
Abstract
Local and global ecological stressors are leading to increased documentation of phase shifts in coral reefs from healthy stony corals to macrophytes. In more rare cases, phase shifts result in sponge, zoantharian or other dominant species. In Ulithi Atoll, Federated States of Micronesia, we have documented an unusual phase shift from reefs with a diverse stony coral assemblage to reefs dominated by a single species of stony coral: Montipora sp.—a coral-to-coral phase shift. This monospecific type of reef lowers fish diversity and biomass, impacting both ecological integrity, and livelihoods of reef-dependent human communities. In this study, we used a genomic approach to characterize such a reef. We assembled a de-novo reference genome and used RAD seq data with thousands of SNPs to determine if different reefs result from sexual or asexual reproduction, if weedy Montipora fragments are transported between islands by human activities, and if there is evidence of natural selection on specific genotypes, thus favoring spreading success. We found that sexual reproduction is predominant in the focal species, that there is no evidence of human-mediated spread, and that some genomic regions might be under selection. While such results eliminate a number of spreading hypotheses, more precise dispersal maps will be important to determine the tempo and mode of ‘invasion’ of Montipora in Ulithi Atoll. This study shows that selection and adaptation may be contributing to the success of a stony coral (e.g., Phase shift). While a stony coral may be successful in a disturbed environment, it does not necessarily provide the type of habitat that is conducive to high fish biomass and coral diversity. These results serve as a cautionary tale for restoration efforts that focus on single species coral resilience rather than ecosystem function.
Similar content being viewed by others

Introduction
Recent years have witnessed an increasing number of coral reef disturbances that in some cases resulted in phase shifts, most notably from diverse hard coral assemblages to macrophyte dominated reefs (Knowlton 1992; Bruno et al. 2009; Hughes 2022). Less frequently, reefs may shift into sponge or zoantharian dominated assemblages (Cruz et al. 2015). In most cases, ecological consequences negatively impact the entire ecosystem, in particular coral and fish assemblages (Done 1992; Halford et al. 2004; Crisp et al. 2022). In response to this ecological downgrading, and the continued decline of coral reefs globally, attention has turned to ‘coral gardening’ and reef restoration, focusing on ‘planting’ resistant corals (Camp et al. 2018; Schmidt-Roach et al. 2020). Our work here describes a ‘strong’ or ‘resistant’ coral that is contributing to a phase shift, with negative ecological consequences, a potential cautionary tale for facilitated restoration.
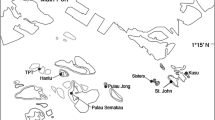
In Ulithi Atoll (Yap State, Federated States of Micronesia), several reefs that exhibited a diverse coral assemblage have slowly shifted into a Montipora monospecific coral dominated state over the past few decades. Local people have proposed that the initial shift may have coincided with Typhoon Ophelia, which occurred in 1960 (Lessa 1964). By then, reefs had already been severely disturbed by major rearrangements that happened during World War II, where several hundred ships, landing crafts, and thousands of US Naval personnel had staged out of the Atoll (Lessa 1966). Currently, some west, south (leeward) and ocean facing reefs, as well as nearby small atolls (turtle islands), are very diverse, while others (especially inside the lagoon in the north and east of the Atoll) are Montipora dominated (Figs. 1, 2) (Crane et al. 2017). This situation is mirrored by fish diversity and biomass, where diverse reefs and Montipora-dominated reefs harbor high or low diversity and abundance of reef fishes, respectively (Crane et al. 2017). In 2015, another typhoon (Maysak) made a direct hit on Ulithi Atoll. In the places of its direct passage, Montipora, which is a very fragile and brittle coral, suffered significant damage, thus reducing its abundance. However, by 2019, it had recovered in most places. For example, percent cover of Montipora (as averaged over 20 quadrats along a 100 m transect) at the site UAR2 in Falalop island was 43.9% in 2014, down to 10.6% in 2015, and back up to 45.5% in 2019. In Federai island (Federai Landing site), those values, for the same years, were 44.5%, 19.6%, and 72.6%. In Yealil (inside site), a somewhat pristine site, minimally impacted by the Typhoon, Montipora has shown a slow, steady increase (11.5%, 12.0%, 17.5%). Preliminary data and field observations show that those trends were maintained by the summer of 2023, with a few notable exceptions.

Pictures of Montipora sp.1 aff. capitata in Ulithi Atoll, Yap State, Federated States of Micronesia. Panel A: close up of the species, Panel B, an emerged colony at extreme low tide, Panel C, a colony, encroaching on a neighboring coral, Panel D, a stand of Montipora sp.1 aff. capitata covering a previously coral diverse reef

Map of the study region showing sampling locations used in this study: Asterisks indicate inhabited islands. Islands in red and in blue captions correspond to outside reefs that have a natural (low) abundance of Montipora, and to lagoonal reefs that are overgrown by Montipora, respectively
Phase-shifts that result in the overwhelming abundance of a single or few species of corals are rare, yet, with climate change and ocean acidification, it is possible for some species of corals to thrive in those conditions and capitalize on changing environmental conditions (Evensen et al. 2015; Henley et al. 2021). It is therefore important to understand the modes of invasion of these corals, and potential consequences to the ecosystem. Suggested that Montipora sp. first spread at landing sites at the four inhabited islands of the Atoll, leading to a conclusion that the breaking of fragile corals associated with anchoring, and the attachment of fragments to anchor lines may have aided the dispersal of Montipora sp. from one island to the next. An alternative possibility is that sexual reproduction may have played a dominant role and led to invasion due to the dispersal of propagules across the Atoll. In the latter case, it might be possible for natural selection to play a role in the success of specific genotypes. In this study, we used genomic tools to identify the patterns of invasion of Montipora sp. in Ulithi. We used RADseq data with hundreds of single nucleotide polymorphisms (SNPs) and a newly assembled genome to determine the mode of invasion of Montipora sp. and the potential role of selection in the system.
Materials and methods
Sampling—A total of 148 coral nubbin samples of Montipora sp. were collected while free diving from shallow coral dominated reefs. Samples were immediately placed in 95% ethanol and kept at ambient temperature until reaching the laboratory where they were stored at − 20 °C.
One sample was collected from Taveuni, Fiji, one sample was collected from the lagoon of Woleai Atoll (eastern Yap state, Micronesia), and one sample from Yap (main) Island, Micronesia. In all three locations, that species was found to be very rare, thus the small sample size. The remaining 145 samples were collected from different locations at Ulithi Atoll. In Ulithi, samples were collected inside the lagoon at all four inhabited islands: Falalop, Asor, MogMog, and Federai (two sites, Federai-Landing, near the Federai village and Federai-South, at the southern end of the island), as well as from the uninhabited islands of Soghlei, Laam, and Yealil (Fig. 2). Samples were also collected outside of the lagoon, on the outer reef slope, at the uninhabited islands of Sohng, Piglelei, and Yealil (Fig. 2, Table 1). Genetic identification revealed that our Ulithi collections encompassed two cryptic species, closely related to Montipora capitata, and are called henceforth Montipora sp.1 aff. capitata and Montipora sp.2 aff. capitata, comprising 141 and 7 samples, respectively. We therefore removed the 7 samples belonging to the rarely collected M. sp.2 aff. capitata species and kept 141 samples for our population genomics analyses (138 samples from Ulithi, and one sample each from Fiji, Woleai, and Yap). (Table 2).
DNA extraction—Library preparation. DNA was extracted from coral nubbins using DNeasy Blood & Tissue kits (Qiagen) according to the manufacturer's protocol. We constructed RAD libraries using a variation of the original protocol with the restriction enzyme SbfI (Miller et al. 2007, 2012; Baird et al. 2008; Longo and Bernardi 2015). The SbfI enzyme cuts sites that contain eight nucleotides, and that sequence seems to be very poorly represented in zooxanthellae, if at all, thus the SbfI cut sites in this experiment are found almost exclusively in the host coral. Initial genomic DNA amount for each individual was 400 ng. Libraries were physically sheared on a Covaris S2 sonicator with an intensity of 5, duty cycle of 10%, cycles/burst of 200 and a cycle time of 30 s. We carried out the final PCR amplification step in 50 μl reaction volumes with 10 amplification cycles. Ampure XP beads (Agencourt) were used for each purification step and size selection. The library was sequenced in a single lane on an Illumina HiSeq 4000 at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley (USA). Then, we applied Perl scripts to trim the raw reads to 92 base pairs (bp) on the 3' end, quality filtered and demultiplex them according to the 6 bp unique barcodes. Reads with Phred scores of < 33 were discarded. The barcodes and restriction site residues (6 bp) were removed from the 5' end, and this resulted in a final sequence length of 80 bp.
RAD analysis—The program STACKS version 2.2 (Catchen et al. 2011, 2013) was used to identify orthologous sequences among individuals. In order to optimize the STACKS protocol, we followed published guidelines (Rochette and Catchen 2017). Briefly, we first identified 20 individuals with the highest sequencing coverage and created a catalog of loci based on these samples (using the ustacks and cstacks components of STACKS). In addition, we optimized the values of the parameters M and n (M and n values were kept identical), by varying them from 1 to 9 as recommended. We found that the optimal value for M and n was 3. We then included the remaining samples using ustacks.
Analysis of population structure—Using the population component in STACKS, we further filtered the dataset by retaining loci which aligned in > 80% of individuals (r command, -r 0.8) in every population. In order to remove paralogs, we used the minor alleles function of STACKS following recommendations (min_mav 0.05). We then generated genepop files using the populations component in STACKS with the write_single_snp option (a single SNP was kept for each locus), which were converted afterward using the program PGDSpider V.2.1.0.3 (Lischer and Excoffier 2012). As will be described below, seven samples turned out to be a different species. We therefore repeated the analyses using 141 samples.
In order to ensure that clones (individuals resulting from asexual reproduction) were not sampled multiple times, kinship was assessed using GENODIVE (Meirmans 2020). Genetic diversity and population genetic metrics were also computed using GENODIVE. We examined genetic structure at neutral loci and outlier loci, separately, to investigate the possibility that different factors have shaped population divergence in unique patterns. Population structure was analyzed using STRUCTURE, PCA, and DAPC approaches. First, structure files from the STACKS populations output were analyzed using a Bayesian approach in STRUCTURE version 2.3.4 (Pritchard et al. 2000). Ten replicate runs were performed for a range of K from one to seven, with 10,000 iterations as the burn-in parameter and 100,000 iterations under the admixture model. The highest likelihood for K was estimated according to the Evanno method (Evanno et al. 2005) implemented in Structure Harvester (Earl and vonHoldt 2012). Second, we performed a principal component (PCA) and a discriminant analysis of principal components (DAPC) analysis (Jombart et al. 2010), which combines the benefits of discriminant and principal component analyses and is particularly useful to study differences between clusters (i.e., sites or populations) as it utilizes a multivariate approach to explore the entire variation in the data while minimizing that within clusters. This analysis was performed using the ADEGENET package in R (Jombart 2008) using the vcf file produced by the STACKS populations output used as an input file. The algorithm find.clusters identified the plausible number of clusters by comparing Bayesian information criterion (BIC) values, and the cross-validation tool xvalDapc determined the number of principal components that were retained.
Outlier loci—Although working with outliers might incorporate a series of shortfalls (Bierne et al. 2011, 2013; Lotterhos and Whitlock 2015), in particular on expanding populations (Lotterhos and Whitlock 2015), they are used to identify the potential diverging effects of selection between populations (Gaither et al. 2015; Longo and Bernardi 2015; Bernardi et al. 2016; Stockwell et al. 2016). Outlier loci were identified using PCADAPT, a method that is particularly robust when applied to expanding populations (Privé et al. 2020). Loci identifiers were then placed in “blacklists” and “whitelists,” which were used to run population scripts again for neutral and outlier analyses, respectively.
In order to determine if the ecological ‘weedy’ behavior of Montipora sp. on disturbed habitats might be associated with natural selection, we used a subset of the samples where two ecological situations are most clear. On one hand, we considered outside reefs that have a natural (low) abundance of Montipora (Songh, Piglelei) and compared them with lagoonal reefs that are overgrown by Montipora (Mogmog, Soghlei, Asor). We pooled those individuals (67 individuals) in two separate populations (i.e., without distinguishing sampling localities within these two areas). We then used PCADAPT to identify outlier loci. This resulted in a set of 86 loci that are potentially under selection (outlier loci). PCADAPT, however, does not distinguish between loci that are under balancing selection (very low Phi st—Φst) and those under directional selection (very high Φst), which is the type of selection this study is most interested in. We therefore used a stringent filter, where only those loci with Φst values higher than 0.1 were kept (67 loci). We then used those loci to analyze our entire dataset of individuals (137 samples).
Genome Fst scans—A Genome-Wide Fst scan analysis was carried out in order to gain insight into how the RADseq loci were distributed across the genome and whether outlier loci showed genomic patterns of colocalization (usually due to genomic rearrangements) or a more random distribution. Taking advantage of the inter-chromosomal synteny in corals, we scaffolded the Ulithi Montipora sp.1 aff. capitata genome (NCBI GenBank assembly accession: PRJNA812941, see Supplementary Materials for the genome assembly) into chromosome-scale pseudomolecules (CSPs) using the chromosome-level Montipora capitata v2 (available at: http://cyanophora.rutgers.edu/montipora) assembly as a reference. CSPs were obtained using RagTag (Alonge et al. 2019, 2021), a tool to order and orient draft assembly sequences to a reference assembly. We increased -i, the minimum grouping confidence score for localization, from the default of 0.2 to 0.5 to prevent low confidence contigs from localizing to CSPs. RADseq loci generated from the STACKS catalog were then aligned to the Montipora sp CSP assembly using BOWTIE2 v2.4.1 (Langmead et al. 2009) with the very-sensitive-local parameter. Locus-specific Fst values were generated with HIERFSTAT package in R using two populations.
Functional analyses—All outlier loci were compared to GenBank entries with BLAST, where E-values of 0.001 and below were kept and recorded (probability of obtaining the same result by chance < 0.001). When protein coding matching sequences were found, their function was classified using KEGG (Kyoto Encyclopedia of Genes and Genomes) assignments (Kanehisa et al. 2007).
Results
Loci and Polymorphism Statistics—We used 148 individuals to create a STACKS catalog of loci. Following optimization of the genotyping catalog in STACKS (Rochette and Catchen 2017), 6582 loci were kept, and of those, 3240 were polymorphic loci that passed all filtering criteria.
Cryptic species—Population structure analyses identified seven individuals as being genetically different than the rest of the samples (Fig. 3). While samples could not be morphologically distinguished, we found that six out of nine samples from Yealil-Outside, and one out of 17 samples collected at Federai landing, belonged to this relatively uncommon species (Fig. 3, blue barred individuals). The fixation index between the two genetic groups was consistent with the presence of two putative cryptic species (Fst = 0.788) (Wu et al. 2018; Christmas et al. 2021). The most common species therefore included the majority of samples (141 individuals).

Structure plot of all samples based on all SNPs (6582 loci, only one SNP was used for each locus). Two genetic clusters (K = 2) are shown; sample locations are labeled below the figure
Kinship analyses—Kinship analyses were performed on 141 Montipora sp.1 aff. capitata individuals to ascertain that collected individuals were not genetically identical due to biased sampling of individuals issued from asexual reproduction. For this analysis, 6919 loci were identified, and of those, 3422 were polymorphic. We found that two individuals collected next to each other were genetically identical (population Yealil-Inside, samples 12 and 13), we therefore removed sample 13 and kept sample 12. All other individuals were genetically unique and were included in subsequent analyses.
Genetic diversity in Ulithi Atoll—The remainder of the analysis was done on 137 Ulithi samples belonging to a single species, Montipora sp.1 aff. capitata. From the original 148 sample file, we removed seven individuals that belonged to Montipora sp.2 aff. capitata, one individual sampled twice (YEI 13) and three individuals collected outside of Ulithi (Fiji, Woleai, and Yap). With this final dataset, we obtained 6918 loci that passed all filtering criteria, and of those, 3422 were polymorphic.
There were no major differences between the genetic characteristics of the different sampling locations in Ulithi. While genetic diversity π (pi) values were very similar among locations (Table 1; average 0.23, st.dev 0.03), observed heterozygosity was particularly low in Falalop (0.096). Sample size there is low; only five individuals were sampled; however, other locations with small sample sizes, such as Yealil-outside (three samples) and Laam (five samples), did not show such low heterozygosity (0.183 and 0.223, respectively). The number of private alleles was very different among localities, the highest being found at Yealil-outside, where 514 private alleles were observed, but based on only three samples.
Population structure analyses—While the sample numbers varied between collection localities, they did not significantly correlate with the number of usable loci, average number of alleles (i.e., levels of polymorphism), and observed heterozygosity (Table 1) for each location, suggesting that performing a comparison between locations with different sample sizes is appropriate.
Neutral loci—As mentioned above, out of 6918 loci, 3422 were found to be polymorphic. Of those, 86 were identified as outliers (see below); thus, we used the remaining 3336 polymorphic neutral loci for this portion of the analysis. As expected, gene flow among locations was very high (average pairwise Fst = 0.025; average pairwise Φst = 0.029). Pairwise Fst’s were slightly elevated when comparing each of Falalop and Federai South to other sampling localities. (Average pairwise Fst’s with other sampling localities were 0.054 and 0.039, respectively.)
STRUCTURE HARVESTER suggests that our data reach the highest likelihood when clustered in two (K = 2) genetic groups (Fig. 4, upper panel). STRUCTURE results reflect the Fst values described above, where sampling localities do not show a clear genetic clustering trend. This is also reflected in the PCA analysis (Fig. 4, lower left panel). The DAPC analysis, however, showed a slight separation of Songh and Federai South from the rest of the samples (Fig. 4, lower right panel).

Analyses of Montipora sp.1 aff. capitata from Ulithi Atoll based on 3336 neutral loci. Structure plot, principal components analysis (PCA), and discriminant analysis of principal components (DAPC) cluster plots are shown in the top, left bottom, and right bottom panels, respectively
Outlier loci— PCADAPT was used on pooled individuals (67 individuals) in two separate populations that represented ‘pristine’ and ‘weedy’ areas of the reef (see Supplementary Figure S1 for the PCA analysis results). Keeping only loci with high Φst values (potential directional selection) retained 67 loci. Those loci were then used to analyze the entire dataset of individuals (137 samples).
As expected, the population differentiation was higher for outlier loci (pairwise average Fst: 0.058, F’st = 0.086). The values were higher when comparing populations in ‘pristine’ vs ‘phase-shifted’ areas (pairwise average Fst: 0.092, F’st = 0.138), and more so when only comparing those original 67 individuals used to identify outlier loci (Fst = 0.153; F’st = 0.231).
This pattern was again mirrored by STRUCTURE, PCA, and DAPC analyses. Indeed, the STRUCTURE analysis (Fig. 5, upper panel) based on 67 outlier loci showed that genetic clusters more clearly partitioned sampling sites compared to the analysis based on neutral loci (Fig. 4, upper panel). The STRUCTURE analysis shows a partition between northern sites (Falalop, Asor, Soghlei, Mogmog) and the southern sites (Songh, Piglelei, Federai and Yealil), with Laam looking like an intermediate, transitional site. Indeed, the northern site individuals display an average 70.00% of the orange genetic cluster (30.00% blue), while the southern individuals comprise 35.24% of the orange genetic cluster (64.76% blue). The most extreme locality is Songh, with individuals comprising an average 88.48% blue genetic cluster (11.52% orange), while individuals from the transition locality Laam comprise a more moderate 39.00% and 61.00% orange and blue clusters, respectively.

Analyses of Montipora sp.1 aff. capitata from Ulithi Atoll based on 86 outlier loci. Structure plot, principal components analysis (PCA), and discriminant analysis of principal components (DAPC) cluster plots are shown in the top, left bottom, and right bottom panels, respectively
The PCA analysis (Fig. 5, lower left panel) does not show any particular population trend, while the DAPC analysis again showed a separation of Songh from the rest of the samples (Fig. 5, lower right panel).
Genome-Fst scans – The goal of the Fst scan analysis was to determine if outlier loci are either clustered in a specific region of the genome, or if there are areas of the genome with structural variants. This analysis showed that loci under selection did not cluster on a particular region of the genome (chromosome), as the highest GenBank matches were found across all 14 Montipora capitata chromosomal regions (Fig. 6).

Genome Fst scan of neutral loci (black and blue dots, alternate with chromosomes) and outlier loci (blue dots)
Functional analyses—When comparing outlier loci with GenBank, we found that out of 67 outlier sequences, 61 had GenBank highest matches with all 14 Montipora capitata chromosomal regions (Table S1). A single outlier was found in each of chromosomes 6 and 13, while 12 outliers matched regions of chromosome 5 of Montipora capitata. Nevertheless, outlier loci were matched with regions found in each of the 14 Montipora capitata chromosomes, indicating that outliers are widespread through the genome. This result is consistent with the Fst genome scan results discussed above (Fig. 6). There were, however, very few protein coding genes that were identified by our searches. Indeed, only three genes matched outlier loci: serine/threonine kinase (identified by KEGG as an environmental information processing protein), ankyrin, and amidase, which are metabolic genes). The fact that only three genes were identified is in part due to the fact that RADseq markers represent a very small percentage of the genome, and also likely due to the fact that coral genomes are still poorly known and annotations remain difficult.
Discussion
In the past few decades, several coral reefs in Ulithi Atoll have experienced a phase shift from a diverse assemblage of corals to a predominantly Montipora dominated state (Crane et al. 2017). These reefs are mostly found inside the lagoon, and often (but not always) at disturbed sites. On the outer reef slopes, Montipora is present but at low density, as is commonly seen in other typical Indo-Pacific reefs (Franklin et al. 2013). The goal of this study was to determine if outer reef slope, ‘outside,’ and lagoonal, ‘inside,’ individuals were genetically different, and if natural selection played a role in the weedy nature of lagoonal colonies.
Presence of multiple species—Due to the complex logistics of working in remote atolls, our study was done free-diving, at shallow depths (up to 5 M). We found that our samples comprised two cryptic species. The less common species is mostly found at the leeward Yealil-Outside site, which is facing the open ocean on a steep outer slope. While poorly represented in our samples, it is possible that the less common species is abundant in deeper water, where we did not collect samples, but would more likely be prevalent in a site such as Yealil-Outside.
A STRUCTURE analysis shows that some genetic material may be shared between the two species. (Individuals contain colors from both genetic clusters, Fig. 2.) This may be due to some low level of introgression via gene flow between species, or some remnants of unsorted loci. The one sample from Fiji does contain some genetic material from the blue cluster (Fig. 2) and is unlikely to have experienced recent gene flow with Ulithi individuals. It is therefore more likely that the small amount of genetic material from alternative genetic clusters, as seen in a few individuals, is the result of unsorted ancestral shared loci.
Genetic diversity—Genetic diversity analysis did not provide strong evidence for any particular trends. In recent years, Montipora has quickly expanded into the island of Falalop, that is at the northeastern edge of the Ulithi Atoll (Fig. 1). The lowest levels of Observed heterozygosity at that location (0.096, Table 1) may reflect that ecological transition, but the low sample size indicates that these data should be interpreted cautiously.
Population structure trends—In general, small geographic scales, particularly in marine environments, do not lend themselves to strong population structure. The fact that only two individuals out of 138 were genetically identical suggests that sexual reproduction is likely to be a dominant feature of Montipora sp.1 aff. capitata in Ulithi. This is counter to the common local belief that Montipora propagated quickly among Ulithi locations due to human movement between islands and unintentional fragmentation due to anchoring activities. This also offers an opportunity for population structure to be observed if propagules do not settle randomly, or if selection hinders or favors given genotypes.
Results presented here show that little genetic structure was observed using neutral loci. This result is consistent with extensive mixing of propagules. In contrast, genetic partitioning was observed for the dataset based on outlier loci, suggesting that some propagules may undergo natural selection. Outlier loci were identified using a subset of samples restricted to outer ‘natural’ reefs and phase-shifted reefs. Yet, when those loci were tested using samples collected at all locations, northern and southern locations partitioned in two genetic clusters (Fig. 5, upper panel), as opposed to just outer ‘pristine’ reefs and phase-shifted reefs. This may suggest that phase-shifted reefs are the result of specific genotypes that are not just restricted to reefs where Montipora-dominated “breakouts” have occurred. Since neutral loci do not show structure, it is likely that propagules disperse extensively around the Atoll. Yet only a subset of them, which carries outlier loci, may survive in specific conditions. Most shifted reefs are within the lagoon and in front of villages. It may be the case that water conditions (high temperatures and nutrients, limited mixing) are the limiting factors that select specific genotypes, allowing Montipora sp. to thrive, while weakening other species of corals, thus shifting the balance among species. Importantly, the type of reef (pristine vs phase-shifted) and localities are correlated, because most phase-shifted reefs happen to be in the north of the Atoll. It is therefore impossible to definitively tease out the potential effects of selection and locality.
Natural selection and local adaptation—Recent developments in genomic approaches provide enhanced power to identify genes under selection and their genomic locations. Yet, seldom do we know the actual function of a given gene. This is even more true when dealing with organisms poorly characterized at the genomic level such as corals. Here, we identified 67 loci that are potentially under directional selection, but only 3 were potentially associated with protein coding genes. It would be instructive to study each gene separately and rank them according to their level of fixation, paying closer attention to those genes that are nearly fixed, a potential indication of stronger selection levels. For example, the two genes that display the highest fixation indexes between outer reefs and phase shifted reefs are the ankyrin-3 and the serine/threonine-protein kinase 36. These are genes that are likely to play an important role in this context, yet little is known about their function in corals.
Conclusion
Phase shifts on coral reefs have been hailed as an important consequence of the global degradation of marine tropical environments. These phase shifts can have consequences to the ecological integrity of reefs and to the people who rely on them (Hoegh-Guldberg et al. 2019; Hughes 2022). Healthy stony corals to macrophyte phase shifts are well known and have been studied extensively (Bruno et al. 2009; Hughes 2022). Yet phase shifts to monospecific coral assemblages have been less well documented. Here, we studied an unusual shift, from a diverse stony coral system, to a system where a single stony coral becomes predominant and vastly modifies the overall ecology of the reef as a result (Crane et al. 2017). It is possible that current abiotic factors (warming and acidification) may favor some corals over others and that this type of shift might become more common in future. Understanding the nature of these shifts, and the potential consequences to ecosystem function, is therefore imperative to our collective understanding of the future of coral reefs, and communities that rely on them.
The use of corals that thrive in changed environments has been seen both as a window to what future reefs will look like, and as potential candidates for reef restoration (Van Oppen et al. 2015; Hughes et al. 2017; Camp et al. 2018; Morikawa and Palumbi 2019; Schmidt-Roach et al. 2020). This study shows that selection and adaptation may be contributing to the success of a stony coral (e.g., phase shift), which may be leading to ecosystem degradation. While a stony coral may be successful in a disturbed environment, it does not necessarily provide the type of habitat that is conducive to high fish biomass and coral diversity. These results serve as a cautionary tale for restoration efforts that focus on single species coral resilience rather than ecosystem function.
References
Alonge M, Soyk S, Ramakrishnan S, Wang X, Goodwin S, Sedlazeck FJ, Lippman ZB, Schatz MC (2019) RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biol 20:224
Alonge M, Lebeigle L, Kirsche M, Aganezov S, Wang X, Lippman ZB, Schatz MC, Soyk S (2021) Automated assembly scaffolding elevates a new tomato system for high-throughput genome editing.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3:e3376
Bernardi G, Azzurro E, Golani D, Miller MR (2016) Genomic signatures of rapid adaptive evolution in the bluespotted cornetfish, a Mediterranean Lessepsian invader. Mol Ecol 25:3384–3396
Bierne N, Welch J, Loire E, Bonhomme F, David P (2011) The coupling hypothesis: why genome scans may fail to map local adaptation genes. Mol Ecol 20:2044–2072
Bierne N, Roze D, Welch J (2013) Pervasive selection or is it … ? why are Fst outliers sometimes so frequent ? Mol Ecol 22:2061–2064
Bruno JF, Sweatman H, Precht WF, Selig ER, Schutte VGW (2009) Assessing evidence of phase shifts from coral to macroalgal dominance on coral reefs. Ecology 90:1478–1484
Camp EF, Schoepf V, Suggett DJ (2018) How can “Super Corals” facilitate global coral reef survival under rapid environmental and climatic change? Glob Change Biol 24:2755–2757
Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH (2011) Stacks : building and genotyping loci de novo from short-read sequences. G3 Genes Genomes Genetics 1:171–182
Catchen JM, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Christmas MJ, Jones JC, Olsson A, Wallerman O, Bunikis I, Kierczak M, Peona V, Whitley KM, Larva T, Suh A, Miller-Struttmann NE, Geib JC, Webster MT (2021) Genetic barriers to historical gene flow between cryptic species of alpine bumblebees revealed by comparative population genomics. Mol Biol Evol 38:3126–3143
Crane NL, Nelson P, Abelson A, Precoda K, Rulmal J, Bernardi G, Paddack M (2017) Atoll-scale patterns in coral reef community structure: Human signatures on Ulithi Atoll. Micronesia Plos ONE 12:e0177083
Crisp SK, Tebbett SB, Bellwood DR (2022) A critical evaluation of benthic phase shift studies on coral reefs. Mar Environ Res 178:105667
Cruz ICS, Loiola M, Albuquerque T, Reis R, de Anchieta CC, Nunes J, Reimer JD, Mizuyama M, Kikuchi RKP, Creed JC (2015) Effect of phase shift from corals to zoantharia on reef fish assemblages. PLoS ONE 10:e0116944
Done TJ (1992) Effects of tropical cyclone waves on ecological and geomorphological structures on the Great Barrier Reef. Cont Shelf Res 12:859–872
Earl DA, vonHoldt BM (2012) Structure harvester: a website and program for visualizing structure output and implementing the Evanno method. Conservation Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Evensen N, Edmunds P, Sakai K (2015) Effects of pCO2 on spatial competition between the corals Montipora aequituberculata and Porites lutea. Mar Ecol Prog Ser 541:123–134
Franklin E, Jokiel P, Donahue M (2013) Predictive modeling of coral distribution and abundance in the Hawaiian Islands. Mar Ecol Prog Ser 481:121–132
Gaither MR, Bernal MA, Coleman RR, Bowen BW, Jones SA, Simison WB, Rocha LA (2015) Genomic signatures of geographic isolation and natural selection in coral reef fishes. Mol Ecol 24:1543–1557
Halford A, Cheal AJ, Ryan D, Williams DMcB, (2004) Resilience to large-scale disturbance in Coral and Fish assemblages on the Great Barrier Reef. Ecology 85:1892–1905
Henley EM, Quinn M, Bouwmeester J, Daly J, Zuchowicz N, Lager C, Bailey DW, Hagedorn M (2021) Reproductive plasticity of Hawaiian Montipora corals following thermal stress. Sci Rep 11:12525
Hoegh-Guldberg O, Pendleton L, Kaup A (2019) People and the changing nature of coral reefs. Regional Studies in Marine Science 30:100699
Hughes TP (2022) Catastrophes, Phase Shifts, and Large-Scale Degradation of a Caribbean Coral Reef. Science 265(5178):1547–1551
Hughes TP, Barnes ML, Bellwood DR, Cinner JE, Cumming GS, Jackson JBC, Kleypas J, Van De Leemput IA, Lough JM, Morrison TH, Palumbi SR, Van Nes EH, Scheffer M (2017) Coral reefs in the Anthropocene. Nature 546:82–90
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y (2007) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36:D480–D484
Knowlton N (1992) Thresholds and Multiple Stable States in Coral Reef Community Dynamics. Am Zool 32:674–682
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25
Lessa WA (1966) Ulithi; a Micronesian design for living. Holt, Rinehart and Winston, New York
Lessa WA (1964) The Social Effects of Typhoon Ophelia (1960) on Ulithi. 47
Lischer HEL, Excoffier L (2012) PGDSpider: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28:298–299
Longo G, Bernardi G (2015) The evolutionary history of the embiotocid surfperch radiation based on genome-wide RAD sequence data. Mol Phylogenet Evol 88:55–63
Lotterhos KE, Whitlock MC (2015) The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol Ecol 24:1031–1046
Meirmans PG (2020) genodive version 3.0: Easy-to-use software for the analysis of genetic data of diploids and polyploids. Mol Ecol Resour 20:1126–1131
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA (2007) Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res 17:240–248
Miller MR, Brunelli JP, Wheeler PA, Liu S, Rexroad CE, Palti Y, Doe CQ, Thorgaard GH (2012) A conserved haplotype controls parallel adaptation in geographically distant salmonid populations. Mol Ecol 21:237–249
Morikawa MK, Palumbi SR (2019) Using naturally occurring climate resilient corals to construct bleaching-resistant nurseries. Proc Natl Acad Sci USA 116:10586–10591
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Privé F, Luu K, Vilhjálmsson BJ, Blum MGB (2020) Performing highly efficient genome scans for local adaptation with R package pcadapt version 4. Mol Biol Evol 37:2153–2154
Rochette NC, Catchen JM (2017) Deriving genotypes from RAD-seq short-read data using Stacks. Nat Protoc 12:2640–2659
Schmidt-Roach S, Duarte CM, Hauser CAE, Aranda M (2020) Beyond reef restoration: next-generation techniques for coral gardening, landscaping, and outreach. Front Mar Sci 7:672
Stockwell BL, Larson WA, Waples RK, Abesamis RA, Seeb LW, Carpenter KE (2016) The application of genomics to inform conservation of a functionally important reef fish (Scarus niger) in the Philippines. Conserv Genet 17:239–249
Van Oppen MJH, Oliver JK, Putnam HM, Gates RD (2015) Building coral reef resilience through assisted evolution. Proc Natl Acad Sci USA 112:2307–2313
Wu W, Ng W-L, Yang J-X, Li W-M, Ge X-J (2018) High cryptic species diversity is revealed by genome-wide polymorphisms in a wild relative of banana, Musa itinerans, and implications for its conservation in subtropical China. BMC Plant Biol 18:194
Acknowledgements
We would like to thank the people of Ulithi, Federated States of Micronesia, for permission to collect Montipora samples, and for their continued collaboration in the studies of their Atoll.
Funding
This work was funded by the Department of the Interior/Office of Insular Affairs (CRI-Ulithi1-2), NOAA (Award # NA15NMF4270336), Revive and Restore (Award #2022–041), and the National Science Foundation (OCE 1546374).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
338_2024_2486_MOESM1_ESM.pdf
Principal components analysis (PCA), obtained using PCADAPT, of samples partitioned in Outside (‘pristine’) and Inside (‘invaded by weedy Montipora’) reefs. (PDF 34 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bernardi, G., Gatins, R., Paddack, M. et al. Genomics of a novel ecological phase shift: the case of a ‘weedy’ Montipora coral in Ulithi, Micronesia. Coral Reefs (2024). https://doi.org/10.1007/s00338-024-02486-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00338-024-02486-9