
Abstract
Sleep is an essential and evolutionarily conserved process that affects many biological functions that are also strongly regulated by cellular metabolism. The interdependence between sleep homeostasis and redox metabolism, in particular, is such that sleep deprivation causes redox metabolic imbalances in the form of over-production of ROS. Likewise (and vice versa), accumulation of ROS leads to greater sleep pressure. Thus, it is theorized that one of the functions of sleep is to act as the brain’s “antioxidant” at night by clearing oxidation built up from daily stress of the active day phase. In this review, we will highlight evidence linking sleep homeostasis and regulation to redox metabolism by discussing (1) the bipartite role that sleep–wake neuropeptides and hormones have in redox metabolism through comparing cross-species cellular and molecular mechanisms, (2) the evolutionarily metabolic changes that accompanied the development of sleep loss in cavefish, and finally, (3) some of the challenges of uncovering the cellular mechanism underpinning how ROS accumulation builds sleep pressure and cellularly, how this pressure is cleared.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sleep is an instrumental biological process in restorative functions, memory consolidation, cognitive abilities, and energy conservation (Appelbaum et al. 2010; Kayaba et al. 2017; Klinzing et al. 2019; Mullington et al. 2000). Using behavioural criteria, sleep has been described in all branches of the animal kingdom from invertebrate octopi, worms, and insects to vertebrates including fish and mammals (Campbell and Tobler 1984; Keene and Duboue 2018). These criteria include reduced mobility, increased arousal threshold, rapid reversibility, place preference/posture, and homeostatic regulation (Campbell and Tobler 1984; Jaggard et al. 2021). Despite the concentrated research on mammals, the current sleep definition lacks an all-inclusive explanation to cover its evolutionary-conserved role, lending support to the hypothesis that sleep serves a fundamental need, and the best way to define sleep would be to identify this need.
The neurological definition of sleep has been established since the 1950–60s to include the main electrophysiological hallmarks of human sleep recorded by polysomnography (PSG), combining electroencephalogram (EEG), electromyogram (EMG of voluntary muscles), electrocardiogram (ECG) and electrooculogram (EOG). Slow-wave sleep (SWS), the deepest stage of non-REM sleep (NREM), is characterized by slow, synchronized neocortical waves and low muscle activity. In contrast to NREM/SWS, Rapid Eye Movement sleep (REM), also known as Paradoxical Sleep (PS), has a core characteristic of muscle atonia with wake-like desynchronized EEG, cardiorespiratory irregularities, tremors, and episodic bursts of rapid eye movements. Until recently, these neurological sleep stages had only been reported in the more evolutionary-recent amniotic vertebrates: mammals and birds (Campbell and Tobler 1984; Leung and Mourrain 2018; Shein-Idelson et al. 2016). Our group has identified analogous cellular signatures of sleep in zebrafish including synchronous slow oscillation and paradoxical sleep activity, suggesting that neural and muscular patterns of NREM-REM sleep emerged at least 450 million years ago (Leung et al. 2019). To advance the understanding of conserved sleep need, the cellular and molecular functions of sleep need to be further studied. Having phylogenetically conserved behavior and neural sleep profile, fish provides a simpler vertebrate model for sleep studies that overcomes the challenges of studying sleep in mammalian systems. Some of these challenges include, but not limited to, animal handling and care, invasiveness of live imaging as well as behavioural tracking, and accessibility of deeper tissues and organs.
To this date, there is no united opinion on the function of sleep. From the earlier sleep research, the theories behind the function of sleep remain similar (Rechtschaffen 1998). Some prominent theories on the function of sleep include: (1) restorative theory, suggesting that sleep repairs the body through increased production of growth hormone, induced muscle repair and strengthened immune system (Rechtschaffen and Bergmann 2002), (2) memory consolidation theory, suggesting different stages of sleep contribute to various aspects of memory formation and organization (Diekelmann and Born 2010), (3) synaptic homeostasis hypothesis, suggesting that sleep is necessary to downscale and reset synaptic strength in the brain, preventing overload and maintaining optimal neuronal function (Tononi and Cirelli 2006), (4) energy conservation theory, suggesting that by reducing activity and metabolic rate during sleep, the body conserves energy that can be used during waking hours (Siegel 2005), and finally (5) brain plasticity and learning theory, suggesting that sleep promotes brain plasticity to enhance learning and adaptation to new experiences (Frank 2006). It must be noted that these theories are not mutually exclusive, and sleep likely serves multiple functions. Furthermore, these theories do not provide an explanation to the evolutionary-conserved roles of sleep.
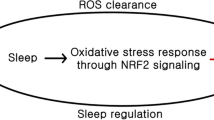
Unquestionably, sleep is not solely for brain but also for the entire body. Chronic sleep deprivation systematically leads to weight gain and metabolic imbalances and sleep deprivation decreases antioxidant levels in rats and humans, leading to oxidative stress (D'Almeida et al. 1998; Everson et al. 2005; Ramanathan et al. 2002; Silva et al. 2004; Trivedi et al. 2017; Van Cauter et al. 2008). As sleep regulation and cellular sleep dynamics are highly conserved, so too may the interplay between sleep and redox homeostasis and the underlying mechanisms be similarly conserved across evolution. A theory for sleep, that also supports a restorative function for sleep, was proposed in which sleep functions essentially as an “antioxidant” for the brain where excess free radicals are removed during sleep (Reimund 1994). Wake periods consist of higher metabolic rates and higher brain activity and a selective metabolic shift occurs during sleep (DiNuzzo and Nedergaard 2017). During sleep, the body’s metabolic rate decreases and this reduction in metabolic activity could contribute to a lower oxidative environment along with an increased antioxidant capacity, decreasing overall free radicals and the oxidized substrates, that accumulated throughout the wake period due to high cellular metabolic activities. Although it is unlikely that the sole function of sleep is to clear oxidation from the brain, this theory points towards a direction for the new definition of sleep: what are the metabolic, particularly redox-related, implications of sleep?
Redox metabolism represents the reduction and oxidation (redox) reactions induced via oxidized molecular oxygen, or reactive oxygen species (ROS). ROS are highly reactive chemicals, enabling electron transfer from one molecule to another. The cellular redox state is determined by the balance between ROS production and the counteracting cellular antioxidant systems. Under physiological redox conditions, the production and elimination of ROS are in homeostasis and redox signalling is in action, during which proteins are post-translationally modified via oxidation to relay signal transduction (Dickinson and Chang 2011; Finkel 2011; Reczek and Chandel 2015). When the production and presence of ROS dominates the antioxidant capacity, cell undergoes oxidative stress, leading to damage to cellular components and causing aging, cancer, neurodegenerative diseases (Davalli et al. 2016; Park et al. 2008; Rama Rao et al. 2018; Weinberg et al. 2019). On the contrary, when ROS production is limited below physiological levels, disruption in redox signalling interferes with variety of cell functions including stem cell maintenance and differentiation, cell migration, axonal growth and guidance (Le Belle et al. 2011; Munnamalai et al. 2014; Somanna et al. 2016; Terzi et al. 2020). Hence, it is critical to maintain physiologically relevant levels of intracellular ROS for health and longevity.
The main cellular sources of ROS are (1) mitochondria, where electron-transport-chain (ETC) leads ROS production through leakage of electrons during oxidative phosphorylation, (2) NADPH oxidases (NOXes), which generate superoxide and hydrogen peroxide (H2O2) through transferring electrons from NADPH donors to oxygen, regulating both immune response and homeostatic ROS signalling (3) peroxisomes, which produce H2O2 as a byproduct during breakdown of metabolic reactions such as breakdown of fatty acids and (4) xanthine oxidase, which generates superoxide and H2O2 as a byproduct of oxidative hydroxylation of hypoxanthine in purine metabolism; (5) cytochrome P450 enzymes, a group of heme monooxygenases, which catalyse the metabolism of endogenous and exogenous molecules by electron transfer through NADPH cofactor, and finally (6) endoplasmic reticulum, which generates ROS in response to misfolded proteins (De Almeida et al. 2022).
The counteracting cellular antioxidant systems include both enzymatic, such as catalase, superoxide dismutase (SOD), glutathione peroxidase, and nonenzymatic, such as glutathione (GSH), vitamin C, vitamin E, members (Haida and Hakiman 2019). In addition, uncoupling proteins (UCPs), located in the inner mitochondrial membrane, prevents ROS accumulation, and lowers the mitochondrial membrane potential, and their overexpression was found to counteract oxidative stress (Barreiro et al. 2009; Hirschenson et al. 2022). Finally, autophagy and/or mitophagy mechanisms act against cellular oxidative stress (Filomeni et al. 2015). Increased ROS production triggers autophagy to mediate clearing of ROS and oxidative damage (Chen et al. 2007). As mitochondria are major source of ROS, chronic impairment of any kind of mitochondrial function overproduces ROS and triggers a self-removal signal, or mitophagy, to eliminate the further oxidative environment (Schofield and Schafer 2021). Redox metabolism is not only maintained by ROS producing enzymes and antioxidants but also by nicotinamide adenine dinucleotide (NAD +) and its metabolites. NAD + and NAD + -related metabolites, NADH, NADP + and NADPH, are crucial in energy metabolism, DNA repair, epigenetic modifications, inflammation and circadian rhythms (Xie et al. 2020). NAD + and its metabolites serve as co-enzymes for redox reactions, relaying oxidative and reductive signalling between molecules. Coupled NAD + /NADH redox exert their main effect in mitochondria, by serving as an electron donor through ETC for oxidative phosphorylation and producing cellular ROS (Li and Sauve 2015), but they also exhibit protective effects by enhancing GSH levels and the activity of antioxidant enzymes (Wang et al. 2014). NADPH, also, serves both as an electron donor for NADPH oxidases, contributing to cellular ROS production, and as a reductive power for antioxidant defence by transferring electrons from enzymatic antioxidants (Bedard and Krause 2007; Bradshaw 2019). The major intracellular redox players are summarized in Fig. 1.

Summary of major intracellular ROS sources and antioxidant systems. The major intracellular ROS sources (upper panel) include: NADPH oxidases, peroxisomes, oxidative phosphorylation in mitochondria, xanthine oxidases, and the endoplasmic reticulum stress response towards unfolded proteins (UPR). The major antioxidant systems (lower panel) include enzymatic (grey): catalase, superoxide dismutase (SOD), glutathione peroxidases; and nonenzymatic (orange): glutathione (GSH), vitamin C, and vitamin E
At night, oxidized molecules are either repaired or replaced in plants (Bechtold et al. 2004). Similarly, in animals, wake and sleep loss elevate mitochondrial ROS in dorsal fan-shaped body (dFB) neurons (Kempf et al. 2019). Like the night phase in plants, sleep has been involved in toxin elimination, DNA repair, and infection defense (Mourrain and Wang 2019). Further, recent studies in flies showed that (1) short-sleeping mutants are extremely sensitive to oxidative stress, suggesting that a key function of sleep is to defend against oxidative stress (Hill et al. 2018); (2) ROS accumulation builds sleep pressure sensed in specific neurons that promote sleep, which in turn dissipate ROS burden and return the mitochondrial NAD + /NADH ratio to baseline (Kempf et al. 2019). Although the exact mechanisms have not been discovered yet, accumulation of ROS during the active phase or day, builds up sleep pressure that is cleared during inactive phase, or night, thus, getting the body and brain ready for the prospective day.
In the forthcoming sections, we will focus on the current knowledge on the relationship between redox metabolism and sleep homeostasis mainly in zebrafish and cavefish, where such processes are well conserved, as vertebrate models for human studies, and discuss the implications of this relationship on the universal definition of sleep.
Behavioural and neural definition of sleep is conserved in fish
Zebrafish are diurnal animals and exhibit a recurring pattern of sleep and wakefulness, similar to humans. Zebrafish experience consolidated periods of sleep at night. This sleep state is characterized behaviourally by reduced locomotor activity, decreased responsiveness to stimuli, and homeostatic regulation (Yokogawa et al. 2007; Zhdanova et al. 2001; Zhdanova. 2006). Zebrafish also have sleep brain dynamics analogous to mammals, including Slow Bursting Sleep (SBS) and Propagating Wave Sleep (PWS). SBS shares many commonalities with NREM-Slow Wave Sleep by exhibiting synchronous, slow and high amplitude oscillations of the telencephalic neurons occurring in an overall background of reduced brain activity, low muscle tone, and reduced but steady cardiovascular activity (Leung et al. 2019). PWS is a sleep state sharing many features with REM/Paradoxical Sleep, including pontine activation, ponto-midbrain-telencephalic wave propagation, rostro-caudal voluntary muscle atonia propagation, heart beat and breathing arrhythmia as well as wake-like activity of the telencephalon (Leung et al. 2019). The regulation of sleep involves at least two key processes in zebrafish as it does in other organisms: (1) the circadian process, which aligns sleep patterns with the natural 24-h day and night cycle, and (2) the homeostatic process, which increases the urge to sleep based on the duration of prior wakefulness. Here, we will engage in discussion on homeostatic regulation of sleep only in relation to redox metabolism.
Sleep-modulating neuropeptides are conserved in zebrafish
Neuropeptides that control sleep–wake states in mammals are conserved in fish as well. Galanin, an important neuropeptide in energy homeostasis and sleep regulation, is also required for homeostatic sleep rebound following sleep deprivation in zebrafish larvae (Martinelli et al. 2021; Reichert et al. 2019). Neuropeptide Y (NPY), another highly conserved neuropeptide, contributes to controlling energy homeostasis, anxiety and sleep; exhibiting dual impact on sleep–wake behaviours in mammals (Hsieh et al. 2013; Shen et al. 2022). In zebrafish, NPY promotes sleep via inhibition of noradrenergic signaling and loss-of-function mutations of NPY resulted in decreased sleep (Singh et al. 2017). Neuromedin U, a key neuropeptide regulating feeding, energy metabolism and insulin secretion, has been shown to inhibit sleep in zebrafish, similar to its function in rats (Chiu et al. 2016; Wren et al. 2002). The hypocretin/orexin neuropeptides are essential for the maintenance of wakefulness and the suppression of REM sleep, and zebrafish possess a functional hcrt-pineal gland circuit, connecting the hcrt and melatonin systems together in sleep consolidation (Appelbaum et al. 2009). In addition to neuropeptides, melatonin, a naturally occurring hormone, is produced in the pineal gland at night and is also required for circadian regulation of zebrafish sleep (Kazimi and Cahill 1999; Zhdanova et al. 2001). Melatonin’s ability to facilitate sleep is evolutionarily conserved as it is widely used as an over-the-counter sleep aid. Finally, adenosine regulates homeostatic sleep by creating sleep pressure and inducing sleep as a result of accumulation during prolonged wakefulness (Wigren et al. 2007). The role of adenosine in sleep homeostasis remains a topic of controversy in different species. In Drosophila, caffeine-mediated reduction and fragmentation of sleep did not depend on adenosine receptor (Wu et al. 2009). Similarly, adenosine receptor knock-out mice did not exhibit any defects in sleep homeostasis (Stenberg et al. 2003). However, more recent evidence in mice suggests that intracellular adenosine mediates sleep homeostasis through glial-neural circuits; while homeostatic sleep drive was enhanced in glial-deficient adenosine, neural-deficient adenosine did not influence sleep drive (Bjorness et al. 2016). In zebrafish, adenosine was reported to trigger sleep and prevent activity during the day but not at night (Mourrain lab unpublished data; Gandhi et al. 2015).
Sleep-modulating neuropeptides, hormones and their connection to redox metabolism
Sleep–wake regulating neuropeptides are implicated in cellular redox metabolism. For instance, elevated NPY enhances cellular redox potential by increasing NADPH and the production of NPY itself is mediated by ROS (Raghuraman et al. 2011; Schwetz et al. 2013). Another example is that the loss of functional galanin causes mitochondrial oxidative stress in mice (Boal et al. 2022). In addition, increased orexin A/hypocretin 1 peptide causes mitochondrial impairment and dysfunction, contributing to increased cellular ROS (Li et al 2020). Lastly, adenosine alleviates oxidative stress by increasing expression levels of an essential antioxidant gene, nuclear factor (erythroid‑derived 2)‑like 2 (Nrf2) (Gholinejad et al. 2018), suggesting that adenosine and oxidative burden could act together to drive sleep and bring redox balance back to homeostatic levels.
A direct relationship between ROS and sleep-regulating neuropeptides remain to be elucidated to better understand the mechanism of action of ROS on these neuropeptides, however, there is evidence from C. elegans studies that mitochondrial-produced ROS can modulate neuropeptide release: in cholinergic motor neurons, neuropeptide-like protein (NLP-21) secretion was inhibited in two-different mutant types, both mutants causing enhanced mitochondrial ROS production via different mechanisms (Zhao et al. 2018). On the other hand, mitochondria-derived ROS increased secretion of neuropeptide FMRP-like peptide (FLP-1) in AIY interneurons, which in turn induced a major antioxidant mechanism, Nrf2, to eliminate excess ROS and ameliorate oxidative stress (Jia and Sieburth 2021). Hence, depending on cell type and upstream activators, ROS can have different effects on neuropeptide secretion. Overall, major cell-to-cell signalling molecules regulating sleep–wake behavior in mammals and zebrafish have also been implicated in oxidative stress/redox homeostasis, which may involve positive as well as negative feedback loops for further regulation of redox control and neuropeptide production and/ or release.
Melatonin is the first hint connecting redox metabolism to sleep function through its functions beyond regulating circadian rhythms. Melatonin is quite a peculiar compound as the function of melatonin has evolved and diversified over 3 billion years. Melatonin is believed to serve as an antioxidative agent, evolving in photosynthetic bacteria and acquiring new roles in circadian regulation and sleep in the present day (Manchester et al. 2015; Tan et al. 2013). While pineal melatonin regulates the circadian rhythms, extra pineal melatonin performs various functions beyond its primary role, which includes acting as an antioxidant, stimulating the production of endogenous antioxidant enzymes, scavenging free radicals, and playing a homeostatic role within mitochondria (Aranda‐Martínez et al. 2022; Gandhi et al. 2015; Zhdanova et al. 2001). It has been shown that melatonin is capable of indirectly scavenging free radicals by regulating the activity and expression of other antioxidant systems in both plant and mammals (Bidabadi et al. 2020; Morvaridzadeh et al. 2020; Nogués et al. 2006). In zebrafish, like in other animals, melatonin also maintains the redox balance by regulating the ratio of reduced glutathione to oxidized glutathione (GSH/GSSG), reducing lipid peroxidation, and enhancing the activity and expression of other antioxidant enzymes such as SOD and catalase (Duarte et al. 2023; Lunkes et al. 2021; Yan et al. 2022). Furthermore, melatonin administration alleviated the reduction in catalase, glutathione peroxidase and SOD activity following sleep-deprivation and enhanced GSH/GSSG ratio levels to normal (Alzoubi et al. 2016). In zebrafish, there is only one study that utilized melatonin treatment in sleep-deprived animals, and showed that melatonin treatment did not rescue the learning performance in zebrafish (Pinheiro-da-Silva et al. 2018). However, this study lacks in-depth biochemical measures at the cellular level to assess any improvement in the cellular effects of sleep deprivation. Finally, in a zebrafish model of Parkinson’s disease (PD), a neurodegenerative disorder where sleep disturbances are among the most prevalent symptoms, melatonin administration restored the sleep–wake cycles of animals with impaired rhythm and dysfunctional mitochondria (Aranda-Martínez et al. 2023; Mattis and Sehgal 2016). This is similar to studies where melatonin administration prevents sleep loss in PD patients and confers protective effects on mitochondria in mice and rat models of PD (Daneshvar Kakhaki et al. 2020; López et al. 2017; Paul et al. 2018). Thus, melatonin has a bipartite role in sleep through circadian and homeostatic redox regulation.
Role of redox metabolism in sleep homeostasis
It is well-documented that during prolonged sleep deprivation, ROS production is elevated in multiple species and in multiple organs including different regions of the brain (hippocampus, frontal cortex, cerebellum, neocortex), liver, heart, gut, and skeletal muscle (Kempf et al. 2019; Rodrigues et al. 2018; Vaccaro et al. 2020; Villafuerte et al. 2015). However, the source of this over-produced ROS following sleep-deprivation is currently unknown. The possible sources are: (1) decreased expression and activity of cellular antioxidant systems (discussed above), or (2) increased production of ROS through major sources, such as mitochondria, that overcome the cellular antioxidant capacity, or (3) both. Mitochondria undergo several ultrastructural and biochemical changes after sleep deprivation. Mitochondrial size and density increases, fusion/fission dynamics are dysregulated, ETC efficiency decreases, and ROS production increases (De Vivo et al. 2016; Flores et al. 2022; Lu et al. 2021). Mitochondria-derived ROS turns on the dFB neurons, which are more excitable with higher sleep pressure in fruit flies. dFB neurons are activated through the voltage-gated potassium channel Shaker and its subunit Hyperkinetic, and loss-of-function mutations in either protein cause insomnia (Bushey et al. 2007; Cirelli et al. 2005; Kempf et al. 2019). Hyperkinetic has a redox-sensing capability via its binding to NADPH in its active site; as sleep pressure increases, mitochondria ETC enhances ROS production, which in turn oxidizes NADPH to NADP + . As a result, Hyperkinetic is more likely converted to its NADP + -bound form, such that A-type potassium current flows through Shaker with slower inactivation, and thus enhancing the activity of sleep-control neurons (Kempf et al. 2019). Similarly, loss of voltage-gated potassium channel kcna2 and Na + /K + pump atp1a3 genes in zebrafish reduced sleep, and it is highly likely that redox metabolism and sleep are mechanically connected in zebrafish as in fruit flies (Barlow et al. 2023; Srdanovic et al. 2017). In zebrafish, intracellular NAD(H) levels were shown to be reduced in the absence of Letm1, a conserved mitochondrial cation exchanger which follows diurnal rhythms together with NAMPT, the key enzyme in NAD + production (Dao et al. 2022). Finally, Parp1, which is both an NAD + consuming and a DNA repair enzyme, can trigger sleep in zebrafish and adult mice. Zada et al., showed that neuronal DNA damage, which builds up during the day, increases sleep pressure and that Parp1 activity promotes sleep to facilitate efficient DNA repair and clearing of the pressure (Zada et al. 2021). Thus, the redox metabolites NAD + /NADH and NADP + /NADPH serve as a link between cellular metabolism and sleep. It has been acknowledged that during the active phase, high cellular activity concentrates ROS from different sources, leading to increased sleep drive. During sleep, antioxidant mechanisms take over to counteract the ROS accumulation and restore the cellular redox balance. When the ROS burden cannot be alleviated because of reduced or lack of sleep, the redox imbalance persists and causes cellular damage. The metabolic regulation of sleep and the redox state is summarized in Fig. 2.

Schematics of redox-related changes during sleep–wake cycle. ROS accumulates from high cellular activity and increased NADH during the wake period (top left), creating sleep pressure (top right) in the form of membrane lipid peroxidation, redox imbalances and DNA damage. The effects of the wake period are ameliorated during the sleep period (bottom left), where DNA damaged during the day period is repaired and membrane lipids and redox systems are replenished and returned to homeostatic levels. Inadequate sleep and sleep deprivation (bottom right) worsen the redox imbalance with lower antioxidant systems (GSH, SOD, catalase) that cannot prevent further ROS accumulation
Astyanax mexicanus: lessons to be learned from fish that sleep less
Astyanax mexicanus, commonly known as the Mexican tetra, exhibits two-different morphotypes: surface- and cave-dwelling populations (Jeffery 2020). These populations have diverged due to their contrasting habitats and environmental conditions. The surface populations reside in rivers and streams with access to nutrients, while the cave populations inhabit underground cave systems, having adapted to scarce nutrient environments (Rohner 2018). The surface populations have retained their pigmentation and functional eyes, allowing them to thrive in lighted environments. In contrast, the cave populations have undergone evolutionary changes, resulting in reduced pigmentation, and degenerated or non-functional eyes, as they inhabit dark cave environments with limited or no access to light (Krishnan and Rohner 2017; Moran et al. 2015). These adaptations reflect the selective pressures acting on each population, such as the surface populations adapting to visual cues and the cave populations relying on other sensory mechanisms for survival in the darkness. The study of these distinct populations provides valuable insights into evolutionary processes, as well as metabolic and molecular adaptations to extreme environments.
Interestingly, cavefish exhibited 80% reduction in their sleep compared to surface-dwelling populations, thus becoming a robust model for understanding evolutionary changes in sleep (Jaggard et al. 2018; Keene and Appelbaum 2019; Yoshizawa et al. 2015). Two interconnected mechanisms have been reported to affect the sleep reduction in cavefish populations: sensory input and the wake-promoting Hcrt neuropeptide. The lateral line of fish consists of mechanosensory organs called neuromasts, and the number, size and sensitivity of neuromasts to sensory stimuli are more profound in cavefish than in surface-dwelling populations (Yoshizawa et al. 2014). Targeted ablation of neuromasts in cavefish enhanced their sleep, suggesting that increased sensory input is responsible for sleep loss (Jaggard et al. 2017). In addition, the Hcrt/orexin network is conserved in Mexican tetra, and loss of Hcrt function in cavefish restores sleep to levels similar to those in surface fish (Jaggard et al. 2018). Furthermore, ablating mechanosensory lateral line reduced Hcrt levels in cavefish, implying that the recognition of sensory cues through the lateral line plays a crucial role in promoting Hcrt signalling (Jaggard et al. 2018), thereby maintaining wakefulness. Despite the given role of sensory responsiveness in sleep reduction, it is noteworthy that lateral line ablation did not have an impact on sleep in four separate populations of cavefish (Jaggard et al. 2017). This finding implies that unique mechanisms likely govern the evolutionary process of sleep loss in independently derived cavefish populations, or there are other commonly evolved mechanisms that are important in sleep regulation, that have yet to be studied.
Cavefish face extreme conditions in their natural habitats such as long periods of nutrient deprivation, which in turn triggers chronic stress. As opposed to other organisms, for which these conditions would be deleterious, cavefish overcome the environmental challenges and maintain physiological health via metabolic adaptations, potentially contributing to reduced sleep phenotype in cavefish. Compared to surface-dwelling populations, cavefish acquired insulin resistance and elevated blood glucose in multiple cave populations to develop metabolic resilience (Riddle et al. 2018). Decreasing sleep duration significantly reduced insulin sensitivity in healthy individuals, and sleep loss is a risk factor for insulin resistance and type 2 diabetes in humans, suggesting insulin-mediated metabolic changes could have impact on sleep regulation (Buxton et al. 2010; Spiegel et al. 2005). Furthermore, redox alterations have bidirectional role in developing insulin resistance: H2O2 has been shown in mice to attenuate insulin resistance while chronic ROS production via mitochondria and NOXes contribute to development of insulin resistance by pro-inflammatory cytokines (Loh et al. 2009; Tiganis 2011). Unlike the surface morphotype, cavefish have lower levels of ROS and enhanced antioxidant activities, which could contribute to reduced need for sleep and weaker oxidative response to stress conditions. Key antioxidant genes involving GSH metabolism were upregulated and the major cellular antioxidant, GSH, as well as vitamin C, were increased in the liver and brain of cave populations, but not in surface-dwelling populations (Krishnan et al. 2020; Medley et al. 2022). Moreover, under stress conditions of prolonged starvation, cavefish exhibited lower cytoplasmic ROS compared to surface fish (Medley et al. 2022), indicating that ROS accumulation is likely to be at a lesser extent in cavefish populations, supporting the hypothesis that ROS accumulation creates sleep pressure and ROS is cleared during sleep.
Concluding remarks
Despite being evolutionarily conserved across all animals, the core physiological function of sleep remains unclear. The imbalance between the antioxidant defense system and the generation of oxidants creates oxidative stress which can further cellular damage and compound adverse issues from chronically impaired sleep such as obesity and even neurodegenerative disorders. Redox metabolism, like sleep, is well conserved such that from plants to animals, it has been found that oxidized molecules are dissipated at night to facilitate processes like DNA repair. Here, we primarily focus on zebrafish, a highly amenable model in which sleep, and metabolic mechanisms are highly conserved, to delineate the reciprocal relationship between sleep homeostasis and redox metabolism.
As mentioned above, it is well documented across species that prolonged sleep deprivation increases susceptibility to oxidative stress in the form of increased mitochondrial ROS which in turn increases sleep pressure. During sleep, this pressure is ameliorated by removing scavenging free radicals with antioxidant enzymes which can be regulated with sleep–wake modulators like melatonin. Concordantly, some species, like the cavefish highlighted in this review, have evolved metabolic evolutionary changes different from their surface-dwelling counterparts to increase their resilience to oxidative stress via lower ROS and higher antioxidant levels. If a key function of sleep is to defend against oxidative stress accumulated during the wake period, these metabolic changes to prevent oxidative stress in cavefish may be contributing to their shorter sleep phenotype and might mean that differences in sleep between animals or species is due in part to metabolic differences. In addition, it is possible that evolution led to shared redox-sensing channels (e.g., Hyperkinetic and Shaker) to mechanically link sleep-active neurons and redox metabolism in order to more efficiently serve a common fundamental need. This fundamental need requires identification to truly define sleep in an explanation that accounts for the many evolutionarily conserved roles of sleep, such as in redox metabolism as we covered here.
In this review, we cover the significance and the mechanisms of redox metabolism and sleep homeostasis and how they are connected. However, there are challenges remaining to pinpoint the cellular mechanisms that explain where and how exactly ROS accumulation builds sleep pressure and how precisely this sleep pressure is cleared. We focus particularly on to explore how this ROS accumulation contributes to sleep pressure buildup and the exact mechanisms by which this sleep pressure is cleared as these questions remain unknown in this group of organisms. Metabolic homeostasis and the cellular redox systems are delicate systems that differ in subcellular compartments and depend on the physiological condition of the animal/cell (e.g., circadian rhythm, cell cycle stage, disease pathology). Due to these finer differences, non-targeted treatments of reactive oxidant radicals which are highly specific in their signalling, may fail as a remedy if subcellular precision at specific timepoints is necessary. Therefore, the study of intracellular mechanisms at the interplay of redox metabolism and sleep–wake regulation remains a difficult endeavor. Future studies may require precise targeting of both systems in tandem to fully elucidate this key function of sleep that maintains metabolic homeostasis.
References
Alzoubi KH, Mayyas FA, Khabour OF, Bani Salama FM, Alhashimi FH, Mhaidat NM (2016) Chronic melatonin treatment prevents memory impairment induced by chronic sleep deprivation. Mol Neurobiol 53:3439–3447
Appelbaum L, Wang GX, Maro GS, Mori R, Tovin A, Marin W, Mourrain P (2009) Sleep-wake regulation and hypocretin-melatonin interaction in zebrafish. Proc Natl Acad Sci USA 106:21942–21947. https://doi.org/10.1073/pnas.906637106
Appelbaum L, Wang G, Yokogawa T, Skariah GM, Smith SJ, Mourrain P, Mignot E (2010) Circadian and homeostatic regulation of structural synaptic plasticity in hypocretin neurons. Neuron 68(1):87–98. https://doi.org/10.1016/j.neuron.2010.09.006
Aranda-Martínez P, Fernández-Martínez J, Ramírez-Casas Y, Guerra-Librero A, Rodríguez-Santana C, Escames G, Acuña-Castroviejo D (2022) The Zebrafish, an outstanding model for biomedical research in the field of melatonin and human diseases. Int J Mol Sci. https://doi.org/10.3390/ijms23137438
Aranda-Martínez P, Fernández-Martínez J, Ramírez-Casas Y, Rodríguez-Santana C, Rusanova I, Escames G, Acuña-Castroviejo D (2023) Chronodisruption and loss of melatonin rhythm, associated with alterations in daily motor activity and mitochondrial dynamics in Parkinsonian Zebrafish, are corrected by melatonin treatment. Antioxidants. https://doi.org/10.3390/antiox12040954
Barlow IL, Mackay E, Wheater E, Goel A, Lim S, Zimmerman S, Rihel J (2023) The zebrafish mutant dreammist implicates sodium homeostasis in sleep regulation. Elife. https://doi.org/10.7554/eLife.87521.1
Barreiro E, Garcia-Martínez C, Mas S, Ametller E, Gea J, Argilés JM, Busquets S, López-Soriano FJ (2009) UCP3 overexpression neutralizes oxidative stress rather than nitrosative stress in mouse myotubes. FEBS Lett 583(2):350–356. https://doi.org/10.1016/j.febslet.2008.12.023
Bechtold U, Murphy DJ, Mullineaux PM (2004) Arabidopsis peptide methionine sulfoxide Reductase2 prevents cellular oxidative damage in long nights. Plant Cell 16:908–919. https://doi.org/10.1105/tpc.015818
Bedard K, Krause K-H (2007) The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87(1):245–313. https://doi.org/10.1152/physrev.00044.2005
Bidabadi SS, Vanderweide J, Sabbatini P (2020) Exogenous melatonin improves glutathione content, redox state and increases essential oil production in two Salvia species under drought stress. Sci Rep 10(6883):1–12. https://doi.org/10.1038/s41598-020-63986-6
Bjorness TE, Dale N, Mettlach G, Sonneborn A, Sahin B, Fienberg AA, Yanagisawa M, Bibb JA, Greene RW (2016) An adenosine-mediated glial-neuronal circuit for homeostatic sleep. J Neurosci 36(13):3709–3721. https://doi.org/10.1523/JNEUROSCI.3906-15.2016
Boal F, Cinato M, Timotin A, Münzberg H, Qualls-Creekmore E, Kramar S, Kunduzova O (2022) Galanin regulates myocardial mitochondrial ROS homeostasis and hypertrophic remodeling through GalR2. Front Pharmacol 13(March):1–8. https://doi.org/10.3389/fphar.2022.869179
Bradshaw PC (2019) Cytoplasmic and mitochondrial NADPH-coupled Redox systems in the regulation of aging. Nutrients. https://doi.org/10.3390/nu11030504
Bushey D, Huber R, Tononi G, Cirelli C (2007) Drosophila hyperkinetic mutants have reduced sleep and impaired memory. J Neurosci 27(20):5384–5393. https://doi.org/10.1523/JNEUROSCI.0108-07.2007
Buxton OM, Pavlova M, Reid EW, Wang W, Simonson DC, Adler GK (2010) Sleep restriction for 1 week reduces insulin sensitivity in healthy men. Diabetes 59(9):2126–2133. https://doi.org/10.2337/db09-0699
Campbell SS, Tobler I (1984) Animal sleep: a review of sleep duration across phylogeny. Neurosci Biobehav Rev 8(3):269–300. https://doi.org/10.1016/0149-7634(84)90054-X
Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB (2007) Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci 120(23):4155–4166. https://doi.org/10.1242/jcs.011163
Chiu CN, Rihel J, Lee DA, Singh C, Mosser EA, Chen S, Prober DA (2016) A zebrafish genetic screen identifies neuromedin u as a regulator of sleep/wake states. Neuron 89(4):842–856. https://doi.org/10.1016/j.neuron.2016.01.007
Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, Tononi G (2005) Reduced sleep in Drosophila Shaker mutants. Nature 434:1087–1092
D’Almeida V, Lobo LL, Hipolide DC, De Oliveira AC, Nobrega JN, Tufik S (1998) Sleep deprivation induces brain region-specific decreases in glutathione levels. NeuroReport 9(12):2853–2856. https://doi.org/10.1097/00001756-199808240-00031
Daneshvar Kakhaki R, Ostadmohammadi V, Kouchaki E, Aghadavod E, Bahmani F, Tamtaji OR et al (2020) Melatonin supplementation and the effects on clinical and metabolic status in Parkinson’s disease: a randomized, double-blind, placebo-controlled trial. Clin Neurol Neurosurg 195:105878. https://doi.org/10.1016/j.clineuro.2020.105878
Dao P, Hajny S, Mekis R, Orel L, Dinhopl N, Tessmar-Raible K, Nowikovsky K (2022) The cation exchanger Letm1, circadian rhythms, and NAD(H) levels interconnect in diurnal zebrafish. Life Sci Alliance 5(9):e202101194
Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D (2016) ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxidative Med Cell Longev. https://doi.org/10.1155/2016/3565127
De Almeida AJPO, De Oliveira JCPL, Da Silva Pontes LV, De Souza Júnior JF, Gonçalves TAF, Dantas SH, De Medeiros IA (2022) ROS: basic concepts, sources, cellular signaling, and its implications in aging pathways. Oxidative Med Cell Longev. https://doi.org/10.1155/2022/1225578
De Vivo L, Nelson AB, Bellesi M, Noguti J, Tononi G, Cirelli C (2016) Loss of sleep affects the ultrastructure of pyramidal neurons in the adolescent mouse frontal cortex. Sleep 39(4):861–874. https://doi.org/10.5665/sleep.5644
Dickinson BC, Chang CJ (2011) Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7(8):504–511. https://doi.org/10.1038/nchembio.607
Diekelmann S, Born J (2010) The memory function of sleep. Nat Rev Neurosci 11(2):114–126. https://doi.org/10.1038/nrn2762
DiNuzzo M, Nedergaard M (2017) Brain energetics during the sleep–wake cycle. Curr Opin Neurobiol 47:65–72. https://doi.org/10.1016/j.conb.2017.09.010
Duarte MB, Medeiros BZ, da Silva Lemos I, da Silva GL, Alano CG, Dondossola ER, Streck EL (2023) Melatonin improves behavioral parameters and oxidative stress in zebrafish submitted to a leucine-induced MSUD protocol. Metab Brain Dis. https://doi.org/10.1007/s11011-023-01220-8
Everson CA, Laatsch CD, Hogg N (2005) Antioxidant defense responses to sleep loss and sleep recovery. Am J Physiol Regul Integr Comp Physiol 288(2):374–383. https://doi.org/10.1152/ajpregu.00565.2004
Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ 22(3):377–388. https://doi.org/10.1038/cdd.2014.150
Finkel T (2011) Signal transduction by reactive oxygen species. J Cell Biol 194(1):7–15. https://doi.org/10.1083/jcb.201102095
Flores CC, Loschky SS, Marshall W, Spano GM, Massaro Cenere M, Tononi G, Cirelli C (2022) Identification of ultrastructural signatures of sleep and wake in the fly brain. Sleep 45(5):1–12. https://doi.org/10.1093/sleep/zsab235
Frank MG (2006) The mystery of sleep function: current perspectives and future directions. Rev Neurosci 17(4):375–392. https://doi.org/10.1515/revneuro.2006.17.4.375
Gandhi AV, Mosser EA, Oikonomou G, Prober DA (2015) Melatonin is required for the circadian regulation of sleep. Neuron 85(6):1193–1199. https://doi.org/10.1016/j.neuron.2015.02.016
Gholinejad M, Anarkooli IJ, Taromchi A, Abdanipour A (2018) Adenosine decreases oxidative stress and protects H2O2-treated neural stem cells against apoptosis through decreasing Mst1 expression. Biomed Rep 8(5):439–446. https://doi.org/10.3892/br.2018.1083
Haida Z, Hakiman M (2019) A comprehensive review on the determination of enzymatic assay and nonenzymatic antioxidant activities. Food Sci Nutr 7(5):1555–1563. https://doi.org/10.1002/fsn3.1012
Hill VM, O’Connor RM, Sissoko GB, Irobunda IS, Leong S, Canman JC, Shirasu-Hiza M (2018) A bidirectional relationship between sleep and oxidative stress in Drosophila. PLoS Biol 16(7):1–22. https://doi.org/10.1371/journal.pbio.2005206
Hirschenson J, Melgar-Bermudez E, Mailloux RJ (2022) The uncoupling proteins: a systematic review on the mechanism used in the prevention of oxidative stress. Antioxidants. https://doi.org/10.3390/antiox11020322
Hsieh YS, Chen PN, Yu CH, Liao JM, Kuo DY (2013) The neuropeptide y Y1 receptor knockdown modulates activator protein 1-involved feeding behavior in amphetamine-treated rats. Mol Brain 6(1):1–12. https://doi.org/10.1186/1756-6606-6-46
Jaggard JB, Robinson BG, Stahl BA, Oh I, Masek P, Yoshizawa M, Keene AC (2017) The lateral line confers evolutionarily derived sleep loss in the Mexican cavefish. J Exp Biol 220(2):284–293. https://doi.org/10.1242/jeb.145128
Jaggard JB, Stahl BA, Lloyd E, Prober DA, Duboue ER, Keene AC (2018) Hypocretin underlies the evolution of sleep loss in the Mexican cavefish. Elife 7:1–22. https://doi.org/10.7554/eLife.32637
Jaggard JB, Wang GX, Mourrain P (2021) Non-REM and REM/paradoxical sleep dynamics across phylogeny. Curr Opin Neurobiol 71:44–51. https://doi.org/10.1016/J.CONB.2021.08.004
Jeffery WR (2020) Astyanax surface and cave fish morphs. EvoDevo 11(1):1–10. https://doi.org/10.1186/s13227-020-00159-6
Jia Q, Sieburth D (2021) Mitochondrial hydrogen peroxide positively regulates neuropeptide secretion during diet-induced activation of the oxidative stress response. Nat Commun. https://doi.org/10.1038/s41467-021-22561-x
Kayaba M, Park I, Iwayama K, Seya Y, Ogata H, Yajima K, Tokuyama K (2017) Energy metabolism differs between sleep stages and begins to increase prior to awakening. Metab Clin Exp 69:14–23. https://doi.org/10.1016/j.metabol.2016.12.016
Kazimi N, Cahill GM (1999) Development of a circadian melatonin rhythm in embryonic zebrafish. Dev Brain Res 117:47–52
Keene AC, Appelbaum L (2019) Sleep in fish models. In: Dringenberg HC (ed) Handbook of behavioral neuroscience, 1st edn, vol 30. https://doi.org/10.1016/B978-0-12-813743-7.00024-4
Keene AC, Duboue ER (2018) The origins and evolution of sleep. J Exp Biol. https://doi.org/10.1242/jeb.159533
Kempf A, Song SM, Talbot CB, Miesenböck G (2019) A potassium channel β-subunit couples mitochondrial electron transport to sleep. Nature 568(7751):230–234. https://doi.org/10.1038/s41586-019-1034-5
Klinzing JG, Niethard N, Born J (2019) Mechanisms of systems memory consolidation during sleep. Nat Neurosci 22(10):1598–1610. https://doi.org/10.1038/s41593-019-0467-3
Krishnan J, Rohner N (2017) cevafish and the basis for eye loss. Philos Trans R Soc B Biol Sci 372(1713):1–10. https://doi.org/10.1016/s0140-6736(02)53944-1
Krishnan J, Persons JL, Peuß R, Hassan H, Kenzior A, Xiong S, Rohner N (2020) Comparative transcriptome analysis of wild and lab populations of Astyanax mexicanus uncovers differential effects of environment and morphotype on gene expression. J Exp Zool Part B Mol Dev Evol 334(7–8):530–539. https://doi.org/10.1002/jez.b.22933
Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Kornblum HI (2011) Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8(1):59–71. https://doi.org/10.1016/j.stem.2010.11.028
Leung LC, Mourrain P (2018) Sleep: short sleepers should keep count of their hypocretin neurons. Curr Biol 28(9):R558–R560. https://doi.org/10.1016/j.cub.2018.03.006
Leung LC, Wang GX, Madelaine R, Skariah G, Kawakami K, Deisseroth K, Mourrain P (2019) Neural signatures of sleep in zebrafish. Nature 571(7764):198–204. https://doi.org/10.1038/s41586-019-1336-7
Li W, Sauve AA (2015) NAD+ content and its role in mitochondria. In: Palmeira CM, Rolo AP (eds) Mitochondrial regulation. Methods in molecular biology, vol 1241. Humana Press, New York, NY https://doi.org/10.1007/978-1-4939-1875-1_4
Li M, Meng Y, Chu B, Shen Y, Liu X, Ding M, Song C, Cao X, Wang P, Xu L, Wang Y, Xu S, Bi J, Xie Z (2020) Orexin-A aggravates cytotoxicity and mitochondrial impairment in SH-SY5Y cells transfected with APPswe via p38 MAPK pathway. Ann Transl Med 8(1):5. https://doi.org/10.21037/atm.2019.11.68
Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Tiganis T (2009) Reactive oxygen species enhance insulin sensitivity. Cell Metab 10(4):260–272. https://doi.org/10.1016/j.cmet.2009.08.009
López A, Ortiz F, Doerrier C, Venegas C, Fernández-Ortiz M, Aranda P, Acuña-Castroviejo D (2017) Mitochondrial impairment and melatonin protection in parkinsonian mice do not depend of inducible or neuronal nitric oxide synthases. PLoS One 12:e0183090. https://doi.org/10.1371/journal.pone.0183090
Lu Z, Hu Y, Wang Y, Zhang T, Long J, Liu J (2021) Topological reorganizations of mitochondria isolated from rat brain after 72 hours of paradoxical sleep deprivation, revealed by electron cryo-tomography. Am J Phys Cell Physiol 321(1):C17–C25. https://doi.org/10.1152/AJPCELL.00077.2021
Lunkes LC, Paiva IM, Egger RC, Braga WF, Alvarez-Leite JI, da Cunha Barreto-Vianna AR, Murgas LDS (2021) Melatonin administration attenuates acute stress by inducing sleep state in zebrafish (Danio rerio). Comp Biochem Physiol Part C Toxicol Pharmacol 246(December 2020):1–7. https://doi.org/10.1016/j.cbpc.2021.109044
Manchester LC, Coto-Montes A, Boga JA, Andersen LPH, Zhou Z, Galano A, Reiter RJ (2015) Melatonin : an ancient molecule that makes oxygen metabolically tolerable. J Pine 59:403–419. https://doi.org/10.1111/jpi.12267
Martinelli I, Timotin A, Moreno-Corchado P, Marsal D, Kramar S, Loy H, Kunduzova O (2021) Galanin promotes autophagy and alleviates apoptosis in the hypertrophied heart through FoxO1 pathway. Redox Biol 40:101866. https://doi.org/10.1016/j.redox.2021.101866
Mattis J, Sehgal A (2016) Circadian rhythms, sleep, and disorders of aging. Trends Endocrinol Metab 27(4):192–203. https://doi.org/10.1016/j.tem.2016.02.003
Medley JK, Persons J, Biswas T, Olsen L, Peuß R, Krishnan J, Rohner N (2022) The metabolome of Mexican cavefish shows a convergent signature highlighting sugar, antioxidant, and ageing-related metabolites. Elife 11:1–25. https://doi.org/10.7554/eLife.74539
Moran D, Softley R, Warrant EJ (2015) The energetic cost of vision and the evolution of eyeless Mexican cavefish. Sci Adv. https://doi.org/10.1126/sciadv.1500363
Morvaridzadeh M, Sadeghi E, Agah S, Nachvak SM, Fazelian S, Moradi F, Heshmati J (2020) Effect of melatonin supplementation on oxidative stress parameters: a systematic review and meta-analysis. Pharmacol Res 161(August):105210. https://doi.org/10.1016/j.phrs.2020.105210
Mourrain P, Wang GX (2019) Sleep: DNA repair function for better neuronal aging? Curr Biol 29(12):R585–R588. https://doi.org/10.1016/J.CUB.2019.05.018
Mullington J, Korth C, Hermann DM, Orth A, Galanos C, Holsboer F, Pollmächer T (2000) Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol 278(4):947–955. https://doi.org/10.1152/ajpregu.2000.278.4.r947
Munnamalai V, Weaver CJ, Weisheit CE, Venkatraman P, Agim ZS, Quinn MT, Suter DM (2014) Bidirectional interactions between Nox2-type NADPH oxidase and the F-actin cytoskeleton in neuronal growth cones. J Neurochem 130(4):526–540. https://doi.org/10.1111/jnc.12734
Nogués MR, Giralt M, Romeu M, Mulero M, Sánchez-Martos V, Rodríguez E, Mallol J (2006) Melatonin reduces oxidative stress in erythrocytes and plasma of senescence-accelerated mice. J Pineal Res 41(2):142–149. https://doi.org/10.1111/j.1600-079X.2006.00344.x
Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Iadecola C (2008) Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. PNAS 105(4):1347–1352. https://doi.org/10.1073/pnas.0711568105
Paul R, Phukan BC, Thenmozhi AJ, Manivasagam T, Battacharya P, Borah A (2018) Melatonin protects against behavioral deficits, dopamine loss and oxidative stress in homocysteine model of Parkinson’s disease. Life Sci 192:238–245. https://doi.org/10.1016/j.lfs.2017.11.016
Pinheiro-da-Silva J, Tran S, Luchiari AC (2018) Sleep deprivation impairs cognitive performance in zebra fish: a matter of fact ? Behav Proc 157(March):656–663. https://doi.org/10.1016/j.beproc.2018.04.004
Raghuraman G, Kalari A, Dhingra R, Prabhakar NR, Kumar GK (2011) Enhanced neuropeptide y synthesis during intermittent hypoxia in the rat adrenal medulla: Role of reactive oxygen species-dependent alterations in precursor peptide processing. Antioxid Redox Signal 14(7):1179–1190. https://doi.org/10.1089/ars.2010.3353
Rama Rao KV, Iring S, Younger D, Kuriakose M, Skotak M, Alay E, Chandra N (2018) A single primary blast-induced traumatic brain injury in a rodent model causes cell-type dependent increase in nicotinamide adenine dinucleotide phosphate oxidase isoforms in vulnerable brain regions. J Neurotrauma 35(17):2077–2090. https://doi.org/10.1089/neu.2017.5358
Ramanathan L, Gulyani S, Nienhuis R, Siegel JM (2002) Sleep deprivation decreases superoxide dismutase activity in rat hippocampus and brainstem. NeuroReport 13(11):1387–1390. https://doi.org/10.1097/00001756-200208070-00007
Rechtschaffen A (1998) Current perspectives on the function of sleep. Perspect Biol Med 41(3):359–390. https://doi.org/10.1353/pbm.1998.0051
Rechtschaffen A, Bergmann BM (2002) Sleep deprivation in the rat: an update of the 1989 paper. Sleep 25(1):18–24. https://doi.org/10.1093/sleep/25.1.18
Reczek CR, Chandel NS (2015) ROS-dependent signal transduction. Curr Opin Cell Biol 33:8–13. https://doi.org/10.1016/j.ceb.2014.09.010
Reichert S, Pavón Arocas O, Rihel J (2019) The neuropeptide galanin is required for homeostatic rebound sleep following increased neuronal activity. Neuron 104(2):370-384.e5. https://doi.org/10.1016/j.neuron.2019.08.010
Reimund E (1994) The free radical flux theory of sleep. Med Hypotheses 43(4):231–233. https://doi.org/10.1016/0306-9877(94)90071-X
Riddle MR, Aspiras AC, Gaudenz K, Peuß R, Sung JY, Martineau B, Rohner N (2018) Insulin resistance in cavefish as an adaptation to a nutrient-limited environment. Nature 555(7698):647–651. https://doi.org/10.1038/nature26136
Rodrigues NR, Macedo GE, Martins IK, Gomes KK, de Carvalho NR, Posser T, Franco JL (2018) Short-term sleep deprivation with exposure to nocturnal light alters mitochondrial bioenergetics in Drosophila. Free Radic Biol Med 120(March):395–406. https://doi.org/10.1016/j.freeradbiomed.2018.04.549
Rohner N (2018) Cavefish as an evolutionary mutant model system for human disease. Dev Biol 441(2):355–357. https://doi.org/10.1016/j.ydbio.2018.04.013
Schofield JH, Schafer ZT (2021) Mitochondrial reactive oxygen species and mitophagy: a complex and nuanced relationship. Antioxid Redox Signal 34(7):517–530. https://doi.org/10.1089/ars.2020.8058
Schwetz TA, Ustione A, Piston DW (2013) Neuropeptide Y and somatostatin inhibit insulin secretion through different mechanisms. Am J Physiol Endocrinol Metab 304(2):211–221. https://doi.org/10.1152/ajpendo.00374.2012
Shein-Idelson M, Ondracek JM, Liaw H, Reiter S, Laurent G (2016) Slow Slow waves, sharp waves, ripples, and REM in in sleeping dragons. Science 352(6285):590–595
Shen YC, Sun X, Li L, Zhang HY, Huang ZL, Wang YQ (2022) Roles of neuropeptides in sleep-wake regulation. Int J Mol Sci 23(9):1–17. https://doi.org/10.3390/ijms23094599
Siegel JM (2005) Clues to the functions of mammalian sleep. Nature 437(7063):1264–1271. https://doi.org/10.1038/nature04285
Silva RH, Abílio VC, Takatsu AL, Kameda SR, Grassl C, Chehin AB, Medrano WA, Calzavara MB, Registro S, Andersen ML, Machado RB, Carvalho RC, Ribeiro RDA, Tufik S, Frussa-Filho R (2004) Role of hippocampal oxidative stress in memory deficits induced by sleep deprivation in mice. Neuropharmacology 46(6):895–903. https://doi.org/10.1016/j.neuropharm.2003.11.032
Singh C, Rihel J, Prober DA (2017) Neuropeptide Y regulates sleep by modulating noradrenergic signaling. Curr Biol 27(24):3796-3811.e5. https://doi.org/10.1016/j.cub.2017.11.018
Somanna NK, Valente AJ, Krenz M, Fay WP, Delafontaine P, Chandrasekar B (2016) The Nox1/4 dual inhibitor GKT137831 or Nox4 knockdown inhibits angiotensin-II-induced adult mouse cardiac fibroblast proliferation and migration. AT1 physically associates with Nox4. J Cell Physiol 231(5):1130–1141. https://doi.org/10.1002/jcp.25210
Spiegel K, Knutson K, Leproult R, Tasali E, Van Cauter E (2005) Sleep loss: a novel risk factor for insulin resistance and type 2 diabetes. J Appl Physiol 99(5):2008–2019. https://doi.org/10.1152/japplphysiol.00660.2005
Srdanovic S, Þorsteinsson H, Friðriksson Þ, Pétursson SÓ, Maier VH, Karlsson KÆ (2017) Transient knock-down of kcna2 reduces sleep in larval zebrafish. Behav Brain Res 326:13–21. https://doi.org/10.1016/j.bbr.2017.02.026
Stenberg D, Litonius E, Halldner L, Johansson B, Fredholm BB, Porkka-Heiskanen T (2003) Sleep and its homeostatic regulation in mice lacking the adenosine A1 receptor. J Sleep Res 12(4):283–290. https://doi.org/10.1046/j.0962-1105.2003.00367.x
Tan D-X, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ (2013) Mitochondria and chloroplasts as the original sites of melatonin synthesis : a hypothesis related to melatonin ’ s primary function and evolution in eukaryotes. J Pineal Res 54:127–138. https://doi.org/10.1111/jpi.12026
Terzi A, Roeder H, Weaver CJ, Suter DM (2020) Neuronal NADPH oxidase 2 regulates growth cone guidance downstream of slit2/robo2. Dev Neurobiol 81(1):3–21. https://doi.org/10.1002/dneu.22791
Tiganis T (2011) Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci 32(2):82–89. https://doi.org/10.1016/j.tips.2010.11.006
Tononi G, Cirelli C (2006) Sleep function and synaptic homeostasis. Sleep Med Rev 10(1):49–62. https://doi.org/10.1016/j.smrv.2005.05.002
Trivedi MS, Holger D, Bui AT, Craddock TJA, Tartar JL (2017) Short-term sleep deprivation leads to decreased systemic redox metabolites and altered epigenetic status. PLoS One 12(7):1–13. https://doi.org/10.1371/journal.pone.0181978
Vaccaro A, Kaplan Dor Y, Nambara K, Pollina EA, Lin C, Greenberg ME, Rogulja D (2020) Sleep loss can cause death through accumulation of reactive oxygen species in the gut. Cell 181(6):1307-1328.e15. https://doi.org/10.1016/j.cell.2020.04.049
Van Cauter E, Spiegel K, Tasali E, Leproult R (2008) Metabolic consequences of sleep and sleep loss. Sleep Med 9(1):S23–S28. https://doi.org/10.1016/S1389-9457(08)70013-3
Villafuerte G, Miguel-Puga A, Murillo Rodríguez E, Machado S, Manjarrez E, Arias-Carrión O (2015) Sleep deprivation and oxidative stress in animal models: a systematic review. Oxidative Med Cell Longev. https://doi.org/10.1155/2015/234952
Wang B, Ma Y, Kong X, Ding X, Gu H, Chu T, Ying W (2014) NAD+ administration decreases doxorubicin-induced liver damage of mice by enhancing antioxidation capacity and decreasing DNA damage. Chem Biol Interact 212(1):65–71. https://doi.org/10.1016/j.cbi.2014.01.013
Weinberg F, Ramnath N, Nagrath D (2019) Reactive oxygen species in the tumor microenvironment: an overview. Cancers 11(8):E1191. https://doi.org/10.3390/cancers11081191
Wigren HK, Schepens M, Matto V, Stenberg D, Porkka-Heiskanen T (2007) Glutamatergic stimulation of the basal forebrain elevates extracellular adenosine and increases the subsequent sleep. Neuroscience 147(3):811–823. https://doi.org/10.1016/j.neuroscience.2007.04.046
Wren AM, Small CJ, Abbott CR, Jethwa PH, Kennedy AR, Murphy KG, Bloom SR (2002) Hypothalamic actions of neuromedin U. Endocrinology 143(11):4227–4234. https://doi.org/10.1210/en.2002-220308
Wu MN, Ho K, Crocker A, Yue Z, Koh K, Sehgal A (2009) The effects of caffeine on sleep in Drosophila require PKA activity, but not the adenosine receptor. J Neurosci 29(35):11029–11037. https://doi.org/10.1523/JNEUROSCI.1653-09.2009
Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, Zou B (2020) NAD+ metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. https://doi.org/10.1038/s41392-020-00311-7
Yan R, Ding J, Wei Y, Yang Q, Zhang X, Huang H, An Y (2022) Melatonin prevents NaAsO2-induced developmental cardiotoxicity in zebrafish through regulating oxidative stress and apoptosis. Antioxidants. https://doi.org/10.3390/antiox11071301
Yokogawa T, Marin W, Faraco J, Pézeron G, Appelbaum L, Zhang J, Mignot E (2007) Characterization of sleep in zebrafish and insomnia in hypocretin receptor mutants. PLoS Biol. https://doi.org/10.1371/journal.pbio.0050277
Yoshizawa M, Jeffery WR, Van Netten SM, McHenry MJ (2014) The sensitivity of lateral line receptors and their role in the behavior of Mexican blind cavefish (Astyanax mexicanus). J Exp Biol 217(6):886–895. https://doi.org/10.1242/jeb.094599
Yoshizawa M, Robinson BG, Duboué ER, Masek P, Jaggard JB, O’Quin KE, Keene AC (2015) Distinct genetic architecture underlies the emergence of sleep loss and prey-seeking behavior in the Mexican cavefish. BMC Biol 13(1):1–12. https://doi.org/10.1186/s12915-015-0119-3
Zada D, Sela Y, Matosevich N, Monsonego A, Lerer-Goldshtein T, Nir Y, Appelbaum L (2021) Parp1 promotes sleep, which enhances DNA repair in neurons. Mol Cell 81(24):4979-4993.e7. https://doi.org/10.1016/j.molcel.2021.10.026
Zhao T, Hao Y, Kaplan JM (2018) Axonal mitochondria modulate neuropeptide secretion through the hypoxic stress response in Caenorhabditis elegans. Genetics 210(1):275–285. https://doi.org/10.1534/genetics.118.301014
Zhdanova IV (2006) Sleep in zebrafish. Zebrafish 3(2):215–226. https://doi.org/10.1089/zeb.2006.3.215
Zhdanova IV, Wang SY, Leclair OU, Danilova NP (2001) Melatonin promotes sleep-like state in zebrafish. Brain Res 903(1–2):263–268. https://doi.org/10.1016/S0006-8993(01)02444-1
Acknowledgements
The Mourrain laboratory is supported by NHLBI HL151576, NINDS NS104950, NIGMS GM136741, NIA AG071787, NICHD HD109861 and the Simons Collaboration on Plasticity and the Aging Brain. The figures were created with Biorender.com.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors indicate that there is no conflict of interest.
Additional information
Communicated by Vladyslav V. Vyazovskiy.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Terzi, A., Ngo, K.J. & Mourrain, P. Phylogenetic conservation of the interdependent homeostatic relationship of sleep regulation and redox metabolism. J Comp Physiol B (2024). https://doi.org/10.1007/s00360-023-01530-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00360-023-01530-4