
Abstract
Weeds are responsible for major crop losses worldwide but can provide beneficial agroecosystem services. This study aimed to elucidate how arbuscular mycorrhizal fungi (AMF) in weeds respond to host identity and conservation agricultural practices. The study was carried out at two locations in Southern Africa during off-season and in-season maize cultivation. Off-season AMF root colonisation, diversity indices and community composition significantly differed among weed species at both locations. Glomus sp. VTX00280 explains most of the AMF community differences. In-season, implementation of conventional tillage with mulching alone (CT + M) or together with crop rotation (CT + M + R) resulted in a 20% increase in AMF colonisation of the constantly occurring weed species, Bidens pilosa (BIDPI) and Richardia scabra (RCHSC), compared with conventional tillage plus rotations (CT + R). The diversity of AMF was highest under no-tillage plus mulching (NT + M). Off-season and in-season AMF structures of both BIDPI and RCHSC were not related, but 39% of the taxa were shared. Structural equation modelling showed a significant effect of the cropping system on weed AMF diversity parameters and weed and maize root colonisation, but no significant influence of weed root AMF traits and maize colonisation was detected on maize yield. This may be explained by the improvement in weed competitive ability, which may have offset the AMF-mediated benefits on yield. Our findings highlight that implementing M and CR to CT and NT positively affected weed AMF colonisation and diversity. The similarity between the off-season and in-season AMF composition of weeds supports the fact that weeds functionally host AMF during the non-crop period.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The increasing demand for food for the continuously growing world population calls for more sustainable management practices that promote yield whilst reducing the impact on the environment and biodiversity (MacLaren et al. 2020). Among these practices, the management of weeds is one of the major challenges worldwide in large- and small-scale agriculture systems. This is especially true in southern Africa under smallholder cropping systems, where weeds account for 10 to 100% of yield losses in cereals, depending on the involved weed species and the level of management (Heyl 2022). In this area, the high losses are mainly due to inadequate weed management practices, such as late weeding caused by a lack of manpower and inadequate associated practices, such as rotation, intercropping and mulching (Silva et al. 2019). On the other hand, successful weed management requires a deep knowledge of the weed communities occurring under different cropping systems, especially under multi-component systems, such as conservation agriculture (CA), where management practices can be implemented in different combinations (Derrouch et al. 2021). Conservation agriculture is based on three main principles: minimum soil disturbance, permanent soil cover and crop diversification (FAO 2019). Modification of cropping systems, especially through changing of cropping sequences and including mulching, altered weed species composition (Koocheki et al. 2009; Zhang et al. 2021).
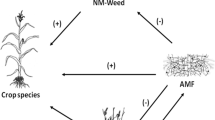
Despite their negative effects on crop productivity due to competition for water, radiation and nutrients, weeds can provide beneficial agroecosystem services (MacLaren et al. 2020; El Omari and El Ghachtouli 2021). Thus, several options, different from full weed eradication, have been suggested with the goal of achieving a trade-off between negative impacts and positive effects resulting from the conservation of weed diversity and functionality. Indeed, a more diverse weed community has recently been shown to be less competitive with many crops, as well as to promote crop health and beneficial bees (e.g. Storkey and Neve 2018; Ferrero et al. 2017; Bretagnolle and Gaba 2015). Regarding winter cereals, Adeux et al. (2019) have recently demonstrated that the reduction in yield loss was better explained by the increase in weed diversity rather than the decrease in weed density. In addition, ecosystem services can be rendered by some weed species, such as black-jack (Bidens pilosa L.), barnyard grass (Echinochloa crus-galli (L) P. Beauv) and black nightshade (Solanum nigrum L.), forming associations (called mycorrhizas) with arbuscular mycorrhizal fungi (AMF) (Veiga et al. 2011; Massenssini et al. 2014) that are taxonomically classified either as a phylum, Glomeromycota (Schüβler et al. 2001; Hibbett et al. 2007; Tedersoo et al. 2018), or as the sub-phylum Glomeromycotina, which together with Mortierellomycotina and Mucoromycotina, make up the phylum Mucoromycota (Spatafora et al. 2016; James et al. 2020; Li et al. 2021). As obligate mutualistic symbionts, AMF acquire nutrients (e.g. phosphorus (P), nitrogen (N), sulphur (S)), through the extraradical mycelium, which acts as an extension of the host root system and transfer them to the host plant in exchange for photosynthetically assimilated carbon (4% to 20% of total fixed C) (Smith and Read 2008; Gavito et al. 2019). Thus, it is expected that the relationship that AMF form through mycelial fungal networks with host plants, such as the mycorrhizal weeds, will increase their growth and thus help them to proliferate through the acquisition of nutrients and water (Wilson and Hartnett 1998; van der Heyde et al. 2017). However, the AMF-weed interaction might not be of the mutualistic type, and this is the case of some ruderal plants, including several agricultural suppressive weeds that respond negatively to AM fungal colonisation (Vatovec et al. 2005; Veiga et al. 2011). Some non-mycorrhizal and mycorrhizal weeds exhibited growth suppression induced by AMF (i.e. reduced biomass, growth rate and survival) through direct antagonistic effects, such as fungal parasitism and defence response of plants, and indirect effects in the triple interaction of AMF-weed-crop species by benefiting the associated mycorrhizal crop (Qiao et al. 2016; El Omari and El Ghachtouli 2021). Moreover, weeds can also become alternative AMF hosts during the off-season (non-crop growing dry periods) (Massenssini et al. 2014). This is particularly important in southern Africa, which is characterised by short crop growing seasons and long winter dry periods within the year. Furthermore, arbuscular mycorrhizas (AM) formed with native weed species can increase their competitive ability against invasive species and hence prevent their dominance (Zhang et al. 2018; El Omari and El Ghachtouli 2021). Despite abundant reports on the AM fungal host preference/specificity in field trials, supporting specialisation between plant and associated AMF community (e.g. Gollotte et al. 2004; Helgason et al. 2007; Martínez-García and Pugnaire 2011; Li et al. 2019), no information is available on the effect of host identity on AM fungal assemblages in off-season and in-season weeds. This effect can also be modulated by agronomical management practices that might affect the weed outcome, ranging from suppression to promotion (Bever 2002; Zhang et al. 2010). This is particularly important in the CA systems of southern Africa where farmers implement the components in different combinations, resulting in different weed communities potentially hosting diversified AM fungal assemblages having differential functions.
Thus, in the present study, we aimed to verify if mycorrhizal weeds occurring under distinct cropping systems could act as hosts of AMF during the off-season and in-season, and if AM fungal assemblages would be affected by host identity and by the CA components (cropping system). We hypothesised that (i) the implementation of all three CA components leads to a promotion of AMF through the increase of AMF colonisation and diversity within weed roots and that these traits are shaped by the identity of the hosting weed species; (ii) the identity of the hosting weed species would also affect the AM fungal colonisation and community composition and diversity of the weeds off-season; (iii) off-season and in-season AM fungal composition of weeds are related. Moreover, we aimed to dissect the potential effects of AM fungal diversity and root colonisation of maize and weeds on maize grain yield. The elucidation of these topics is necessary to set up optimal weed control strategies with the goal of looking for an equilibrium between the control of damage caused by weeds and the conservation of biodiversity, ecosystem functioning and soil quality.
Material and methods
Experimental field locations
The experiment was done at two sites namely the Domboshawa Training Centre (DTC) and the University of Zimbabwe (UZ). The geographical location and climate of the two locations are given in Table 1. Soil sampling was carried out in November 2018 before maize sowing. Soil properties and the corresponding analytical methods are given in Table 1. The experiments at the two locations started in the summer crop growing season of 2013, and in this study, we report data collected in the 2019 growing seasons only. The climate of the two sites is classified as warm temperate with dry winters and hot summers (Kottek et al. 2006). Between the two sites, DTC had the highest average daily temperature of 29.0 °C and rainfall of 630 mm, whilst UZ had an average daily temperature of 27.5 °C and rainfall of 383 mm during the study period (Fig. S1).
Experimental set-up and crop management
The experiments consisted of eight treatments (referred to as the cropping system hereafter), which were arranged in a randomised complete block design (RCBD), and these were as follows:
-
i.
Conventional tillage (CT)—land preparation was done through digging with a hand hoe to simulate ploughing. Maize was sown as a monocrop in either riplines created using a Magoye (DTC) or basins (UZ) created afterwards. Crop residues were removed from the field after harvesting.
-
ii.
Conventional tillage plus mulch (CT + M)—land preparation and crop sowing as in treatment (i). Crop residues were retained on the soil surface at a rate of 2.5 t ha−1 at the beginning of the season.
-
iii.
Conventional tillage plus rotation (CT + R)—land preparation was done as in treatment (i), but maize was rotated with cowpea (Vigna unguiculata (L.) Walp.) in 1-year rotations.
-
iv.
Conventional tillage plus mulch and rotation (CT + M + R)—land preparation was done through digging with a hand hoe to simulate ploughing, maize was rotated with cowpea in 1-year rotations and crop residues were retained on the soil surface at a rate of 2.5 t ha−1 at the beginning of the season.
-
v.
No-tillage (NT)—no soil inversion was done, and maize was sown as a monocrop in either riplines created using a Magoye (DTC) or basins (UZ) created afterwards. Crop residues were removed from the field after harvesting.
-
vi.
No-tillage plus mulch (NT + M)—no soil inversion was done, and maize was sown as a monocrop in either riplines created using a Magoye (DTC) or basins (UZ) created afterwards. Crop residues were retained on the soil surface at a rate of 2.5 t ha−1 at the beginning of the season.
-
vii.
No-tillage plus rotation (NT + R)—land preparation and crop residue management were done as in treatment (v), maize was sown in rotation with cowpea and crop residues were removed at harvest.
-
viii.
No-tillage plus mulch and rotation (NT + M + R); referred to as CA herein—land preparation was done as in treatment (v), maize was rotated with cowpea in 1-year rotations and crop residues were retained on the soil surface at a rate of 2.5 t ha−1 at the beginning of the season.
All treatments were replicated four times. For treatments involving rotation, plots were split into half and maize was sown in a 1-year rotation with cowpea, with phases of the rotation present in each year. The in-plots measured 12 m × 6 m (72 m2). The maize rows were spaced at 90 cm and the maize plants at 25 cm. The cowpea rows were spaced at 45 cm whilst the cowpea plants were spaced at 25 cm. The maize population was 44,444 plants ha−1 whilst the cowpea population was 88,888 plants ha−1. Both maize and cowpea received a basal fertiliser in the form of compound D (7:14:7 NPK) at the rate of 11.6 kg N ha−1, 10.1 kg P ha−1 and 9.6 kg potassium (K) ha−1 at sowing, and maize further received a top-dressing fertiliser in the form of ammonium nitrate (NH4NO3) at the rate of 46 kg N ha−1, split applied 4 and 7 weeks after sowing. Weeds were controlled by spraying glyphosate (N-(phosphono-methyl) glycine) at the rate of 1.025 L active ingredient ha−1 at the beginning of the season. Weeds were then manually controlled using hand hoes whenever weeds were 10 cm tall or 10 cm in diameter for stoloniferous weeds.
Maize yield assessment
For each plot, maize plants were harvested from four rows that were 5 m long (i.e. an area of 18 m2). Maize cobs and stover were separated and a fresh weight of 10 cobs was recorded. These cobs were air-dried for 4 weeks and reweighed for dry weight. The moisture content of the grain was determined, and the yield was expressed at 12.5% moisture content. Maize stover was dried, and the stover was determined on a dry weight basis.
AM fungal root colonisation of weeds and maize and intraradical AMF diversity and community composition
Assessment of AM fungal root colonisation of maize and weeds
To assess if weeds can act as alternative hosts of AMF during the off-season (pre-season; which was at 30 days before crop sowing) and during the season (at 60 days after crop sowing, which corresponds to the silking stage (R1); hereafter indicated as anthesis) and to also assess the effect of host identity (weed species) and cropping system, we determined AM fungal root colonisation and molecularly characterised the diversity and community compositions within the roots of weeds. For the pre-season, we identified five previously reported mycorrhizal weed species that were growing at the edges of the experimental field in the winter dry period. These weed species were as follows: Bidens pilosa L. (Asteraceae) (BIDPI), Cynodon nlemfuensis Vanderyst (Poaceae) (CYNNL), Erigeron sumatrensis Retz. (ERISU), Melinis repens (Willd.) Zizka (Poaceae) (RHYRE) and Richardia scabra L. (Rubiaceae) (RCHSC). For the anthesis assessment, we identified the species from the list of weeds collected in the pre-season present in all experimental plots. Two species (i.e. BIDPI and RCHSC) were common to all cropping systems at UZ, whereas at DTC, no common species were found across cropping systems. For both the pre-season and anthesis samplings, we randomly collected the roots of four plants (replicates) for each species at both locations (Fig. S2). At anthesis, the four plants were collected from each plot (four replicates per cropping system), and then mixed to form one composite sample per plot. For maize, we assessed if cropping systems influenced the percentage of AM fungal root colonisation by sampling four plants from each plot at the anthesis stage, i.e. the pollination stage (R1). The percentage of AM fungal root colonisation and root length containing arbuscules and vesicles were assessed using the magnified intersections method of McGonigle et al. (1990) after clearing and staining (Phillips and Hayman 1970). Details are given in Section 1 of the Supplementary Materials and Methods.
Molecular analysis
Plant DNA was extracted from 0.02 g of fine roots of the weeds, collected at pre-season and anthesis (pre-season: a total of 20 samples, four replicates per five plant species; anthesis: 48 samples, three replicates per two plant species per eight cropping systems only at the UZ location), using the DNeasy® Plant Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer’s instructions. Taking into consideration the patchy distribution of AMF within roots, weed root fragments, i.e. 20 mg used for DNA extraction, were chosen by randomly sampling from fine roots and then selecting those having good AM fungal colonisation. Root pieces (2–3 cm long) were mounted on slides in water and observed under a Zeiss Jenamed2 microscope with tungsten and UV lamps. Filter combinations used for fluorescence microscopy were BPF510 Excitation BPF475 (× 3)/Barrier G247, G245 (Ames et al. 1982; Merryweather and Fitter 1991). Root samples showing a detectable level of autofluorescence were selected for DNA extraction. The extracted genomic DNA was quantified by a Nanodrop spectrophotometer (Thermo Fisher Scientific, Vantaa, Finland) and then stored at − 20 °C for further analyses. The DNA was amplified using an amplicon-specific polymerase chain reaction (PCR). A two-step nested PCR approach was used with two primer pairs to amplify the small subunit ribosomal RNA (SSU) fragments. In the first step, the forward primer AML1 (5′-ATC AAC TTT CGA TGG TAG GAT AGA-3′) and reverse primer AML2 (5′-GAA CCC AAA CAC TTT GGT TTCC-3′) (Lee et al. 2008) were used, and in the second step, the forward primer WANDA-ill (5′-TCG TCG GCA GCG TCA GAT GTG TAT AAG AGA CAG ANN NHN NNW NNN HGC AGC CGC GGT AAT TCC AGCT-3′) (Dumbrell et al. 2011) and reverse primer AML2-ill (5′-GTC TCG TGG GCT CGG AGA TGT GTA TAA GAG ACA GGA ACC CAA ACA CTT TGG TTT CC-3′) (Lee et al. 2008) were used (the adaptors for the Illumina reaction are in bold type). Both PCR reactions were performed in a 25-µl volume using 1 µl of the genomic DNA template (undiluted DNA at 13.5 ± 1.2 ng µl−1), 1.25 U µl−1 of GoTaq® Hot Start Polymerase (Promega Corporation, WA, USA), 0.2 µM of each primer, 0.2 mM of dNTPs, 2 mM of MgCl2 and 1 × reaction buffer. The PCR cycle for both steps involved an initial denaturation at 95 °C for 2 min followed by 25 cycles of 94 °C for 30 s, 59 °C for 45 s, 72 °C for 1 min 30 s and 72 °C for 10 min. All PCR reactions were carried out using an S1000 Thermal Cycler™ (BioRad, Hercules, CA, USA). The quality of the PCR products was checked by gel electrophoresis using 2% agarose gel in 1 × TBE buffer and then purified with magnetic beads (Agencourt AMPure® XP, Beckham Coulter, USA) and freshly prepared 80% ethanol. The concentration of DNA was then quantified using fluorimetry (Invitrogen™ Qubit™ 4 fluorometer) by the Qubit4™ 1 × dsDNA High-Sensitivity Assay Kit (Invitrogen, Thermo Fisher Scientific, CA, USA), following the manufacturer’s instructions. The cleaned and quantified amplicons of each library were then adjusted to an equimolar ratio of 10 ng µl−1 for the addition of dual-index barcodes using the Nextera® XT DNA library preparation kit (Illumina Inc., CA, USA). For more information on the dual-indexing procedure, please refer to Section 2 of the Supplementary Materials and Methods. The generated metabarcoding libraries were run on an Illumina MiSeq sequencer at the University of York (UK), loading a 12-pM final library concentration with 20% PhiX library spike-in (Illumina) and an Illumina MiSeq V3 600 cycle sequencing kit.
Bioinformatics analyses
Raw sequence data were processed and analysed using the QIIME2 (2018.11) pipeline and plugins (Bolyen et al. 2019). Demultiplexed forward and reverse paired-end reads were joined using the ‘-fastq_mergepairs’ of the USEARCH plugin (Edgar 2010). Out of the 1,867,193 reads exposed to merging, 89% (1,662,165 reads) were successfully merged and 47% (874,286 reads) were aligned with zero differences. Primer sequences were trimmed off from the sequences using the cutadapt plugin 1.18 with Python 3.5.5, and 1,659,726 valid sequences were obtained after optimisation. The average read length was approximately 250 base pairs (bp) based on the maximum expected error (MaxEE). The command USEARCH ‘fastq_eestats2’ was used to check sequence quality and, based on the percentage MaxEE, reads were truncated at the drop-off point of 250 bp using the USEARCH ‘fastq_filter’ command. Quality-filtered reads were dereplicated using the USEARCH ‘fastx_uniques’ command, and operational taxonomic units (OTUs) were generated by clustering reads at a 97% similarity threshold using the USEARCH ‘cluster_otus’ command. During the process, chimeric sequences and singletons were also removed. The resulting OTUs were assigned to virtual taxa (VT) using the MaarjAM database (https://maarjam.botany.ut.ee). All representative sequences (143 in total) were submitted to the NCBI sequence read database (submission number SUB10794739), and these correspond to the accession numbers OM049043–OM049185. The representative sequences were aligned using the MAFFT online service (Katoh et al. 2019), and a Neighbour-Joining tree was built using MEGA11 (Tamura et al. 2021), following the bootstrap test of phylogeny with 1000 bootstraps. The substitution model used was the Kimura 2-parameter with uniform rates among sites, pairwise deletion and 7 threads.
Calculations and statistical analyses
Weed community diversity and AMF diversity in weed roots
Data analyses were done separately for the clay and sand locations. Community diversity of AMF within weed roots was also computed using Shannon’s diversity (H′), Pielou’s evenness (J') and richness (S) based on VT counts. Shannon-Weiner index (Shannon and Weaver 1949) was calculated as:
where H′ is the Shannon-Weiner diversity index, Pi is the proportion of individuals belonging to the ith VT and S is the total number of VT.
Pielou’s evenness index (Pielou 1969) was calculated as the ratio of observed diversity to maximum diversity as follows:
where H′ is the Shannon’s diversity, and Hmax or lnS is the maximum Shannon diversity in which all present VT appear in equal abundances for a community, and S is the number of observed VT. Evenness values range between 0 and 1, representing absolute dominance and equal VT abundance, respectively.
AM fungal data were assessed for normality and where necessary, data were fourth-root transformed before further analyses. The effect of host weed species identity, cropping systems and their interaction (treated as fixed factors) on H′, J′ and S were assessed using linear mixed models using the ‘lme4’ package (Bates et al. 2015) in the R environment (R Core Team 2021). Replicates were included in the analyses as a random factor. The means and standard errors of back-transformed data were reported herein. F-tests were used to test the significance of fixed effects, and where means were significantly different, the mean comparison procedure was used to contrast them based on the Tukey’s tests (P < 0.05) using the ‘emmeans’ package (Lenth 2019) in R.
Weed and maize root colonisation by AMF
For AM fungal root colonisation percentage of weeds and maize, data were checked for normality and fourth-root transformed before analysis. For weed data, mixed models were used to assess the effects of host species identity, cropping systems and their interaction (fixed effects) on the percentage of AM fungal colonisation, and root length containing arbuscules and vesicles. For maize data, mixed models were used to assess the effects of cropping systems on the colonisation rate. In both cases, replicates were included as random factors. The significance of the fixed effects was tested using F-tests and where means were significantly different, they were contrasted using a mean comparison procedure following Tukey’s tests (P < 0.05). However, the back-transformed means and standard errors were reported.
Intraradical AM fungal community composition
To analyse the effect of weed species identity and cropping system on AMF community structure in weed roots, we used type III permutational multivariate analysis of variance (PERMANOVA). As a semiparametric multivariate test, PERMANOVA generates pseudo-F ratios and P values using the Monte Carlo permutation P(MC) test by permutating the resemblance measures (Anderson 2001). In our analyses, 999 permutations were employed.
AM fungal species relative abundances were fourth-root transformed, and a resemblance matrix was constructed based on the Bray–Curtis dissimilarity index (Bray and Curtis 1957) before carrying out PERMANOVA. Where group differences in community composition were detected, similarity percentage analysis (SIMPER) was done to detect the species responsible for 70% of the differences by calculating the percentage contribution of the species to the total effects. Further, we carried out a permutation test for homogeneity of multivariate dispersions (PERMDISP) on each significant factor level. This test is used as a measure of multivariate beta diversity to check whether the significant group differences observed in PERMANOVA were also not influenced by differences in the dispersion of group objects from the group centroid (alpha diversity).
Principal coordinates analysis (PCoA) was then performed to visualise relevant patterns in the data. Finally, we used the RELATE procedure based on Spearman rank correlation to test if there was a relationship between the AM fungal communities observed in the roots of BIDPI and RCHSC collected during the pre-season and the anthesis periods at UZ using the Primer 7 (with PERMANOVA +) software (called the RELATE test). All the multivariate analyses were performed using Primer 7 with PERMANOVA + software (Anderson 2001; Clarke et al. 2014). Finally, Venn diagrams were constructed to show the shared and unique AM fungal taxa within the roots of BIDPI and RCHSC weed species collected at UZ at pre-season and anthesis (BIDPI pre-season vs. BIDPI anthesis; RCHSC pre-season vs. RCHSC anthesis) and between BIDPI and RCHSC at anthesis. The diagrams were built using Venny 2.1 (Oliveros 2015).
Relationship between cropping systems, weed AMF diversity, maize AM fungal colonisation, weed AM fungal colonisation and maize grain yield
To assess the effect pathway of weed AMF diversity parameters (H′, J′, and S′, maize AM fungal colonisation, and weed AM fungal colonisation on maize grain yield, we used piecewise structural equation modelling (pSEM). The pSEM analysis was based on multiple regression and was done using the ‘piecewiseSEM’ package in R (Lefcheck 2016). Models were fit using linear models, and variables were standardised for the effects to be directly comparable, and for each pathway, a standardised coefficient (λ) was estimated. In the models, we also calculated the covariance of weed AM fungal H′ and J′; weed AM fungal H′ and S; and RCHSC and BIDPI colonisation rate. Model fits were estimated by the Fisher’s C test.
Results
Effect of cropping systems and host identity on AM fungal root colonisation of weeds and on AM fungal diversity and community structure in weed roots
After blast matching of OTUs against the MaarjAM database, 143 AM fungal virtual taxa (VT) were retrieved, and these belonged to seven families, namely Acaulosporaceae, Archaesporaceae, Claroideoglomeraceae, Diversisporaceae, Gigasporaceae, Glomeraceae and Paraglomeraceae (Fig. S3). Of all the VT, 57% belonged to the Glomeraceae, 10% to Acaulosporaceae and 10% to Gigasporaceae (Fig. S3).
At pre-season, the percentage of AM fungal root colonisation and of root length containing arbuscules and vesicles significantly differed among weed species at DTC (sandy location) (Fig. 1a). CYNNL showed an AM fungal root colonisation significantly lower than ERISU and RCHSC (12% vs 25%). Arbuscules and vesicles were higher within RHYRE as compared to other weed species, whilst RCHSC had the lowest percentage of arbuscules and CYNNL and ERISU the lowest percentage of vesicles (Fig. 1a). At UZ (clay location), CYNNL and RHYRE showed a significantly lower AM fungal root colonisation (18%) compared with BIDPI and ERISU (30%) (Fig. 1b). Arbuscules differed among weed species, with the RCHSC having the highest percentage and RHYRE the lowest (Fig. 1b). At anthesis, we found only two plant species (BIDPI and RCHSC) over the five pre-season mycorrhizal weeds that were common to all cropping systems at UZ, whereas no common plant species were found at DTC. Indeed, at the UZ location, the cropping system and weed identity had significant effects on AM fungal root colonisation (Figs. 1c, d). The implementation of CT with mulching alone (CT + M) or together with crop rotation (CT + M + R) resulted in a higher AM fungal root colonisation (50%) with respect to the implementation of crop rotation alone (CT + R) (30%) (Fig. 1c). Moreover, RCHSC had a higher AM fungal root colonisation than BIDPI (45% vs. 36%) (Fig. 1d). Percentage of vesicles differed among the two weed species with the RCHSC having higher colonisation (11%) as compared to that of BIDPI’s (8%) (Fig. 1e). The interaction of cropping system and weed identity had a significant effect on the percentage of arbuscules, with RCHSC under CT + R and NT + R having the highest rate (Fig. 1f).

Effect of weed species identity on the percentage of arbuscular mycorrhizal (AM) fungal root colonisation and of root length containing arbuscules and vesicles of five weed species collected from the experimental boundaries at the Domboshawa Training Centre (DTC; sandy location) (a) and the University of Zimbabwe (UZ; clay location) (b) during the off-season (pre-season). AM fungal root colonisation as affected by different cropping systems (c), weed species (d), vesicles as affected by weed species (e) and arbuscules as affected by the interaction of cropping system and weed species (f) at UZ during the in-season (anthesis) in 2019. Abbreviations of the weed species based on European and Mediterranean Plant Protection Organization coding are BIDPI, Bidens Pilosa; CYNNL, Cynodon nlemfuensis; ERISU, Erigeron sumatrensis; RHYRE, Melinis repens; RCHSC, Richardia scabra. Abbreviations of the cropping systems are CT, conventional tillage; CT + M, CT plus mulch; CT + R, CT plus crop rotation; CT + M + R, CT plus mulch and rotation; NT, no-tillage; NT + M, NT plus mulch; NT + R, NT plus rotation; NT + M + R, NT plus mulch and rotation. Values are means ± SE of four replicates for each cropping system per season. Bars with different letters are significantly different from each other based on P values: **P < 0.01; *P < 0.05 (see Table 2)
At pre-season, the AM fungal communities showed significantly different H′ and J′ among weed species at both locations (Table 2). The weed species BIDPI consistently exhibited the highest H′ and J′ (Figs. 2a–d). At anthesis, whilst no differences were observed in terms of AM fungal diversity indices between BIDPI and RCHSC, the cropping system had a significant effect on H′ and S, with the NT + M system showing higher values at both locations as compared with CT, CT + R and NT + M + R (Figs. 2e, f). Interestingly, under the NT + M system, AMF had also a higher S than under NT + M + R (Figs. 2e, f).

Effect of species identity on arbuscular mycorrhizal (AM) fungal community Shannon diversity (H′) and Pielou (J') evenness during the off-season (pre-season) at the Domboshawa Training Centre (DTC; sandy location) (a and b) and at the University of Zimbabwe (UZ; clay location) (c and d), and effect of cropping system on AM fungal Shannon diversity index (H′) and taxonomic richness (S) at UZ in-season (anthesis) (e and f). Abbreviations of the weed species based on European and Mediterranean Plant Protection Organization coding are BIDPI, Bidens pilosa; CYNNL, Cynodon nlemfuensis; ERISU, Erigeron sumatrensis; RHYRE, Melinis repens; RCHSC, Richardia scabra. Abbreviations of the cropping systems are CT, conventional tillage; CT + M, CT plus mulch; CT + R, CT plus crop rotation; CT + M + R, CT plus mulch and rotation; NT, no-tillage; NT + M, NT plus mulch; NT + R, NT plus rotation; NT + M + R, NT plus mulch and rotation. Values are means ± SE of four replicates for each cropping system per season. Bars with different letters are significantly different from each other based on P values reported in Table 2
Based on the PERMANOVA results, weed species collected at pre-season hosted different AM fungal communities at both locations, whereas at anthesis, only the cropping system significantly shaped the AM fungal communities (Table 3). This is also supported by the PCoA plots explaining 63% and 50% of the total variance in pre-season at DTC and UZ, respectively, where the group centroids of the AM fungal communities retrieved at pre-season within the roots of the weed species were clearly separated on the ordination space (Figs. 3a, c). Similarly, the PERMANOVA results at anthesis were supported by the PCoA plot, explaining 41% of the total variance, in which the group centroids of the AM fungal communities retrieved in the cropping systems were clearly separated in the ordination space (Fig. 3e). This occurred despite the high percentage of AMF common to all cropping systems (core community: 24% among CT-based systems and 26% among NT-based systems; 60% between CT-based and NT-based systems) (Fig. S4). For both pre-season and anthesis data, the distances of the group object to their centroid did not significantly differ, supporting similar alpha diversity (Figs. 3b, d, f). The SIMPER analyses revealed that AM fungal taxa, such as Glomus sp. VTX00280, explained most of the AM fungal community differences in most of the weed species collected at pre-season in both locations (Figs. 4a, b). However, at DTC, Glomus sp. VTX00092 and Glomus sp. VTX00264 also showed high contributions to the AM fungal community differences (up to 30% in the CYNLE) (Fig. 4a). As for the weeds collected during the anthesis period at the UZ, although the AM fungal taxa, such as Acaulospora sp. VTX00028, Claroideoglomus sp. VTX00193, Dentiscutata sp. VTX00255, Gigaspora sp. VTX00039 and Glomus sp. VTX00112, strongly contributed to the community differences among cropping systems (Fig. 4c).

Principal coordinates analysis (PCoA) based on Bray–Curtis distance dissimilarity of fourth-root transformed AMF community relative abundances. Plots show the AM fungal differences among weed species and cropping systems at the Domboshawa Training Centre (DTC; sandy location) (a) and at the University of Zimbabwe (UZ; clay location) (c and e) (see Table 2). Permutational dispersion (PERMDISP) tests on the same data matrices at DTC and UZ (b and d weed species; f cropping system) represented by the distances of the objects from the centroid and standard error (SE). Abbreviations of the weed species based on European and Mediterranean Plant Protection Organization coding are BIDPI, Bidens pilosa; CYNNL, Cynodon nlemfuensis; ERISU, Erigeron sumatrensis; RHYRE, Melinis repens; RCHSC, Richardia scabra. Abbreviations of the cropping systems are CT, conventional tillage; CT + M, CT plus mulch; CT + R, CT plus crop rotation; CT + M + R, CT plus mulch and rotation; NT, no-tillage; NT + M, NT plus mulch; NT + R, NT plus rotation; NT + M + R, NT plus mulch and rotation. Bars with different letters are significantly different based on the reported P-permutational values (Pperm)

Similarity percentages analysis (SIMPER) identifying the arbuscular mycorrhizal (AM) fungal taxa that were responsible for the AM fungal community differences among weed species at the Domboshawa Training Centre (DTC; sandy location) (a) and the University of Zimbabwe (UZ; clay location) (b) and among cropping systems at UZ (c). The listed species explain approximately 70% of the contribution. Abbreviations of the weed species based on European and Mediterranean Plant Protection Organization coding are BIDPI, Bidens Pilosa; CYNNL, Cynodon nlemfuensis; ERISU, Erigeron sumatrensis; RHYRE, Melinis repens; RCHSC, Richardia scabra. Abbreviations of the cropping systems are CT, conventional tillage; CT + M, CT plus mulch; CT + R, CT plus crop rotation; CT + M + R, CT plus mulch and rotation; NT, no-tillage; NT + M, NT plus mulch; NT + R, NT plus rotation; NT + M + R, NT plus mulch and rotation
An analysis comparing the AM fungal communities retrieved from the roots of the same weed species (i.e. BIDPI and RCHSC) collected at pre-season with those collected at anthesis revealed different AM fungal community compositions (Rho = 0.268; P > 0.05) (Fig. S4a). However, some AM fungal taxa were common to the weed species. For example, in terms of AM fungal composition, BIDPI sampled at pre-season had 36% of the same AM fungal taxa as BIDPI sampled at anthesis (Fig. S4b). Similarly, RCHSC sampled at pre-season and anthesis showed 42% of common AM fungal taxa (Fig. S4c). Finally, the comparison between the AM fungal community composition of the two weed species at anthesis showed that 53% of the retrieved AM fungal taxa were shared, 47% were unique to BIDPI, and no taxa were unique to RCHSC (Fig. S4d).
Effect of cropping system on maize productivity and relationship with weed AMF diversity, maize AM fungal colonisation and weed AM fungal colonisation
Data on maize productivity and AM fungal root colonisation has already been reported by Mhlanga et al. (2022) and hence will not be reported herein, but we refer the reader to the aforementioned paper. In this current analysis, we will use the same data to relate to weed AMF diversity, maize AM fungal colonisation and weed AM fungal colonisation.
Structural equation modelling resulted in an overall significant fit (Fig. 5) (Fisher’s C = 30.63; P-value = 0.242; DF = 26). The cropping system had a significant and positive influence on weed AMF diversity (J′), weed AMF richness (J′), maize AMF root colonisation and colonisation of BIDPI and RCHSC (Fig. 5) (Table 4). All the investigated factors did not have a significant influence on maize grain yield. The cropping system NT + M resulted in the highest path coefficient estimates for H, S, and RCHSC, whilst CT + M had the highest coefficient for BIDPI AMF root colonisation (Table 4). The cropping system NT + M + R had the highest maize AMF root colonisation and grain yield path coefficients, whilst the CT systems had the least grain yield coefficient. Weed AMF diversity showed a positive correlation with weed AMF evenness (λ = 0.74) and weed AMF richness (λ = 0.93).

Structural equation model (SEM) (path analysis) showing the effect of cropping systems on AMF diversity (Shannon diversity (H′)), AMF evenness (Pielou evenness (J′)) and AMF richness (Margalef richness (S) in weed roots and maize, Bidens pilosa (BIDPI) and Richardia scabra (RCHSC) AM fungal root colonisation on grain yield at the University of Zimbabwe (UZ; clay location). The black lines represent positive influence, whilst the red lines represent negative influence. Solid lines and dashed lines represent significant (P < 0.05) and non-significant (P > 0.05) influences, respectively. Standardised path coefficients are reported for each effect pathway
Discussion
Host species identity and cropping system determine AM fungal colonisation, diversity and community structure within the roots of weeds
Despite the potential negative effect of weeds on crop productivity and production costs, weeds can offer agroecosystem services beneficial to crops (MacLaren et al. 2020; El Omari and El Ghachtouli 2021). One of the services that could be provided by weeds is hosting AMF and supporting AM fungal diversity during the off-season (pre-season) in an area surrounding the crop fields, when crops are absent, or during the season (anthesis) along with the crops. This can signify the abundance of AMF in the soil (Barceló et al. 2020). The five mycorrhizal weeds collected during the off-season differed in the percentage of AM fungal root colonisation and of root length containing arbuscules and vesicles. Similarly, at anthesis, RCHSC showed a higher AM fungal root colonisation than BIDPI. These differences may be attributed to the host specificity of AMF, the selectivity or mycorrhizal dependency of the host weeds (Eom et al. 2000; Yang et al. 2012; Ciccolini et al. 2016; Säle et al. 2022). This specificity was also revealed by the different AM fungal community compositions and diversity indices observed within the weed root systems at pre-season. However, the unexplained variance in the PCoA analyses highlights that other factors, in addition to host specificity, play a role in shaping AM fungal community composition. Glomus sp. VTX00280, explaining most of the AM fungal community differences, suggests for the first time that host specificity during dry periods is mainly driven by a unique VT. This indicates that under stress conditions (off-season), the differential supply of host C to the most functional VT promotes its prevalence in roots, improving plant tolerance against dry conditions (Kiers et al. 2011; Omirou et al. 2013). Accordingly, it was previously demonstrated that some species of AMF are less sensitive to water stress than others (Bahadur et al. 2019). However, in the absence of drought stress, the host specificity observed in BIDPI and RCHSC at pre-season was no longer detectable in terms of the AM fungal community composition during the cropping cycle.
Between the two species (BIDPI and RCHSC) that were sampled in the plots at anthesis, the highest AM fungal colonisation was observed in the CT-based systems, either with mulch alone or with mulch and rotation. Despite that intensive tillage was previously observed to reduce AM fungal root colonisation in different crops (Castillo et al. 2006; van der Heyde et al. 2017; Mhlanga et al. 2022), our data suggest that mulching preserves soil moisture and promotes the proliferation of AMF, leading to increased root colonisation (Wilkes et al. 2021). Moreover, mulching added to NT increased AM fungal diversity and richness. This is in agreement with Lu et al. (2018) observing that NT with crop straw retention promoted soil AM fungal diversity with respect to CT. As previously reported, NT and mulching improved soil hydrothermal conditions (Lal 2000) and positively affected fungal diversity (Brito et al. 2012; Piazza et al. 2019; Pellegrino et al. 2020). Moreover, at anthesis, in our study, crop rotation reduced the AM fungal diversity within the roots of BIDPI and RCHSC. Earlier studies have demonstrated that crop rotation increased or did not modify AM fungal diversity (Oehl et al. 2003, Hijri et al. 2006). The positive effect on AMF diversity was found with an extensive crop rotation including a perennial grass-clover mixture (Oehl et al. 2009). Thus, the decrease of AM fungal diversity we found in the systems with crop rotation compared to maize mono-cropping could be linked to the cowpea selection for a few dominant AM fungal species compared to maize (Johnson et al. 2013; Alaux et al. 2021). Since different CA practices result in different weed communities (Mhlanga et al. unpublished results), this gives different AM fungal communities a higher chance of being promoted within a system. Indeed, tillage alters the seedbank and its vertical distribution, the germination, predation and viability and dispersal of weed seeds and the weed community composition and diversity (Nichols et al. 2015). Moreover, crop residues can affect seed germination via physical and chemical changes in the seed environment, whilst rotating crops change the selection pressures, precluding one weed from repeatedly establishing itself. Overall, the implementation of mulching either in NT or CT systems modified the AM fungal community composition as compared to the other systems, despite the high percentage of the core AM fungal taxa. Thus, mulching also plays a crucial role in shaping AM fungal assemblages, as well as in improving AM fungal colonisation and diversity. In accordance, mulching has recently been highlighted as a major driver of improving the stability and resilience of maize-based rainfed systems in southern Africa (Kodzwa et al. 2020; Mhlanga et al. 2021). Most of the AM fungal taxa retrieved from weed roots during the crop cycle belonged to Glomeraceae. Indeed, species of this family, such as Funneliformis mosseae, have a short life cycle that may reduce their sensitivity to discontinuous plant presence and disruption of the extraradical mycelia by frequent tillage (Oehl et al. 2003; Pellegrino et al. 2020). However, our data evidenced among the retrieved Glomus taxa a large variability of response to tillage. As an example, Glomus sp. VTX00112 was abundant under the NT-based systems and was rare under CT systems, whereas Glomus VTX00132 showed the opposite behaviour. These results support the high functional variability within the family Glomeraceae, as previously reported in some studies (Avio et al. 2006; Munkvold et al. 2004). In contrast to previous studies that found Gigasporaceae propagating from intact mycelia to be abundant under NT systems but scarce under intensive tillage (Daniell et al. 2001; Pellegrino et al. 2020), Gigaspora sp. VTX00039 largely occurred under conventionally tilled systems. Our results can be supported by some studies (Schalamuk and Cabello 2010; Hart and Reader 2004) stating that Gigasporaceae is less sensitive to soil disturbance than Glomeraceae because after disturbance, some hyphal fragments lose viability due to cytoplasmatic leakage, whereas spores are not greatly affected, and Gigasporaceae colonise roots primary from spores. Thus, there is still conflicting evidence on the ability of Glomeromycota families to use propagules type and to reconnect once they are disrupted by tillage (De La Providencia et al. 2005).
Our study applied a nested PCR approach using the primer pair AML1/AML2 and the primer pair WANDA-ill/AML2-ill (Lee et al. 2008). Recently, Suzuki et al. (2020) evaluated primer pairs’ suitability for AM fungal community assessment by comparing five approaches, three targeting the 18S rRNA gene (one using the AM fungal-specific primer pair AMV5.4NF/AMDGR (Sato et al. 2005); a nested PCR approach using the AM fungal-specific primer pair AML1/AML2 (Lee et al. 2008) and the N-AMV5.4NF/AMDGR primer set; a nested PCR approach using the AM fungal-specific primer pair AML1/AML2 and the NS31/AML2 primer set (Simon et al. 1992; Lee et al. 2008)), one targeting the 28S rRNA gene (using the AMF-specific primer pair Glo454/NDL22 (van Tuinen et al. 1998)) and one targeting the ITS region (using the fungal universal primer pair ITS1-F KYO1/ITS2-KYO1 (Toju et al. 2012)). AM fungal detection rate ranged from 98% with nested AMV5.4NF/AMDGR to 0.04% with ITS1-F KYO1/ITS2-KYO1 (Suzuki et al. 2020). For the NS31/AML2 approach, similar to the one applied in this study, the AM fungal detection rate was high (87%) and gave a high number of unique sequences, great phylogenetic diversity and low evenness. Moreover, AMF community composition detected by single AMV5.4NF/AMDGR and NS31/AML2 was relatively similar at the genus level (Suzuki et al. 2020), although nested PCR has been shown to affect AMF community analysis (Yu et al. 2015).
Since the off-season and in-season AM fungal composition of the two weeds occurring in all cropping systems were similar, this finding supports the fact that weeds functionally host AMF during the dry periods, playing key roles in the proliferation of AMF during the cropping cycle. Thus, weeds could be crucial for the maintenance of an active pool of beneficial fungi able to colonise and connect plants in cropping systems, potentially stimulating crop defence pathways (Nerva et al. 2022) under the drought conditions characterising the study area. This evidence reinforces the ecological role played by weeds in the agroecosystem. In addition, the similarity in composition between off-season (sampled at the edge of the experimental field) and in-season weeds (sampled inside the plots) supports the fact that the common mycorrhiza network is able to connect plants and transfer nutrients and signals at a long distance (Barto et al. 2012; Bennett et al. 2016).
Since weeds inside plots were controlled by glyphosate at the beginning of the season, the residual effect of glyphosate may have had an effect on the weed community. Glyphosate in soil dissipates almost completely 30 days after application under high temperatures, which are normally recorded in our study area (Bento et al. 2016). However, the main metabolite of glyphosate, aminomethylphosphonic acid (AMPA), can persist in soil, as has been detected at 20% of the applied glyphosate rate after 30 days of glyphosate application (Bento et al. 2016; Guijarro et al. 2018). Following Guijarro et al. (2018), the glyphosate exposure history affected the rate of persistence as the herbicide was degraded rapidly with long-term exposure and slowly when glyphosate was never applied to the soil. Thus, in our experiment, the persistence of glyphosate is likely to be negligible at 60 days after maize sowing, when in-season weeds were sampled. In contrast, the metabolite AMPA can be detected in the soil after long-term glyphosate application, although its concentration at in-season sampling time can be hypothesised to be low (degradation time for 90% of the initial concentration (DT90) between 88 and 148 days), according to Bento et al. (2016) and Guijarro et al. (2018), and not affected by tillage treatments, as reported by Okada et al. (2019). Moreover, since AMF can sporulate already at the early plant growth stage by draining host C during the plant development (Harinikumar and Bagyaraj 1989), the manual removal of weeds inside the plots at the vegetative phase that does not involve the complete elimination of all mycorrhizal roots is not likely to affect weed-mediated AM fungal propagule abundance in soil. This is also confirmed by a study comparing the effect of different methods of weed control, including manual weeding, on spore number and AMF root colonisation of several weeds, including B. pilosa and maize (Ramos-Zapata et al. 2012).
Relationship between cropping systems, weed AMF diversity, maize AM fungal colonisation weed AM fungal colonisation and maize grain yield
As expected, weed AMF diversity showed a positive correlation with weed AMF evenness and weed AMF richness (van der Heijden et al. 1998). However, although the cropping system directly affected all AMF traits in weeds (i.e. diversity, evenness, richness and AM fungal colonisation) and maize AM fungal root colonisation, contrary to our hypothesis, all these traits did not significantly influence maize grain yield. Although we expected that the diversity of AMF in weed roots would result in the improvement of the yield of associated maize plants through the improvement of AMF-mediated traits, this effect may have been masked by other factors. Mycorrhizal weeds also benefit from the mutual association with AMF, and these underground interactions may improve the invasiveness and competitiveness of weeds against maize plants; thus, this competition may have neutralised the AMF-mediated benefits on crops (Massenssini et al. 2014; Callaway et al. 2004). On the other hand, since we only identified two weed species that were common to all cropping systems and used these to assess AMF community response, this may have limited the resolution at which we dissected the relationship. This would mean that it is necessary to molecularly characterise the AM fungal community in maize roots and relate these to communities in the roots of more weed species.
Conclusion
Arbuscular mycorrhizal fungi are important in agricultural systems as they assist their host plants in taking up nutrients through the extraradical mycelium whilst obtaining photosynthetic assimilates from the host plant. Since AMF are obligate mutualistic symbionts, they require a host for their proliferation. In southern Africa, where short crop growing seasons are experienced, understanding how weed communities host AMF as alternative hosts is important in agroecosystem management since this symbiosis determines the promotion of biodiversity and hence ecosystem services. Here, for the first time, we assessed if mycorrhizal weeds surrounding the experimental fields and, among these weeds, those commonly found in all experimental plots would act as hosts of AMF during the off-season and during the season, respectively, and if AM fungal assemblages would be affected by host identity and the different combinations of CA components. In this work, we have also shown that weeds growing during the dry off-season can host AMF, and that, although the AM fungal community composition in the dry winter period was not predictive of the composition at anthesis, a large proportion of AM fungal taxa were shared between sampling stages. This is a novel finding indicating that weeds in off-season can exert a functional role during the dry periods since they represent the pool for later AM fungal colonisation of crops. Moreover, we demonstrated for the first time that host specificity is modulated by drought conditions, usually occurring at our site in the off-season period, inducing the plants under severe stress to select the most functional AMF. Finally, the models describing the response of maize yield to weed AMF traits and maize AM fungal colonisation showed no significant influence. This absence of influence may reflect that the competitive ability of the weeds was improved, hence overshadowing the anticipated AMF-mediated benefits to maize productivity. It may also reflect the absence of a link between the AMF that colonised the weeds and that colonised the maize crops. Overall, our findings suggest that drought-resistant weeds, growing off-season along the field borders, can act as AMF hosts during the dry season when there are no crops in the field, and part of this AMF community is carried over into the fields. These new insights support the need to find an equilibrium between the control of weeds and the maintenance of their diversity to guarantee crop yield and AMF-mediated ecosystem services. For example, farmers could consider adopting cropping systems that result in less competitive and diverse weed communities instead of complete eradication of weeds to conserve biodiversity and improve ecosystem services. However, further research needs to focus on the assessment of the effect of weeds on maintaining or increasing the AM fungal propagules in the soil during the non-cropping period and on the AMF shared among in-season and off-season weeds and crops. Finally, the AM fungal community composition should be studied by applying innovative sequencing methods (i.e. the third-generation long-read sequencing technology) that allow for improved specificity and enhanced resolution compared to Illumina sequencing. This, together with the assessment of other weed AMF-mediated services (i.e. soil structure and nutrient retention), would allow us to understand the link between AMF, weeds, crop growth and nutrient uptake.
Data availability
Data used in this study are stored in a public data repository and can be made available upon reasonable request following data-sharing regulations. The R scripts used in data analyses are available from the corresponding author upon request. Sequences generated in this study were uploaded to the NCBI database (submission number SUB10794739) and accession numbers OM049043–OM049185.
References
Adeux G, Vieren E, Carlesi S, Bàrberi P, Munier-Jolain N, Cordeau S (2019) Mitigating crop yield losses through weed diversity. Nat Sustain 2:1018–1026. https://doi.org/10.1038/s41893-019-0415-y
Alaux PL, Mison C, Senés-Guerrero C, Moreau V, Manssens G, Foucart G, Cranenbrouck S, Declerck S (2021) Diversity and species composition of arbuscular mycorrhizal fungi across maize fields in the southern part of Belgium. Mycorrhiza 31:265–272. https://doi.org/10.1007/s00572-020-01007-0
Ames RN, Ingham ER, Reid CPP (1982) Ultra-violet induced autofluorescence of arbuscular mycorrhizal root infection: an alternative to clearing and staining methods for assessing infections. Can J Microbiol 28:351–355. https://doi.org/10.1139/m82-052
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26:32–46. https://doi.org/10.1046/j.1442-9993.2001.01070.x
Avio L, Pellegrino E, Bonari E, Giovannetti M (2006) Functional diversity of arbuscular mycorrhizal fungal isolates in relation to extraradical mycelial networks. New Phytol 172:347–357. https://doi.org/10.1111/j.1469-8137.2006.01839.x
Bahadur A, Batool A, Nasir F, Jiang S, Mingsen Q, Zhang Q, Pan J, Liu Y, Feng H (2019) Mechanistic insights into arbuscular mycorrhizal fungi-mediated drought stress tolerance in plants. Int J Mol Sci 20:1–18. https://doi.org/10.3390/ijms20174199
Bàrberi P, Bocci G, Carlesi S, Armengot L, Blanco-Moreno JM, Sans FX (2018) Linking species traits to agroecosystem services: a functional analysis of weed communities. Weed Res 58:76–88. https://doi.org/10.1111/wre.12283
Barceló M, van Bodegom PM, Tedersoo L, den Haan N, Veen GF(, Ostonen I, Trimbos K, Soudzilovskaia NA (2020) The abundance of arbuscular mycorrhiza in soils is linked to the total length of roots colonized at ecosystem level. PLoS ONE 15:e0237256. https://doi.org/10.1371/journal.pone.0237256
Barto EK, Weidenhamer JD, Cipollini D, Rillig MC (2012) Fungal superhighways: do common mycorrhizal networks enhance below ground communication? Trends Plant Sci 17:633–637. https://doi.org/10.1016/j.tplants.2012.06.007
Bennett JA, Cahill JF, van der Heijden M (2016) Fungal effects on plant-plant interactions contribute to grassland plant abundances: evidence from the field. J Ecol 104:755–764. https://doi.org/10.1111/1365-2745.12558
Bates D, Maechler M, Bolker B, Walker S (2015) Fitting linear mixed-effects models using lme4. J Stat Softw 67:1–48. https://doi.org/10.18637/jss.v067.i01
Bento CPM, Yang X, Gort G, Xue S, van Dam R, Zomer P, Mol HGJ, Ritsema CJ, Geissen V (2016) Persistence of glyphosate and aminomethylphosphonic acid in loess soil under different combinations of temperature, soil moisture and light/darkness. Sci Total Environ 572:301–311. https://doi.org/10.1016/j.scitotenv.2016.07.215
Bever JD (2002) Host-specificity of AM fungal population growth rates can generate feedback on plant growth. Plant Soil 244:281–290. https://doi.org/10.1023/A:1020221609080
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37:852–857. https://doi.org/10.1038/s41587-019-0209-9
Bray JR, Curtis JRT (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27:325–349. https://doi.org/10.2307/1942268
Bremner JM, Mulvaney CS (1982) Nitrogen - total. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2, chemical and microbiological properties agronomy monograph. American Society of Agronomy, Madison, pp 595–624
Bretagnolle V, Gaba S (2015) Weeds for bees? A review. Agron Sustain Dev 35:891–909. https://doi.org/10.1007/s13593-015-0302-5
Brito I, Goss MJ, De Carvalho M (2012) Effect of tillage and crop on arbuscular mycorrhiza colonization of winter wheat and triticale under Mediterranean conditions. Soil Use Manag 28:202–208. https://doi.org/10.1111/j.1475-2743.2012.00404.x
Callaway RM, Thelen GC, Barth S, Ramsey PW, Gannon JE (2004) Soil fungi alter interactions between the invader Centaurea maculosa and North American natives. Ecology 85:1062–1071. https://doi.org/10.1890/02-0775
Castillo CG, Rubio R, Rouanet JL, Borie F (2006) Early effects of tillage and crop rotation on arbuscular mycorrhizal fungal propagules in an Ultisol. Biol Fertil Soils 43:83–92. https://doi.org/10.1007/s00374-005-0067-0
Ciccolini V, Ercoli L, Davison J, Vasar M, Öpik M, Pellegrino E (2016) Land-use intensity and host plant simultaneously shape the composition of arbuscular mycorrhizal fungal communities in a Mediterranean drained peatland. FEMS Microbiol Ecol 92:1–13. https://doi.org/10.1093/femsec/fiw186
Clarke KR, Gorley RN, Somerfield PJ, Warwick RM (2014) Change in marine communities: an approach to statistical analysis and interpretation, 3rd edn. PRIMER-E Ltd, Plymouth, UK: Plymouth Marine Lab. pp 262
Daniell TJ, Husband R, Fitter AH, Young JPW (2001) Molecular diversity of arbuscular mycorrhizal fungi colonising arable crops. FEMS Microbiol Ecol 36:203–209. https://doi.org/10.1111/j.1574-6941.2001.tb00841.x
De La Providencia IE, De Souza FA, Fernández F, Delmas NS, Declerck S (2005) Arbuscular mycorrhizal fungi reveal distinct patterns of anastomosis formation and hyphal healing mechanisms between different phylogenic groups. New Phytol 165:261–271. https://doi.org/10.1111/j.1469-8137.2004.01236.x
Derrouch D, Chauvel B, Cordeau S, Dessaint F (2021) Functional shifts in weed community composition following adoption of conservation agriculture. Weed Res 62:103–112. https://doi.org/10.1111/wre.12517
Dumbrell AJ, Ashton PD, Aziz N, Feng G, Nelson M, Dytham C, Fitter AH, Helgason T (2011) Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol 190:794–804. https://doi.org/10.1111/j.1469-8137.2010.03636.x
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
El Omari B, El Ghachtouli N (2021) Arbuscular mycorrhizal fungi-weeds interaction in cropping and unmanaged ecosystems: a review. Symbiosis 83:279–292. https://doi.org/10.1007/s13199-021-00753-9
Eom AH, Hartnett DC, Wilson GWT (2000) Host plant species effects on arbuscular mycorrhizal fungal communities in tallgrass prairie. Oecologia 122:435–444. https://doi.org/10.1007/s004420050050
FAO (2019) Conservation Agriculture. Available online at: http://www.fao.org/conservation-agriculture/overview/what-is-conservation-agriculture/en/ (accessed August 2019)
Ferrero R, Lima M, Davis AS, Gonzalez-Andujar JL (2017) Weed diversity affects soybean and maize yield in a long-term experiment in Michigan, USA. Front Plant Sci 8:236–236. https://doi.org/10.3389/fpls.2017.00236
Gavito ME, Jakobsen I, Mikkelsen TN, Mora F (2019) Direct evidence for modulation of photosynthesis by an arbuscular mycorrhiza-induced carbon sink strength. New Phytol 223:896–907. https://doi.org/10.1111/nph.15806
Gollotte A, van Tuinen D, Atkinson D (2004) Diversity of arbuscular mycorrhizal fungi colonising roots of the grass species Agrostis capillaris and Lolium perenne in a field experiment. Mycorrhiza 14:111–117. https://doi.org/10.1007/s00572-003-0244-7
Guijarro KH, Aparicio V, De Gerónimo E, Castellote M, Figuerola EL, Costa JL, Erijman L (2018) Soil microbial communities and glyphosate decay in soils with different herbicide application history. Sci Total Environ 634:974–982. https://doi.org/10.1016/j.scitotenv.2018.03.393
Hart MM, Reader RJ (2004) Do arbuscular mycorrhizal fungi recover from soil disturbance differently? Trop Ecol 45:97–112
Helgason T, Merryweather JW, Young JPW, Fitter AH (2007) Specificity and resilience in thearbuscular mycorrhizal fungi of a natural woodland community. J Ecol 95:623–630. https://doi.org/10.1111/j.1365-2745.2007.01239.x
Heyl A (2022) Important weeds in maize. https://www.up.ac.za/sahri/article/1810372/important-weeds-in-maize. Accessed 8 Mar 2022
Harinikumar KM, Bagyaraj DJ (1989) Effect of cropping sequence, fertilizers and farmyard manure on vesicular-arbuscular mycorrhizal fungi in different crops over three consecutive seasons. Biol Fertil Soils 7:173–175. https://doi.org/10.1007/BF00292578
Hibbett DS, Binder M, Bischoff JF, Blackwell M, Cannon PF, Eriksson OE, Huhndorf S, James T, Kirk PM, Lücking R, Lumbsch HT, Lutzoni F, Matheny PB, McLaughlin DJ, Powell MJ, Redhead S, Schoch CL, Spatafora JW, Stalpers JA, Vilgalys R, Aime MC, Aptroot A, Bauer R, Begerow D, Benny GL, Castlebury LA, Crous PW, Dai YC, Gams W, Geiser DM, Griffith GW, Gueidan C, Hawksworth DL, Hestmark G, Hosaka K, Humber RA, Hyde KD, Ironside JE, Kõljalg U, Kurtzman CP, Larsson KH, Lichtwardt R, Longcore J, Miadlikowska J, Miller A, Moncalvo JM, Mozley-Standridge S, Oberwinkler F, Parmasto E, Reeb V, Rogers JD, Roux C, Ryvarden L, Sampaio JP, Schüssler A, Sugiyama J, Thorn RG, Tibell L, Untereiner WA, Walker C, Wang Z, Weir A, Weiss M, White MM, Winka K, Yao YJ, Zhang N (2007) A higher-level phylogenetic classification of the Fungi. Mycol Res 111:509–547. https://doi.org/10.1016/j.mycres.2007.03.004
Hijri I, Sýkorová Z, Oehl F, Ineichen K, Maeder P, Wiemken A, Redecker D (2006) Communities of arbuscular mycorrhizal fungi in arable soils are not necessarily low in diversity. Mol Ecol 15:2277–2289. https://doi.org/10.1111/j.1365-294X.2006.02921.x
IUSS Working Group WRB (2015) World Reference Base on Soils (World Soil Resources Reports No. 106), International soil classification system for naming soils and creating legends for soil maps. FAO-ISRIC, Rome, Italy
James TY, Stajich JE, Hittinger CT, Rokas A (2020) Toward a fully resolved fungal tree of life. Annu Rev Microbiol 74. https://doi.org/10.1146/annurev-micro-022020-051835
Johnson JM, Houngnandan P, Kane A, Sanon KB, Neyra M (2013) Diversity patterns of indigenous arbuscular mycorrhizal fungi associated with rhizosphere of cowpea (Vigna unguiculata (L.) Walp.) in Benin. West Africa Pedobiologia 56:121–128. https://doi.org/10.1016/j.pedobi.2013.03.003
Katoh K, Rozewicki J, Yamada KD (2019) MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform 20:1160–1166. https://doi.org/10.1093/bib/bbx108
Kiers ET, Duhamel M, Yugandhar B, Mensah AJ, Oscar F, Verbruggen E, Fellbaum CR, Kowalchuk GA, Hart MM, Bago A, Palmer TM, West SA, Vandenkoornhuyse P, Jansa J, Bücking H (2011) Reciprocal rewards stabilize cooperation in the mycorrhizal symbiosis. Science 333:880–882. https://doi.org/10.1126/science.1208473
Kodzwa JJ, Gotosa J, Nyamangara J (2020) Mulching is the most important of the three conservation agriculture principles in increasing crop yield in the short term, under sub humid tropical conditions in Zimbabwe. Soil till Res 197:104515. https://doi.org/10.1016/j.still.2019.104515
Koocheki A, Nassiri M, Alimoradi L, Ghorbani R (2009) Effect of cropping systems and crop rotations on weeds. Agron Sustain Dev 29:401–408. https://doi.org/10.1051/agro/2008061
Kottek M, Grieser J, Beck C, Rudolf B, Rubel F (2006) World map of the Köppen-Geiger climate classification updated. Meteorol Z 15:259–263. https://doi.org/10.1127/0941-2948/2006/0130
Lal R (2000) Mulching effects on soil physical quality of an alfisol in western Nigeria. Land Degrad Dev 11:383–392. https://doi.org/10.1002/1099-145X(200007/08)11:4%3C383::AID-LDR393%3E3.0.CO;2-6
Lee J, Lee S, Young JP (2008) Improved PCR primers for the detection and identification of arbuscular mycorrhizal fungi. FEMS Microbiol Ecol 65:339–349. https://doi.org/10.1111/j.1574-6941.2008.00531.x
Lefcheck J (2016) piecewiseSEM: Piecewise structural equation modeling in R for ecology, evolution, and systematics. Methods Ecol Evol 7:573–579. https://doi.org/10.1111/2041-210X.12512
Lenth R (2019) emmeans: estimated marginal means, aka least-squares means. R package version 1.3.4. https://CRAN.R-project.org/package=emmeans
Li M, Jordan NR, Koide RT, Yannarell AC, Davis AS (2019) Interspecific variation in crop and weed responses to arbuscular mycorrhizal fungal community highlights opportunities for weed biocontrol. Appl Soil Ecol 142:34–42. https://doi.org/10.1016/j.apsoil.2019.05.016
Li Y, Steenwyk JL, Chang Y, Wang Y, James TY, Stajich JE, Spatafora JW, Groenewald M, Dunn Casey W, Hittinger CT, Shen XX, Rokas A (2021) A genome-scale phylogeny of the kingdom fungi. Curr Biol 31:1653.e5-1665.e5. https://doi.org/10.1016/j.cub.2021.01.074
Lu X, Lu X, Liao Y (2018) Effect of tillage treatment on the diversity of soil arbuscular mycorrhizal fungal and soil aggregate-associated carbon content. Front Microbiol 9:1–10. https://doi.org/10.3389/fmicb.2018.02986
MacLaren C, Storkey J, Menegat A, Metcalfe H, Dehnen-Schmutz K (2020) An ecological future for weed science to sustain crop production and the environment. A Review Agron Sustain Dev 40:24. https://doi.org/10.1007/s13593-020-00631-6
Merryweather JW, Fitter AH (1991) A modified method for elucidating the structure of the fungal partner in a vesicular-arbuscular mycorrhiza. Mycol Res 95:1435–1437. https://doi.org/10.1016/S0953-7562(09)80399-7
Martínez-García LB, Pugnaire FI (2011) Arbuscular mycorrhizal fungi host preference and site effects in two plant species in a semiarid environment. Appl Soil Ecol 48:313–317. https://doi.org/10.1016/j.apsoil.2011.04.003
Massenssini AM, Bonduki VHA, Tótola MR, Ferreira FA, Costa MD (2014) Arbuscular mycorrhizal associations and occurrence of dark septate endophytes in the roots of Brazilian weed plants. Mycorrhiza 24:153–159. https://doi.org/10.1007/s00572-013-0519-6
McGonigle TP, Miller MH, Evans DG, Fairchild GL, Swan JA (1990) A new method which gives an objective measure of colonization of roots by vesicular-arbuscular mycorrhizal fungi. New Phytol 115:495–501. https://doi.org/10.1111/j.1469-8137.1990.tb00476.x
McLean EO (1982) Soil pH and lime requirement. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2, chemical and microbiological properties agronomy monograph, vol 9, 2nd edn. American Society of Agronomy, Madison, pp 199–224
Mhlanga B, Ercoli L, Pellegrino E, Onofri A, Thierfelder C (2021) The crucial role of mulch to enhance the stability and resilience of cropping systems in southern Africa. Agron Sustain Dev 41:29. https://doi.org/10.1007/s13593-021-00687-y
Mhlanga B, Pellegrino E, Thierfelder C, Ercoli L (2022) Conservation agriculture practices drive maize yield by regulating soil nutrient availability, arbuscular mycorrhizas, and plant nutrient uptake. Field Crops Res 277:108403. https://doi.org/10.1016/j.fcr.2021.108403
Munkvold L, Kjøller R, Vestberg M, Rosendahl S, Jakobsen I (2004) High functional diversity within species of arbuscular mycorrhizal fungi. New Phytol 164:357–364. https://doi.org/10.1111/j.1469-8137.2004.01169.x
Nelson DW, Sommers LE (1982) Total carbon, organic carbon and organic matter. In: Page AL, Miller RH, Keeney DR (eds) Methods of soil analysis, part 2, chemical and microbiological properties agronomy monograph. American Society of Agronomy, Madison, pp 539–579
Nerva L, Giudice G, Quiroga G, Belfiore N, Lovat L, Perria R, Volpe MG, Moffa L, Sandrini M, Gaiotti F, Balestrini R, Chitarra W (2022) Mycorrhizal symbiosis balances rootstock-mediated growth-defence tradeoffs. Biol Fert Soils 58:17–34
Nichols V, Verhulst N, Cox R, Govaerts B (2015) Weed dynamics and conservation agriculture principles: a review. Field Crop Res 183:56–68. https://doi.org/10.1016/j.fcr.2015.07.012
Oehl F, Sieverding E, Ineichen K, Mäder P, Boller T, Wiemken A (2003) Impact of land use intensity on the species diversity of arbuscular mycorrhizal fungi in agroecosystems of central Europe. Appl Environ Microbiol 69:2816–2824. https://doi.org/10.1128/AEM.69.5.2816-2824.2003
Oehl F, Sieverding E, Ineichen K, Mader P, Wiemken A, Boller T (2009) Distinct sporulation dynamics of arbuscular mycorrhizal fungal communities from different agroecosystems in long-term microcosms. Agr Ecosyst Environ 134:257–268. https://doi.org/10.1016/j.agee.2009.07.008
Okada E, Costa JL, Bedmar F (2019) Glyphosate dissipation in different soils under no-till and conventional tillage. Pedosphere 29:773–783. https://doi.org/10.1016/S1002-0160(17)60430-2
Oliveros JC (2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. https://bioinfogp.cnb.csic.es/tools/venny/index.html
Omirou M, Ioannides IM, Ehaliotis C (2013) Mycorrhizal inoculation affects arbuscular mycorrhizaldiversity in watermelon roots, but leads to improved colonization and plant response under water stress only. Appl Soil Ecol 63:112–119. https://doi.org/10.1016/j.apsoil.2012.09.013
Pellegrino E, Gamper HA, Ciccolini V, Ercoli L (2020) Forage rotations conserve diversity of arbuscular mycorrhizal fungi and soil fertility. Front Microbiol 10:2969. https://doi.org/10.3389/fmicb.2019.02969
Phillips JM, Hayman DS (1970) Improved procedures for clearing roots and staining parasitic and vesicular-arbuscular mycorrhizal fungi for rapid assessment of infection. Trans Br Mycol Soc 55:158–161. https://doi.org/10.1016/S0007-1536(70)80110-3
Piazza G, Ercoli L, Nuti M, Pellegrino E (2019) Interaction between conservation tillage and nitrogen fertilization shapes prokaryotic and fungal diversity at different soil depths: evidence from a 23-year field experiment in the Mediterranean area. Front Microbiol 10:1–20. https://doi.org/10.3389/fmicb.2019.02047
Pielou EC (1969) An introduction to mathematical ecology. Wiley-Interscience Publications, New York. pp. 294. https://doi.org/10.4039/Ent103288-2
Qiao X, Bei S, Li H, Christie P, Zhang F, Zhang J (2016) Arbuscular mycorrhizal fungi contribute to overyielding by enhancing crop biomass while suppressing weed biomass in intercropping systems. Plant Soil 406:173–185. https://doi.org/10.1007/s11104-016-2863-8
Ramos-Zapata JA, Marrufo-Zapata D, Guadarrama P, Carrillo-Sánchez L, Hernández-Cuevas L, Caamal-Maldonado A (2012) Impact of weed control on arbuscular mycorrhizal fungi in a tropical agroecosystem: a long-term experiment. Mycorrhiza 22:653–661. https://doi.org/10.1007/s00572-012-0443-1
R Core Team (2021) R: A language and environment for statistical computing. RFoundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/. Version 4.1.0
Säle V, Sieverding E, Oehl F (2022) Growth responses of three European weeds on different AMF species during early development. Plants 11:2020. https://doi.org/10.3390/plants11152020
Sato K, Suyama Y, Saito M, Sugawara K (2005) A new primer for discrimination of arbuscular mycorrhizal fungi with polymerase chain reaction-denature gradient gel electrophoresis. Grassl Sci 51:179–181. https://doi.org/10.1111/j.1744-697X.2005.00023.x
Schalamuk S, Cabello M (2010) Arbuscular mycorrhizal fungal propagules from tillage and no-tillage systems: possible effects on Glomeromycota diversity. Mycologia 102:261–268. https://doi.org/10.3852/08-118
Schüβler A, Schwarzott D, Walker C (2001) A new fungal phylum, the Glomeromycota: phylogeny and evolution. Mycol Res 105:1413–1421. https://doi.org/10.1017/S0953756201005196
Shannon CE, Weaver W (1949) The mathematical theory of communication. University of Illinois Press, Urbana. https://doi.org/10.1002/j.1538-7305.1948.tb01338.x
Silva JV, Baudron F, Reidsma P, Giller KE (2019) Is labour a major determinant of yield gaps in sub-Saharan Africa? A study of cereal-based production systems in Southern Ethiopia. Agric Syst 174:39–51. https://doi.org/10.1016/j.agsy.2019.04.009
Simon L, Lalonde M, Bruns TD (1992) Specific amplification of 18S fungal ribosomal genes from vesicular-arbuscular endomycorrhizal fungi colonizing roots. Appl Environ Microbiol 58:291–295
Smith SE, Read D (2008) 1 - The symbionts forming arbuscular mycorrhizas. In: Smith SE, Read D (eds) Mycorrhizal symbiosis (Third Edition). Academic Press, London, pp 13–41
Storkey J, Neve P (2018) What good is weed diversity? Weed Res 58:239–243. https://doi.org/10.1111/wre.12310
Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML, Bonito G, Nicolas C, Grigoriev I, Gryganskyi A, James TY, O’Donnell K, Roberson RW, Taylor TN, Uehling J, Vilgalys R, White MM, Stajich JE (2016) A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108:1028–1046. https://doi.org/10.3852/16-042
Suzuki K, Takahashi K, Harada N (2020) Evaluation of primer pairs for studying arbuscular mycorrhizal fungal community compositions using a MiSeq platform. Biol Fertil Soils 56:853–858. https://doi.org/10.1007/s00374-020-01431-6
Tamura K, Stecher G, Kumar S (2021) MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol Biol Evol 38:3022–3027. https://doi.org/10.1093/molbev/msab120
Tedersoo L, Sánchez-Ramírez S, Kõljalg U, Bahram M, Döring M, Schigel D, May T, Ryberg M, Abarenkov K (2018) High-level classification of the fungi and a tool for evolutionary ecological analyses. Fungal Divers 90:135–159. https://doi.org/10.1007/s13225-018-0401-0
Toju H, Tanabe AS, Yamamoto S, Sato H (2012) High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 7:e40863
van der Heijden MG, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-Engel R, Boller T, Wiemken A, Sanders IR (1998) Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature 396:69–72. https://doi.org/10.1038/23932
van der Heyde M, Ohsowski B, Abbott LK, Hart M (2017) Arbuscular mycorrhizal fungus responses to disturbance are context-dependent. Mycorrhiza 27:431–440. https://doi.org/10.1007/s00572-016-0759-3
van Tuinen D, Jacquot E, Zhao B, Gollotte A, Gianinazzi-Pearson V (1998) Characterization of root colonization profiles by a microcosm community of arbuscular mycorrhizal fungi using 25S rDNA-targeted nested PCR. Mol Ecol 7:879–887
Vatovec C, Jordan N, Huerd S (2005) Responsiveness of certain agronomic weed species to arbuscular mycorrhizal fungi. Renew Agric Food Syst 20:181–189. https://doi.org/10.1079/RAF2005115
Veiga RSL, Jansa J, Frossard E, van der Heijden MGA (2011) Can arbuscular mycorrhizal fungi reduce the growth of agricultural weeds? PLoS ONE 6:27825. https://doi.org/10.1371/journal.pone.0027825
Wilkes TI, Warner DJ, Edmonds-Brown V, Davies KG, Denholm I (2021) Zero tillage systems conserve arbuscular mycorrhizal fungi, enhancing soil glomalin and water stable aggregates with implications for soil stability. Soil Syst 5:1–13. https://doi.org/10.3390/soilsystems5010004
Wilson GWT, Hartnett DC (1998) Interspecific variation in plant responses to mycorrhizal colonization in tallgrass prairie. Am J Bot 85:1732–1738. https://doi.org/10.2307/2446507
Yang H, Zang Y, Yuan Y, Tang J, Chen X (2012) Selectivity by host plants affects the distribution of arbuscular mycorrhizal fungi: evidence from ITS rDNA sequence metadata. BMC Evol Biol 12:50. https://doi.org/10.1186/1471-2148-12-50
Yu G, Fadrosh D, Goedert JJ, Ravel J, Goldstein AM (2015) Nested PCR biases in interpreting microbial community structure in 16S rRNA gene sequence datasets. PLoS ONE 10:e0132253
Zhang Q, Yang R, Tang J, Yang H, Hu S, Chen X (2010) Positive feedback between mycorrhizal fungi and plants influences plant invasion success and resistance to invasion. PLoS ONE 5:12380. https://doi.org/10.1371/journal.pone.0012380
Zhang F, Li Q, Yerger EH, Chen X, Shi Q, Wan F (2018) AM fungi facilitate the competitive growth of two invasive plant species, Ambrosia artemisiifolia and Bidens pilosa. Mycorrhiza 28:703–715. https://doi.org/10.1007/s00572-018-0866-4
Zhang H, Hobbie EA, Feng P, Niu L, Hu K (2021) Can conservation agriculture mitigate climate change and reduce environmental impacts for intensive cropping systems in North China Plain? Sci Total Environ 806:151194. https://doi.org/10.1016/j.scitotenv.2021.151194
Acknowledgements
The PhD fellowship for BM was funded by the Scuola Superiore Sant’Anna. All the authors are grateful to the International Maize and Wheat Improvement Centre for funding the setup and running of the experimental trials and to donors of the MAIZE CGIAR Research Program (www.maize.org) who supported the trials until 2018. The authors are thankful to the DSCATT project for co-funding the CIMMYT step-trials since 2018. The DSCATT project (N° AF 1802-001, N° FT C002181) is funded by the Agropolis Foundation (through the ‘Programme Investissement d’Avenir’ Labex Agro, funding ANR-10-LABX-0001-01) and by the TOTAL Foundation, as part of a sponsorship agreement. The financial support of Project PSR 2014-2020—Misura 16.2 PS-GO 2017—Progetto AGROCIRCOLIVE—CUP ARTEA 831728 is gratefully acknowledged. Special thanks go to the technical teams at the two experimental locations, led by Sign Phiri and Herbert Chipara, who assisted in data collection.
Funding
Open access funding provided by Scuola Superiore Sant'Anna within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
E.P. and B.M.: AM fungal experimental idea and set up of the AM fungal experiment. C.T.: CA experiment idea and set up of omission trials. C.T., B.M., L.E. and E.P.: coordination of data collection. B.M., E.P. and G.P.: formal data analysis. B.M. and EP: writing the original manuscript draft. E.P., L.E., C.T. and G.P.: writing, review and editing. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
All ethics committees of the organisations with which the authors are affiliated have no objections to the publication of this work.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Blessing Mhlanga and Elisa Pellegrino contributed equally to this work.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mhlanga, B., Ercoli, L., Piazza, G. et al. Occurrence and diversity of arbuscular mycorrhizal fungi colonising off-season and in-season weeds and their relationship with maize yield under conservation agriculture. Biol Fertil Soils 58, 917–935 (2022). https://doi.org/10.1007/s00374-022-01678-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-022-01678-1