
Abstract
Background
Species delimitation is a challenging but essential task in conservation biology. Morphologically similar species are sometimes difficult to recognize even after examination by experienced taxonomists. With the advent of molecular approaches in species delimitation, this hidden diversity has received much recent attention. In addition to DNA barcoding approaches, analytical tools based on the multi-species coalescence model (MSC) have been developed for species delimitation. Musa itinerans is widely distributed in subtropical Asia, and at least six varieties have been documented. However, the number of evolutionarily distinct lineages remains unknown.
Results
Using genome resequencing data of five populations (making up four varieties), we examined genome-wide variation and found four varieties that were evolutionary significant units. A Bayesian Phylogenetics and Phylogeography (BP&P) analysis using 123 single copy nuclear genes support three speciation events of M. itinerans varieties with robust posterior speciation probabilities; However, a Bayes factor delimitation of species with genomic data (BFD*) analysis using 1201 unlinked single nucleotide polymorphisms gave decisive support for a five-lineage model. When reconciling divergence time estimates with a speciation time scale, a modified three-lineage model was consistent with that of BP&P, in which the speciation time of two varieties (M. itinerans var. itinerans and M. itinerans var. lechangensis) were dated to 26.2 kya and 10.7 kya, respectively. In contrast, other two varieties (M. itinerans var. chinensis and M. itinerans var. guangdongensis) diverged only 3.8 kya in the Anthropocene; this may be a consequence of genetic drift rather than a speciation event.
Conclusion
Our results showed that the M. itinerans species complex harbours high cryptic species diversity. We recommend that M. itinerans var. itinerans and M. itinerans var. lechangensis be elevated to subspecies status, and the extremely rare latter subspecies be given priority for conservation. We also recommend that the very recently diverged M. itinerans var. chinensis and M. itinerans var. guangdongensis should be merged under the subspecies M. itinerans var. chinensis. Finally, we speculate that species delimitation of recently diverged lineages may be more effective using genome-wide bi-allelic SNP markers with BFD* than by using unlinked loci and BP&P.
Similar content being viewed by others

Background
Species are the basic units of biodiversity, and precise species delimitation is essential for unbiased biodiversity estimation [1]. While there is no perfect species concept, it is generally agreed that species should be delimited as evolutionarily distinct lineages, usually evident by significant morphological, genetic, or niche differentiation [2, 3]. Today, the vast majority of species are still recognized based on morphological differences alone, and many genetically distinct but morphologically similar species remain undetected. These cryptic species make up a significant proportion of hidden diversity: one estimate suggest that about 60% of 408 newly described mammal species between 1993 and 2009 are cryptic species [4]. The presence of cryptic species is related to both the intrinsic properties of organisms and with extrinsic environmental conditions [1, 3], thus making the discovery of cryptic species extremely challenging.
With the advent of cheap and rapid DNA sequencing, species delimitation using molecular data has become active and fruitful [5]. Until recently, cryptic species were identified using molecular data mainly based on estimating reciprocal monophyly or genetic distances. Comparing DNA barcodes (such as the matK and rbcL genes in plants and the COI gene in animals) using the Automatic Barcode Gap Discovery (ABGD) program is an efficient and popular means of cryptic species identification [6,7,8]. This method, which relies on a ‘barcode gap’ within and between taxa, however, becomes impossible if a prior reference library is unavailable or if the barcode gap is unclear. In addition to these simple pairwise distance threshold methods, multi-species coalescent (MSC)-based methods tracks the genealogical histories of samples back to common ancestors and identify possible evolutionarily independent lineages using both Bayesian and maximum likelihood (ML) methods [9]. Accommodating the uncertainties in gene trees, the Bayesian Phylogenetics and Phylogeography program (BPP or BP&P) jointly estimates the posterior probability distributions of different species delimitation models and relevant parameters, including coalescent times and population sizes [10,11,12,13]. This program has been constantly updated and has been widely used in species delimitation studies of various taxonomic groups, including plants [14], birds [15], and insects [16]. However, it ignores ongoing gene flows between populations, and is prone to lump distinct species into one species [17]. Divergence with gene flow is very common in incipient speciation [18, 19], and complicates species delimitation. Although a phylogeographic model test program that considers gene flow in a flexible model space has been developed (i.e. Phylogeographic Inference using Approximate Likelihoods, PHRAPL) [20, 21], the authors themselves have pointed out that PHRAPL may not be as powerful as BP&P in delimiting species with deep divergence times or weak migration rates.
So far, species delimitation studies using coalescent theory with Bayesian or likelihood methods have generally been limited to using datasets consisting of dozens of loci [14]. This amount of data is insufficient for the detection of some shallowly diverged lineages [22]. With the ever-decreasing cost of high-throughput sequencing and improved computational power, genomic data has become more accessible for species delimitation studies. Restriction site associated DNA sequencing, (RADseq) methods [23], for example, provide ample random single nucleotide polymorphisms (SNPs) and have been used to generate genome-wide datasets for species delimitation in non-model organisms [24, 25]. To circumvent the computational challenges inherent in such genomic-enabled species delimitation approaches, species trees are often inferred directly from biallelic markers (e.g., SNP or AFLP data) without genes trees [26]. For instance, the program Bayes Factor Delimitation (*with genomic data) (BFD*) estimates the probability of allele frequency change across ancestor/descendent nodes, and obtains a posterior distribution for the species tree, species divergence times, and effective population sizes simultaneously [27].
Musa itinerans Cheesman (Musaceae) is a giant perennial monocotyledonous herb named after its long rhizome. It is also a wild relative of the edible banana progenitors, M. acuminata and M. balbisiana, and is distributed throught Southeast Asia and subtropical China. Musa itinerans is morphologically highly variable and as many as six varieties have been proposed, some of which are varieties that were formerly considered to be subspecific [28]. The distribution of these varieties spans over a large area within the monsoonal climate in subtropical China. Since it is tolerant to both frost and drought [28], M. itinerans is one of the species in the genus Musa that is most resistant to Fusarium wilt disease, the Tropical Race 4′ (Fusarium oxysporum f. sp. cubense race 4, Foc-TR4) [29]. Wild M. acuminata species are well-known for having diverse subspecies, some of which are associated with early banana cultivars [30]; in contrast, many banana cultivars show a lack of genetic diversity mainly due to clonal propagation over many generations, that has made them vulnerable to a variety of diseases. Hence, because of its close relationship to banana and its tolerance to a diverse range of biotic and abiotic stresses, M. itinerans holds great promise for the improvement of important agronomic traits in banana breeding. To aid in the conservation of genetic resources of M. itinerans, it’s important to ascertain the cryptic diversity present in this highly variable species complex. Recognizing the presence of cryptic species also provides opportunities to understand the evolutionary and ecological processes driving the diversification of the genus Musa [1].
The M. itinerans species complex is composed of six morphologically differentiated varieties, and a draft genome for Musa itinerans var. itinerans has been previously reported [31]. In this study, we sampled four varieties across different latitudes in South China and obtained genome-wide SNP data by genome resequencing. Using this data, we try to answer the following specific questions: (1) Are the varieties represent independent evolutionary lineages or only products of phenotypic plasticity? (2) What’s the real taxonomic status of these lineages under the MSC species delimitation framework?
Results
Genome-wide polymorphisms among different morphological species
Resequencing of 24 M. itinerans individuals from five populations generated a total of 2.75 billion filtered pair-end reads (249.7 Gb of filtered bases), and these short reads were mapped against the reference genome of M. itinerans (http://banana-genome-hub.southgreen.fr/organism/Musa/Itinerans) with a mean unique mapping depth of 15.5, and coverage of 86.9%, (Additional file 1: Table S1). After SNP calling, 9,402,402 SNPs of the 336,835,601 effectively mapped sites passed filtering our criteria.
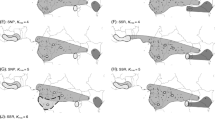
Using a data of 7,940,468 SNP sites without missing genotype, a variational Bayesian inference method implemented in the program fastSTRUCTURE [32] with logistic priors was used to estimate the optimal ancestral components of individuals from different geographic populations. When K = 2, samples of M. itinerans var. itinerans (abbreviated as ‘Mit’ hereafter) from the HN population were separated from the remaining continental populations (i.e. YC, LC: M. itinerans var. guangdongensis, ‘Mgd’; CH: M. itinerans var. chinensis, ‘Mch’; and BX: M. itinerans var. lechangensis, ‘Mlc’). When K was increased to 3, the variety Mlc (BX) clustered out as a distinct lineage. At K = 4, four varieties (Mit, Mlc, Mch, and Mgd) were distinguishable from each other. At K = 5, the two allopatric populations of Mgd were further divided (Fig. 1c). Principal component analysis (PCA) showed strong population structuring (Tracy-Widom statistics: P < 1 × 10–12), with Mit and other mainland varieties separated by the first eigenvector, followed by Mlc being clustered out from other varieties by the second eigenvector (Fig. 1d). According to the optimal clusters given by fastSTRUCTURE, we used K = 4 and estimated genome-wide diversities using the two common statistics, θπ [33] and Tajima’s D [34]. We found that the two marginally distributed varieties Mit (mean θπ = 4.6 × 10− 3) and Mlc (mean θπ = 4.3 × 10− 3) harbored significantly fewer variation than other varieties (mean θπ = 5.1 × 10− 3, and 6.4 × 10− 3 for Mch and Mgd respectively, P < 2.2 × 10− 16, Mann Whitney U-test; Fig. 2, Additional file 1: Figure S1 and S2). The overall positive genome-wide Tajima’s D values for all varieties (the median value of Dmit = 0.69, Dmgd = 0.87, Dmch = 0.55, Dmlc = 0.94, Fig. 2 and Additional file 1:Figure S1 and S2) indicated that M. itinerans probably experienced population contractions in the past.

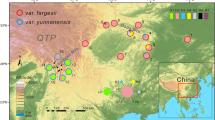
Sampling information and population structure. (a) Sampling locations of Musa itinerans varieties used in this study: LC, Lecang, Guangdong; CH, Conghua, Guangdong; YC, Guangdong; HN, Hainan; The distributions of M. itinerans are outlined by green circles; (b) Morphological characters of M. itinerans varieties; (c) Plots of membership clusters for varieties of M. itinerans using genome wide single nucleotide polymorphism data implemented with fastSTRUCTURE. (d) Principal components analysis of four geographic populations of M. itinerans based on the same genome-wide dataset

Genome diversity among the four varieties of Musa itinerans. (a) The distributions of average pairwise nucleotide diversity θπ, Tajima’s D, Wright’s fixation index FST and absolute genetic divergence Dxy across chromosome 1 with an overlapped window size of 20 kb and a step size of 2 kb for four varieties of Musa itinerans; (b) Boxplots shown for the overall θπ, Tajima’s D, Wright’s fixation index FST and absolute genetic divergence Dxy for four varieties of M. itinerans
The values of genome-wide population differentiation FST [35] among the four varieties ranged between 0.14 and 0.41, with Mit showing higher differentiation with any of the other varieties, which was consistent with the genome-wide absolute genetic divergence Dxy [36] (mean Dxy = 0.0008–0.0009; Table 1). Dxy showed lower levels of variation among different variety pairs (Fig. 2, Additional file 1: Figure S1 and S2), as it is insensitive to current levels of polymorphism within species and reflects the net divergence since their common ancestor [37].
Analyses of the D-statistics calculated from the genome-wide dataset revealed that historical gene flow occurred between different varieties (Table 2). Low values of absolute D-statistic were found in comparisons of Mlc-Mch (D = 0.04) and Mlc-Mit (D = − 0.08) respectively, which indicating infrequent gene flow that may have facilitated the formation of Mlc as a distinct lineage. In contrast, higher absolute D values (− 0.15, − 0.19) suggested that significant gene flow has occurred between the Mit and Mch varieties after divergence. Considering their current allopatric distribution, this historical gene flow may have occurred before the formation of the Qiongzhou Strait (about 10.3 kya), which isolated Hainan island from the continent [31].
Bayesian species delimitation
We performed the BP&P analysis at K = 5, that is composed of Mit, Mch, Mlc, and two allopatric populations of Mgd. Based on this pattern of clustering, the Bayesian species tree estimation yielded 97 distinct species trees, of which the top 40 species trees constituting a 95% credibility set of tree topologies. The majority-rule consensus tree is almost star-shaped, suggesting that these varieties diverged very recently. We used the tree with maximum posterior probability of 0.16 (Fig. 3a), in which two geographical populations of Mgd were monophyletic and most closely related to the variety Mch, followed by Mlc. Using this five-lineage phylogeny as a guide tree, the posterior probabilities of different models and the posterior distribution of the parameters τs and θs for each model were calculated using the rjMCMC algorithm. The five-lineage and four-lineage models yielded posterior probabilities of 0.54, and 0.36, respectively. Thus, the maximum a posterior probability (MAP) model uses a five-lineage model as the guide tree (Fig. 3b). Nonetheless, the posterior speciation probability of the node for the two geographic populations of Mgd (YC, LC) was 0.54, far below the conservative threshold of 0.95, showing weak evidence for a split of this variety. The posterior speciation probability of the node with Mch and Mgd was 0.90, somewhat below the threshold of 0.95, indicating that it is possible to plausibly lump the two varieties (Mch and Mgd) together. Considering the high number of loci (123 genes) used in our coalescence analysis, this is likely an indication of recent divergence of these varieties rather than insufficient variation detected in our study. Two other varieties Mit and Mlc, seemed to be well resolved lineages (i.e. the posterior speciation probability was 1.00 for Mit, and 0.99 for Mlc). Overall, high posterior speciation probabilities supported the three-species delimitation scenario by two speciation events among the four varieties. Using the same gamma prior θ ~ G (2, 1000) for population sizes and divergence time with τ0 ~ G (1, 10,000) at the root to estimate the parameters in the multi-species coalescent model for the MAP tree, the posterior mean of the population size parameter θs ranged from 0.0013 to 0.0180, and population size-scaled divergence time at the root was 7.0 × 10− 5 (95% confidence interval: 5.0 × 10− 5 ~ 9.0 × 10− 5), and other nodes ranged between 2.0 × 10− 5 and 9.0 × 10− 5.

Species delimitation for Musa itinerans varieties using Bayesian phylogenetics and phylogeography (BP&P) based on 123 single copy nuclear loci. (a) Specie tree estimation: the top four species trees and their posterior probabilities with a total probability of 0.5, and population sizes with Gamma priors θ ~ G (2, 1000) for all populations and divergence times with Gamma priors τ ~ G (2, 2000) for the root age. The abbreviations for different varieties or geographical populations are as follow: Mit: Musa itinerans var. itinerans; Mlc: Musa itinerans var. lechangensis; Mch: Musa itinerans var. chinensis; Mgd1: Musa itinerans var. guangdongensis (population Yangchun, Guangdong); Mgd2: Musa itinerans var. guangdongensis (population Lechang, Guangdong); (b) species delimitation on guide tree: above and below the branches are the posterior speciation probabilities, population sizes are shown on every node, the 95% highest posterior density (HPD) for divergence time are in the brackets and are highlighted using horizontal grey bars. Two geographical populations of M. itinerans var. guangdongensis were lumped with weak posterior probability, the splits of other varieties were supported with high posterior probabilities
Bayesian factor delimitation methods also gave decisive support for the five-lineage model with an BF value of 81.4, well above the threshold of 6, supports the hypothesis that the two allopatric geographic populations of Mgd show evolutionary divergence and are therefore evolutionarily distinct lineages (Table 3). Bayesian analyses of the 1201 unlinked SNPs yielded a well-resolved species tree for all currently recognized varieties (Fig. 4, all nodes have a posterior probability of 1.0). The divergence age at the root of the species tree was estimated to be 0.00034 (95% CI: 0.00018 ~ 0.00048). Using a generation time of one year and base substitution rate of 1.30 × 10− 8 substitution rate per site per year [38], we estimate that this divergence occurred approximately 26.2 kya (13.8 ~ 36.9 kya), i.e. during the Late Pleniglacial period, when drastic climatic fluctuations and associated habitat alterations profoundly altered the speciation rates in marginal tropical areas. In Hainan, the final formation of the Qiongzhou Strait during the mid-Holocene (7.0 ~ 10.5 kya) [39] may have further facilitated the divergence between Mit and its continental counterparts. The speciation time of Mlc also date to the Holocene (Mlc: 10.7 kya, 95% CI: 3.1 ~ 22.3 kya), and this variety has the most northerly distribution in South China. Moreover, it is the most frost-tolerant variety and may show some degree of ecological speciation. The divergence time of Mch and Mgd was dated to 3.8 kya (95% CI: 1.5 ~ 9.2 kya). Finally, the divergence of the two allopatric populations Mgd was dated to 3.1kya (95% CI: 0.1 ~ 4.6 kya), and we speculate that this very recent divergence should is more likely a consequence of genetic drift in the Anthropocene rather than a result of speciation. By lumping the two geographical populations of Mgd together, and merging the two varieties of Mgd and Mch, both the BP&P and BFD* approaches agreed on aa consensus species delimitation scenario for the varieties of M. itinerans.

Bayesian factor species delimitation for Musa itinerans varieties using 1201 unlinked loci. (a) the five taxa species tree determined by the most marginal likelihood estimates of different species models. Bayesian posterior probability, ancestral population size, divergence time are separated by two slashes; the 95% highest posterior density (HPD) for divergence times are highlighted using horizontal grey bars; (b) DensiTree shown for all trees of the Markov chain Monte Carlo method with a burn-in of 5000 trees, and higher levels of uncertainty are represented by lower densities
Discussion
Four M. itinerans varieties are evolutionarily significant units
In this study, genome-wide SNP data was used to reveal the cryptic diversity within the M. itinerans species complex. All four sampled M. itinerans varieties were shown to be genetically divergent and to represent evolutionarily distinct lineages. This species is one of the most widely distributed wild relatives of banana in the subtropical region, moreover, it harbors great intraspecific genetic diversity and has potential use for the improvement of disease resistance in banana [29]. So far, 7–8 varieties of M. itinerans with distinct morphological characters have been documented by taxonomists [28, 40,41,42]. However, it is unclear how many of these are genetically distinct lineages. ‘Variety’ is a taxonomic rank below ‘subspecies’ [43] and is commonly used when range-wide populations of a species exhibit recognizable morphological differences, often in response to fluctuation environments. On the other hand, evolutionarily significant units (ESUs) are populations that do not undergo frequent genetic exchange, and hence should display reciprocal monophyly and significant divergence of allele frequencies at nuclear loci [44]. In conservation biology, the recognition of ESUs is relevant to defining conservation priorities and strategies.
Genetic component inferences based on a variational Bayesian framework and on a principal component analysis provided compelling evidence that the four varieties found in South China are distinct evolutionary lineages. The four varieties showed significant genetic differentiation (FST: 0.14 ~ 0.40), which was further validated by the BP&P and BFD* species delimitation analyses. The BP&P analysis supported the three-lineage model (Mit, Mlc, and Mch + Mgd1 + Mgd2), while the BFD* analysis supported the five-lineage model (Mit, Mlc, Mch, Mgd1, and Mgd2). However, the BFD* analysis also provided estimated divergence times between Mch and Mgd, as well as between the two allopatric Mgd populations, that are very recent - 3.8 and 3.1 kyr respectively. These divergence times are beyond a reasonable time scale for speciation. Considering both observations, we propose that two varieties, Mit in Hainan and Mlc in northern Guangdong, be elevated in rank to subspecies. According to Häkkinen’s descriptions [28], Mit was easily distinguished from the other varieties by its long creeping rhizome, which often extends as far as 2 m away from the mother plant; In addition to its narrow distribution, Mlc also presents with obtuse female buds and small purple blotches on its pseudostems. Hence, it seems that rhizomes and suckers can be used as diagnostic traits for the two subspecies. The other two varieties, Mch and Mgd, should be merged and named as the subspecies M. itinerans subsp. chinensis according to their publication date. Mch was found to significantly differ from the other varieties by its denser suckers and the presence of large red-brown blotches on its pseudostems. Mgd is recognized by its green to purple fruit peel when ripe [28]. These traits may be due only to morphological plasticity of a single subspecies, M. itinerans ssp. chinensis. The subspecies Mlc, which has a sporadic distribution, was proposed to be categorized as a ‘Vulnerable’ (VU) species for conservation. This subspecies has been observed to be capable of withstanding frost damage [28], highlighting the importance of its conservation as an important genetic resource. Mlc diverged from the ancestor of Mgd and Mch about 10.7 kya (95% CI: 3.1 ~ 22.3). This divergence was followed by population contractions (as indicated by a positive Tajima’s D), indicating that the Mlc may be a remnant from a larger population in the past.
Unlinked biallelic SNPs or multiple unlinked loci in shallow species delimitation?
We used two analytical tools based on the MSC model to test possible species delimitation schemes for the varieties of M. itinerans. The BP&P method favoured a three-lineage model, whereas the BFD* approach initially supported a five-lineage model; the main difference was the BFD* approach identified the very recently diverged Mgd and Mch varieties as distinct lineages. However, when reconciling the divergence time estimates with plausible time scales of speciation, the BFD* approach also supported to a three-lineage model. The relative power of the different methods involves many factors, including the number of unlinked loci or SNPs, the prior assignments of individuals, and the prior settings of parameters space. In this study, the same cluster assignments were used in both approaches, and the parameters were optimized according to the same polymorphism estimates, leaving the numbers of markers used in the two datasets as the major variable that differed between the two approaches. The BP&P method is known to be more conservative and may be prone to species lumping when divergence is recent [15], as observed in the case here with the Mch and Mgd varieties. The BP&P approach has been shown to be capable of validating species boundaries between well-differentiated species using even one locus [45], and this is of great advantage in revealing cryptic species diversity in poorly resolved taxa. For recent speciation events, hundreds or thousands might improve the power of BP&P where computational resources are available, but such cost is not readily achieved in non-model organisms [22]. In our study, the performance of the BFD* approach using 1201 unlinked SNPs was comparable to using 123 unlinked loci. Since the cost of obtaining this many unlinked SNPs using RADseq was lower than collecting 123 unlinked loci in a non-model organism, it may be most efficient to use the bi-allelic markers with the BFD* method for species delimitation in recently diverged lineages. With ever-decreasing costs, the genotyping of thousands of loci using RADseq may someday become a routine practice and would therefore facilitate the discovery of cryptic species present after recent speciation events. Such an approach is also of paramount importance for the conservation of understudied CWRs.
Population structure or ESUs?
Population genetic structures persist in all taxa along their geographic and/or environmental barriers. One puzzle in species delimitation is how to distinguish population structure from real speciation events of divergent lineages [46]. To avoid misidentification structured populations as separate species, external information (i.e. a priori hypothesis or knowledge) is essential to interpret the genetic data and provide a basis on which to judge whether differences corresponding to different species rather than different populations of a single species. In this study, although the Mlc variety of the BX population and the Mgd variety of the LC population are sympatric, their divergence was found to be much higher than the genetic differentiation between the two allopatric Mgd populations. This result - the opposite of what might be expected from the isolation by distance model - indicated that the genetic divergence observed in our study was between ESUs rather than merely between populations separated by distance. In addition to obvious morphological differences, the two varieties differed remarkably in habitat preference (i.e. warm and humid vs. cold and dry microhabitats) [26].
Conclusion
Overall, classification of plants within the genus Musa is difficult even for skilled taxonomists, and new taxa have been reported mainly based on morphological characters [40,41,42, 47,48,49]. This study demonstrated the feasibility of using high-throughput sequencing data, in association with biological information such as morphological characters, biogeographic history, and ecological differentiation for the delimitation of ESUs within the genus Musa. The currently recognized M. itinerans var. itinerans and M. itinerans var. lechangensis should be elevated in rank as subspecies for better management and conservation of these taxa as CWRs genetic resources. Species delimitation using molecular approaches can free the taxonomists from being entangled with subtle character differences that may mask differences in real. However, species delimitation using molecular approaches should be cautious and consider biologically meaningful data, such as morphological characters, biogeographic history, and ecological differentiation. As the case in our BFD* analysis, the estimated divergence time for the Mch and Mgd varieties was not a reasonable time scale for real speciation to have taken place.
Methods
Sample collection, resequencing, and filtering of raw reads
According to the assigned variety scheme present at the outset of our study [28], we sampled four varieties of M. itinerans from five populations across South China (Fig. 1a and b, Table 4). Fresh leaves were harvested in the field and dried using silica-gel. Total genomic DNA was extracted using the standard CTAB extraction method. A library with 500 bp insertion size for each individual plant was prepared using Paired-End Sample Prep Kits (Illumina, UK) according to the manufacturer’s recommendations, libraries were then sequenced on the HiSeq2000 platform. Using NGSQCToolkit version 2.3.3 [50], raw reads were discarded with excessive (> 10%) ‘N’s or over 40% of the bases with PHRED quality score below 7 were discarded. Raw data has been deposited in the NCBI Sequence Read Archive under BioProject ID 312694 with accession number SRR6382516~SRR6382539.
Read alignment, variant calling, and variant filtering
For the convenience of calculating or visualizing genome wide diversity statistics, we anchored and oriented the assembled scaffolds of M. itinerans against 12 linkage groups of the M. acuminata genome based on the collinearity between them, and the remaining scaffold or contigs were treated as one group and ordered by size. Filtered paired-end reads per sample were aligned to the updated reference genome using the BWA-mem version 0.7.12 [51] algorithm with default options. The sam files were converted into bam format, then sorted and indexed using SAMtools version 1.3.1 [52],. Prior to variant calling, MarkDuplicates in Picard (http://broadinstitute.github.io/picard) was used with default options to mark duplicated reads resulting from PCR. After removing reads with mapQ scores below 30 and trimmed lengths less than 30 base pairs (bp), the remaining reads were headed with sample information using Picard-tools, and local realignments around indels were implemented using the IndelRealigner tool packaged in GATK version 3.7.0 [53,54,55]. Each filtered and aligned file was subjected to variant-calling using the HaplotypeCaller function in GATK, and joint genotyping (by combining all of the above outputs for SNP and indel VCFs) was implemented using GenotypeGVCFs in GATK. Finally, joint variants were filtered using VariantFiltration in GATK using the default settings.
Population structure and principal component analysis
Using vcftools version 0.1.14 [56], the SNP variants among the 12 linkage groups of three populations were selected with options ‘--maf 0.05’ and were further LD-pruned to minimize the linkage disequilibrium of the sites using the option ‘--indep-pairwise 50 5 0.2’ in PLINK version 1.0.9 [57]. Using this filtered genotype SNP data set, posterior inferences of population structure for these varieties were implemented based on a variational Bayesian framework with the python program fastSTRUCTURE.The Smartpca program packaged in the eigensoft version 6.1.3 [58] was used to perform principal component analysis on the same set of variants using the default parameters.
Nucleotide diversity and population divergence
A sliding window approach (20-kb overlapped windows in 2-kb steps) was used to quantify genome-wide variation among inferred genetic clusters. We calculated the average pairwise nucleotide diversity, θπ, Tajima’s D, Wright’s fixation index FST, and the absolute genetic divergence, Dxy, using PopGenome version 2.24 [59] or custom Perl scripts. To distinguish between the relative role of lineage sorting and introgressions in the current divergence patterns, an extended D-statistics four-population test (ABBABABA2) was conducted using the software package ANGSD-wrapper [60, 61]. Z-scores above 3 was used to reject the null hypothesis of no significant introgression between populations.
Species delimitation
According to recommendations by Carstens et al. [62], two distinct methodological approaches were used to detect cryptic lineages in varieties of M. itinerans, i.e. BP&P and the Bayes factor delimitation method with genomic data (BFD*) [26, 27]. In a previous study, we identified 1201 single copy nuclear genes of M. itinerans and other eight related species [29], and different subsets of this data were used in our analyses.
The BP&P3.3a program is a full likelihood-based implementation of the MSC model, and it uses a reversible-jump Markov chain Monte Carlo (rjMCMC) method to evaluate competing delimitation models [10]. It collapses or splits the nodes in the guide species tree according to the nodes posterior probabilities in the guide tree and alternative competing delimitation models [12, 17, 62]. The program consists of four modules: in module A00, it generates the posterior distribution of species divergence times (τs) and population sizes (θs) under the MSC model with a fixed species phylogeny; in module A01, the species tree is estimated with fixed assignments and species delimitation; in module A10, species delimitation is implemented on a guide tree;and in module A11, the species tree estimation and species delimitation are integrated. According to our genome-wide estimates of nucleotide diversity θπ and absolute divergence time Dxy for the varieties of M. itinerans studied here, a small population size with gamma prior θ ~ G (2, 1000) for all populations and a shallow divergence time with τ0 ~ G (1, 10,000) at the root of the species tree were assigned, with Dirichlet priors for other divergence time parameters. A total of 2,000,000 iterations (sample interval of 4) with a burn-in of 1000 was implemented for each run. In this study, we followed a multi-steps analysis. Each step was conducted with ten replicates, and convergence was evaluated across replicates. In addition, the convergence of estimation of model parameters was evaluated with the effective sample size (ESS ≥ 200). Because of the computation burden presented by a multi-species coalescence model of thousands of loci, only 123 loci were randomly selected and used for the BP&P analysis.
The BFD* method was performed using SNAPP version 1.3.0 [26, 27]. This program estimates the marginal likelihood of competing models with different numbers of species and individual assignments, and ranks model fit among runs by Bayes factor. The two essential assumptions of this method were that there was no gene flow among lineages and that multiple unlinked loci are used in the coalescence model. Thus, we excluded samples with admixture proportions over 5%; and for the 1201 single copy loci, only sites with maximum calling depth were used to avoid linkage disequilibrium. In our species delimitation model, we lumped or split varieties or geographic populations according to the current classification of varieties and population structure/clustering. The marginal likelihood of each model was estimated via path sampling using 48 steps each with different levels of power-posterior, an alpha of 0.3, and an MCMC chain length of 100,000 with a pre-burn-in of 100,000 [26].
Abbreviations
- BFD * :
-
Bayes factor delimitation of species with genomic data
- BP&P:
-
Bayesian Phylogenetics and Phylogeography program
- CI:
-
Confidence Interval
- ESUs:
-
Evolutionarily significant units
- kya:
-
Thousands of years ago
- MAP:
-
Maximum a posterior probability model
- MSC:
-
Multi-species coalescence model
- PHRAPL:
-
Phylogeographic Inference using Approximate Likelihoods
- RADseq:
-
Restriction site associated DNA marker sequencing
- SNPs:
-
Single nuclear polymorphisms
References
Bickford D, Lohman DJ, Sodhi NS, Ng PKL, Meier R, Winker K, et al. Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2007;22:148–55.
Su X, Wu G, Li L, Liu J. Species delimitation in plants using the Qinghai-Tibet plateau endemic Orinus (Poaceae: Tridentinae) as an example. Ann Bot. 2015;116:35–48.
Eisenring M, Altermatt F, Westram AM, Jokela J. Habitat requirements and ecological niche of two cryptic amphipod species at landscape and local scales. Ecosphere. 2016;7:1–13.
Ceballos G, Ehrlich PR, Cebados G, Ehrlich PR. Discoveries of new mammal species and their implications for conservation and ecosystem services. Proc Natl Acad Sci U S A. 2009;106:3841–6.
Flot JF. Species delimitation’s coming of age. Syst Biol. 2015;64:897–9.
Hebert PDN, Penton EH, Burns JM, Janzen DH, Hallwachs W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc Natl Acad Sci U S A. 2004;101:14812–7.
Steven GN, Subramanyam R. Testing plant barcoding in a sister species complex of pantropical Acacia (Mimosoideae, Fabaceae). Mol Ecol Resour. 2009;9:172–80.
Tyagi K, Kumar V, Singha D, Chandra K, Laskar BA, Kundu S, et al. DNA barcoding studies on Thrips in India: cryptic species and species complexes. Sci Rep. 2017;7:4898.
Fujita MK, Leaché AD, Burbrink FT, McGuire JA, Moritz C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol Evol. 2012;27:480–8.
Yang Z, Rannala B. Bayesian species delimitation using multilocus sequence data. Proc Natl Acad Sci U S A. 2010;107:9264–9.
Rannala B, Yang Z. Improved reversible jump algorithms for Bayesian species delimitation. Genetics. 2013;194:245–53.
Yang Z, Rannala B. Unguided species delimitation using DNA sequence data from multiple loci. Mol Biol Evol. 2014;31:3125–35.
Rannala B, Yang Z. Efficient Bayesian species tree inference under the multi-species coalescent. Syst Biol. 2017;66:823–42.
Carstens BC, Satler JD. The carnivorous plant described as Sarracenia alata contains two cryptic species. Biol J Linn Soc. 2013;109:737–46.
McKay BD, Mays HL, Wu Y, Li H, Te YC, Nishiumi I, et al. An empirical comparison of character-based and coalescent-based approaches to species delimitation in a young avian complex. Mol Ecol. 2013;22:4943–57.
Lin YP, Edwards RD, Kondo T, Semple TL, Cook LG. Species delimitation in asexual insects of economic importance: the case of black scale (Parasaissetia nigra), a cosmopolitan parthenogenetic pest scale insect. PLoS One. 2017;12:e0175889.
Yang Z. The BP&P program for species tree estimation and species delimitation. Curr Zool. 2015;61:854–65.
Pinho C, Hey J. Divergence with gene flow: models and data. Annu Rev Ecol Evol Syst. 2010;41:215–30.
Martin SH, Dasmahapatra KK, Nadeau NJ, Slazar C, Walters JR, Simpson F, et al. Heliconius and sympatric speciation. Genome Res. 2013;23:1817–28.
Jackson ND, Carstens BC, Morales AE, O’Meara BC. Species delimitation with gene flow. Syst Biol. 2017;66:799–812.
Jackson ND, Morales AE, Carstens BC, O’Meara BC. PHRAPL: Phylogeographic inference using approximate likelihoods. Syst Biol. 2017;66:1045–53.
Hime PM, Hotaling S, Grewelle RE, O’Neill EM, Voss SR, Shaffer HB, et al. The influence of locus number and information content on species delimitation: an empirical test case in an endangered Mexican salamander. Mol Ecol. 2016;25:5959–74.
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376.
Chattopadhyay B, Garg KM, Kumar AKV, Doss DPS, Rheindt FE, Kandula S, et al. Genome-wide data reveal cryptic diversity and genetic introgression in an oriental cynopterine fruit bat radiation. BMC Evol Biol. 2016;16:41.
Beheregaray LB, Pfeiffer LV, Attard CRM, Sandoval-Castillo J, Domingos FMCB, Faulks LK, et al. Genome-wide data delimits multiple climate-determined species ranges in a widespread Australian fish, the golden perch (Macquaria ambigua). Mol Phylogenet Evol. 2017;111:65–75.
Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, Roychoudhury A. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol Biol Evol. 2012;29:1917–32.
Leaché AD, Fujita MK, Minin VN, Bouckaert RR. Species delimitation using genome-wide SNP data. Syst Biol. 2014;63:534–42.
Häkkinen M, Wang H, Ge XJ. Musa itinerans (Musaceae) and its intraspecific taxa in China. Nord J Bot. 2008;26:317–24.
Li W, Dita M, Wu W, Hu G, Xie J, Ge XJ. Resistance sources to fusarium oxysporum f. Sp. cubense tropical race 4 in banana wild relatives. Plant Pathol. 2015;64:1061–7.
Perrier X, De Langhe E, Donohue M, Lentfer C, Vrydaghs L, Bakry F, et al. Multidisciplinary perspectives on banana (Musa subsp.) domestication. Proc Natl Acad Sci U S A. 2011;108:11311–8.
Wu W, Yang YL, He WM, Rouard M, Li WM, Xu M, et al. Whole genome sequencing of a banana wild relative Musa itinerans provides insights into lineage-specific diversification of the Musa genus. Sci Rep. 2016;6:31586.
Raj A, Stephens M, Pritchard JK. FastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics. 2014;197:573–89.
Nei M, Li WH. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci U S A. 1979;76:5269–73.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Wright S. Evolution in mendelian populations. Bull Math Biol. 1990;52:241–95.
Nei M. Molecular evolutionary genetics. New York: Columbia University Press; 1987.
Cruickshank TE, Hahn MW. Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol Ecol. 2014;23:3133–57.
Ma JX, Bennetzen JL. Rapid recent growth and divergence of rice nuclear genomes. Proc Natl Acad Sci U S A. 2004;101:12404–10.
Zhao H, Wang L, Yuan J. Origin and time of Qiongzhou Strait. Mar Geol Quat Geol. 2007;27:33–9.
Häkkinen M, Chinglong Y, Ge X. A new combination and a new variety of Musa itinerans (Musaceae). Acta Phytotaxon Geobot. 2010;61:41–8.
Chiu HL, Shii CT, Yang TYA. A new variety of Musa itinerans (Musaceae) in Taiwan. Novon. 2011;21:405–12.
Chiu HL, Shii CT, Yang TYA. Musa itinerans var. chiumei (Musaceae), a new addition to the Taiwan Flora. Taiwania. 2015;60:133–6.
Clausen R. On the use of the terms "subspecies" and "variety". Rhodora. 1941;43:157–67.
Moritz C. Defining “evolutionarily significant units” for conservation. Trends Ecol Evol. 1994;9:373–5.
Yang Z, Rannala B. Bayesian species identification under the multispecies coalescent provides significant improvements to DNA barcoding analyses. Mol Ecol. 2017;26:3028–36.
Sukumaran J, Knowles LL. Multispecies coalescent delimits structure, not species. Proc Natl Acad Sci U S A. 2017;114:1607–12.
Häkkinen M. Musa voonii, a new Musa species from northern Borneo and discussion of the section callimusa in Borneo. Acta Phytotax Geobot. 2004;55:79–88.
Häkkinen M. Musa chunii Häkkinen, a new species (Musaceae) from Yunnan, China and taxonomic identity of Musa rubra. J Syst Evol. 2009;47:87–91.
Chen WN, Häkkinen M, Ge XJ. Musa ruiliensis (Musaceae, section Musa), a new species from Yunnan, China. Phytotaxa. 2014;172:109–16.
Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7:e30619.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
DePristo MA, Banks E, Poplin RE, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next- generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Levy-moonshine A, Jordan T, et al. From FastQ data to high confidence varant calls: the Genonme analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:1–33.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Price AL, Price AL, Patterson NJ, Patterson NJ, Plenge RM, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9.
Pfeifer B, Wittelsbürger U, Ramos-Onsins SE, Lercher MJ. PopGenome: an efficient swiss army knife for population genomic analyses in R. Mol Biol Evol. 2014;31:1929–36.
Patterson N, Moorjani P, Luo Y, Mallick S, Rohland N, Zhan Y, et al. Ancient admixture in human history. Genetics. 2012;192:1065–93.
Korneliussen TST, Albrechtsen A, Nielsen R, Nielsen R, Paul J, Albrechtsen A, et al. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics. 2014;15:1–13.
Carstens BC, Pelletier TA, Reid NM, Satler JD. How to fail at species delimitation. Mol Ecol. 2013;22:4369–83.
Acknowledgements
The authors thank Dr. Wei-Ming He, and Dr. Yu-lan Yang at BGI for their helpful suggestions on data analysis. We also thank the two reviewers for their insightful suggestions on our raw manuscript.
Funding
This study was financially supported by the National Natural Science Foundation of China (No. 31261140366).
Availability of data and materials
The genome assembly and annotation file have been uploaded to the Banana Genome Hub (http://banana-genome-hub.southgreen.fr/organism/Musa/Itinerans), and the raw data has been deposited in the NCBI Sequence Read Archive under BioProject ID 312694 with accession number SRR6382516~SRR6382539.
Author information
Authors and Affiliations
Contributions
XJG collected plant material, and designed the project. WW, WLN, and JXY, and WML, analyzed the data. WW, WLN, and XJG wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Summary of sequencing depth and coverage for each sample accession of Musa itinerans used in this study. Figure S1. The distributions of overall genome-wide polymorphisms for four varieties of Musa itinerans. Data shown for with overlapped window size of 20 kb and step size of 2 kb. Figure S2. The distributions of average pairwise nucleotide diversity θπ, and Tajima’s D, Wright’s Fixation index FST and absolute genetic divergence Dxy across chromosome 2 ~ 12. (PDF 4614 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wu, W., Ng, WL., Yang, JX. et al. High cryptic species diversity is revealed by genome-wide polymorphisms in a wild relative of banana, Musa itinerans, and implications for its conservation in subtropical China. BMC Plant Biol 18, 194 (2018). https://doi.org/10.1186/s12870-018-1410-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-018-1410-6