988
Sang PF, Wang H, Wang M, Hu C, Zhang JS, Li XJ, Zhu F (2015) NEDD4-1 and PTEN expression in keloid scarring. Genet Mol Res 14(4):13467–13475. https://doi.org/10.4238/2015.October.28.7
Smpokou P, Fox VL, Tan WH (2015) PTEN hamartoma tumour syndrome: early tumour development in children. Arch Dis Child 100(1):34–37. https://doi.org/10.1136/archdischild-2014-30599
Dhamija R, Weindling SM, Porter AB, Hu LS, Wood CP, Hoxworth JM (2018) Neuroimaging abnormalities in patients with Cowden syndrome: retrospective single-center study. Neurol Clin Pract 8(3):207–213. https://doi.org/10.1212/CPJ.0000000000000463
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature.
Author information
Authors and Affiliations
Contributions
Conceptualization: J.M.V., M.C.C.M., P.P.M.; clinical data collection: J.M.V., M.J.R., M.C.C.M., P.P.M.; genetic analysis: M.I.G., E.M.V., S.M.L.; formal analysis and investigation: J.M.V., N.G.U.; writing—original draft preparation: J.M.V.; figures and tables preparation: J.M.V., N.G.U.; writing—reviewing and editing: all authors; supervision: M.C.C.M., P.P.M. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Informed consent statement
Informed consent was obtained from the parents of all subjects involved in the study.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martín-Valbuena, J., Gestoso-Uzal, N., Justel-Rodríguez, M. et al. PTEN hamartoma tumor syndrome: Clinical and genetic characterization in pediatric patients. Childs Nerv Syst 40, 1689–1697 (2024). https://doi.org/10.1007/s00381-024-06301-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-024-06301-2