
Abstract
Coral reefs are biodiversity hotspots, places of high endemicity and provide essential services to billions of people globally. With increasing threats to these reefs worldwide, there is a need to implement faster, more efficient ways to monitor spatial and temporal patterns of biodiversity. Environmental DNA (eDNA) metabarcoding offers a promising tool to address this issue, as it has revolutionized our ability to monitor biodiversity from complex environmental samples such as seawater. However, the capacity for eDNA to resolve fine scale shifts in community composition across habitats in seascapes is yet to be fully explored. Here, we applied eDNA metabarcoding using the rRNA 18S Universal eukaryote assay to explore differences in community profiles between samples collected from the lagoon and reef slope habitats across more than 170 km of the Ningaloo Coast World Heritage Area in Western Australia. We recovered 2061 amplicon sequence variants that comprised of 401 taxa spanning 14 different metazoan phyla such as cnidarians, poriferans, molluscs, algae, worms, and echinoderms. Our results revealed strong clustering of samples by habitat type across the length of the reef. Community dissimilarity (beta diversity) between samples collected from the reef slope and lagoon habitats was high and was driven largely by a strong rate of spatial turnover, indicating a distinct set of taxa representing each reef zone community. We also detected a strong pattern of isolation by distance within our slope samples, suggesting that communities are spatially stratified across the length of the reef. Despite high connectivity due to regular flushing of the lagoon environment, our results demonstrate that metabarcoding of seawater eDNA from different habitats can resolve fine scale community structure. By generating multi-trophic biodiversity data, our study also provided baseline data for Ningaloo from which future changes can be assessed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coral reefs are experiencing increasingly frequent, and larger scale impacts due to a range of direct and indirect human activities (Hughes et al. 2018). Notably, marine heatwave events induced by global warming have caused extensive coral bleaching and mass mortality across most tropical waters (Baker et al. 2008; Hughes et al. 2018). These widespread declines in both coral cover and diversity (Moore et al. 2012; Descombes et al. 2015) have been mirrored by declines in the diversity and abundance of associated biota such as reef fishes (Jones et al. 2004; Pratchett et al. 2011). The biodiversity value of coral reefs is profound as they support an estimated one million species globally, hence declines in coral cover and diversity threaten the health of coastal habitats and the species they sustain (Jones et al. 2004; Knowlton et al. 2010; Descombes et al. 2015). Corals and their holobiont form the calcium carbonate framework of reefs and fill significant roles related to the biogeochemical cycling of littoral zones (Rädecker et al. 2015; Gong et al. 2020). The species composition of corals drives key ecosystem function on reefs, and stressors that alter these configurations threaten the functioning and stability of such ecosystems (Richardson et al. 2020). Consequently, there is an urgent need for documenting baselines of biodiversity that can be used to assess the extent and severity of anthropogenic impacts, provide recovery targets, and increase reef resilience. Such baselines would ideally provide comprehensive data on all components of the reef community across trophic levels, rather than focussing on indicator or keystone species.
Traditional methods of coral reef monitoring (e.g. manta tows, underwater visual and photo benthic surveys, Automated Underwater Vehicles (AUVs), towed video systems, Baited Remote Underwater Stereo-Video Systems (BRUVS)) typically only focus on targeted components of reef communities and are generally time, cost, and labour intensive (Stat et al. 2017). The inconspicuous reef cryptobiome largely made up of small cryptic invertebrates, as well as nocturnal species, are underrepresented in traditional surveys due to difficulties of detection (Pearman et al. 2016; Carvalho et al. 2019). As a result, such monitoring techniques rarely survey holistically across all trophic levels, particularly when surveying large areas of reefs.
Environmental DNA (eDNA) metabarcoding, which enables multispecies detection from an environmental sample such as seawater, may promise greater resolution for whole-ecosystem surveying by increasing detectability of the biosphere that can go unnoticed using traditional methods (Stat et al. 2017; West et al. 2021). This method has become a rapid assessment tool for biodiversity, able to provide broad coverage across taxa. Although a multi-assay survey has been highlighted as the optimal approach (coined “Tree of Life”) to generate the highest taxonomic resolution, it significantly increases costs and time required, as well as technical complexity (Stat et al. 2017; Eble et al. 2020). As such, a rapid universal metabarcoding survey (RUMS; DiBattista et al. 2019) that employs a single broad-coverage assay allows managers to efficiently scale up monitoring to include a much wider range of taxa. Environmental DNA RUMS have been used to investigate shifts in community structure across a depth gradient on a coral reef (DiBattista et al. 2019), between Caribbean islands (Bakker et al. 2019), tropical habitats (Nguyen et al. 2020) or MPA zones (Gelis et al. 2021); however, there are limited applications of the technique to explore finer scale differences in community structure across reef habitat zones (West et al. 2021).
The World Heritage Ningaloo Coast stretches 280 km along the Western Australian coastline and is one of the world’s largest fringing coral reef systems. It is composed of a series of fringing reefs connecting shallow lagoons to the reef flat and outer reef slope broken by with reef channels that allow water flow out of the lagoon (Vanderklift et al. 2020). Ningaloo Reef is thought to be less threatened than the Great Barrier Reef due to limited historical commercial fishing activity, minimal nutrient run-off, and low levels of human coastal development (Cassata and Collins 2008; Johansson et al. 2010). However, the reef has nonetheless experienced a recent increase in frequency of coral bleaching (Moore et al. 2012; Depczynski et al. 2013; Gilmour et al. 2019). Thermal anomalies such as marine heatwaves are predicted to increase in frequency and severity (Ateweberhan et al. 2013; Oliver et al. 2018) and will likely impact Ningaloo in the near future (Babcock et al. 2021). Therefore, a large-scale eDNA biodiversity assessment can provide important baseline data, with assessments of disturbance-related shifts in fine scale community structure that can be used to inform adaptive management actions. Here, we applied a RUMS approach using seawater eDNA to characterize the marine metazoan (and protozoan macroalgae) biodiversity along Ningaloo. We also explored the power of eDNA metabarcoding to resolve differences in community profiles between the sheltered lagoon and exposed reef slope environments. Our results contribute to growing evidence of the power of eDNA metabarcoding in teasing apart habitat-specific localized signals and its ability in generating comprehensive profiles of biodiversity.
Materials and methods
Field sampling
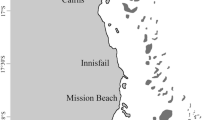
In May 2019, a total of 120 seawater samples were collected at 14 different localities along Ningaloo Reef, spanning approximately 172 km and 2 degrees of latitude along the Western Australian coastline (Fig. 1). We collected near-surface (1–2 m depth) seawater samples from two distinct habitats along the reef; the backreef lagoon and the exposed reef slope. For shallow lagoon samples, five replicates of 1 L samples were collected in Nalgene bottles by snorkellers (site depth < 6 m) from three different locations at five different sites across the reef (n = 75 samples total, 15 samples per lagoon site). The five replicate samples were collected at a single location within a 5 m radius of each other and at each site the substrate was categorized as intermittent coral bommies and structures separated by sand patches with moderate coral cover. The water bottles were kept on ice in a cooler during sampling and stored at ~ 4 °C until filtering. On the reef slope, five replicate samples were collected using a livewire CTD Niskin rosette system, repeated at nine sites approximately ~ 20 km (10 nautical miles) apart along the western edge of the outer reef at a site depth range of 11 to 22 m (n = 45 samples total) (Fig. 1). For each replicate sample collected via CTD Niskin rosettes, we used a small volume of the sample to thoroughly rinse out the individual Nalgene bottles prior to transferring the sample into the bottle and storing at ~ 4 °C until filtering. To track contamination, five tap water controls were also taken by collecting a 1 L sample using the sample bottles post-bleach rinse bucket filled with freshwater originating from the vessel’s desalination system. All samples were filtered using sterile single-use 250 ml funnel EZ-Fit filtration units with 0.45 μm mixed cellulose ester membranes and an EZ-Fit Manifold base (3-place) from Merck Millipore (Merck Group, Burlington, USA). Gloves were changed and the manifold base was sterilized with a 10% bleach solution between the filtering of samples from different sites. Following filtration, the filter papers were rolled and kept in cryogenic tubes at − 20 °C until transport and storage at − 80 °C. All subsequent laboratory work was conducted at the Trace and Environmental DNA (TrEnD) laboratory at Curtin University, Australia.

Map of sampling locations and visual depiction of the sampling methodology for the lagoon and slope habitats. Slope sites are A: NW Cape, B: Tantabiddi, C: Milyering, D: Osprey, E: Widerabandi, F: Point Edgar, G: Point Cloates, H: Bateman Bay and I: Bruboodjoo. Lagoon sites are a: Tantabiddi, b: Norwegian Bay, c: Point Cloates, d: Stanley Pool and e: Coral Bay (Table S1)
Sample processing and sequencing
DNA was extracted from half of the filter membranes using the Qiagen DNeasy Blood & Tissue kit with the following modification to the protocol: 360 μL of ATL buffer and 40 μL of proteinase K for an overnight incubation at 56 °C (Qiagen, Hilden, Germany). Negative extraction controls were included to check for cross contamination. The resulting supernatant was loaded into the QIAcube DNA extraction robot for automated DNA extraction (Qiagen). Quantitative PCR (qPCR) employed the nuclear rRNA 18S Universal eukaryote assay (V1-3 hypervariable region; 18S_uni_1F: 5’ – GCCAGTAGTCATATGCTTGTCT - 3’ and 18S_uni_400R: 5’ – GCCTGCTGCCTTCCTT - 3’; Pochon et al. 2013). Quantitative PCR amplification was performed on duplicates of each sample (seawater, extraction, negative (no template) and positive controls) and using fusion tagged primers; unique sample-specific tag combinations (6–8 bp) that allow separation of reads post-sequencing. Each qPCR reaction was made up to 25 μL containing: 1X AmpliTaq Gold PCR buffer (Life Technologies), 2 mM MgCl2 (Fisher Biotec, Wembley, Australia), 0.4 μM dNTPs (Astral Scientific, Caringbah, Australia), 0.1 mg BSA (Fisher Biotec), 0.6 μL of 5X SYBR Green (Life Technologies), 1U AmpliTaq Gold DNA Polymerase (Life Technologies), 0.4 μM of forward and reverse fusion tags, 2 μL of eDNA template (at optimized dilution) and UltraPure Distilled Water (Life Technologies, Carlsbad, USA). Thermocycler conditions were as follows: initial denaturation at 95 °C for 5 min followed by 45 cycles of 95 °C for 30 s, 52 °C for 30 s, 72 °C for 45 s, 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s and a final extension of 72 °C for 10 min. All pre-PCR and post-PCR steps were performed in separate laboratories using UV-sterilized cabinets at TrEnD laboratory, Curtin University. Amplicons were pooled to approximate equimolar ratios into Mini Pools (MPs) based on the samples qPCR ΔRn values, after which size distribution and DNA concentration were verified using the QIAxcel Advanced System (Qiagen). Based on the QIAxcel results, the MPs were blended into a final equimolar library, which was size selected to a size range of 175–600 bp using Pippin Prep (Sage Science, Beverley, USA). The size-selected library was then purified using the QIAquick PCR purification kit (Qiagen), before fluorometric quantitation using Qubit 4.0 (Thermo Fisher Scientific). The final library (2 nM) was sequenced on a MiSeq 500-cycle V2 chemistry paired-end run (Illumina, San Diego, USA) in a post-PCR laboratory at TrEnD, Curtin University, Australia.
Bioinformatics and data analyses
We used the Greenfield Hybrid Analysis Pipeline (GHAP) to demultiplex raw reads based on the unique sample-specific tags assigned during qPCR using the DemultiplexCustomBC function (Greenfield 2017) with only exact matches retained for further processing. Reads were quality filtered and clustered into ASVs using DADA2, which removes the arbitrary sequence similarity threshold component of OTU-based clustering (Callahan et al. 2016). The ASVs were then curated using the LULU algorithm with default parameters (Frøslev et al. 2017). The resulting ASVs were queried against NCBI’s GenBank nucleotide databases (downloaded on 28/07/2021) using BLASTn on the KAYA HPC housed at the University of Western Australia. Taxonomic assignments were processed using the lowest common ancestor (LCA) step of the eDNAFlow pipeline (Mousavi-Derazmahalleh et al. 2021). All taxonomic assignments required a 100% query coverage and over 98% identity match to a reference sequence for a species-level assignment to be made. Genus-level assignments required a 97% identity match, family-level a 95% match, and order-level a 90% match. Taxonomic hits that were detected in the field tapwater and/or laboratory controls were deemed to be potential contaminants and were excluded from the datasets (Table S2).
All analyses used R Studio (v3.5.3) and figures were made using the ggplot2 R package (Wickham 2016). All analyses used a binary-transformed (presence/absence) matrix, except for the ANCOM-BC analysis which used the raw eDNA counts matrix. We used the metaMDS function of the vegan R package to perform nonmetric multidimensional scaling (nMDS) plots using the Jaccard binary dissimilarity index (Oksanen et al. 2020). We used the adonis function from the vegan R package to conduct a permutational multivariate analysis of variance (PERMANOVA) of Jaccard dissimilarities using 9999 permutations (Oksanen et al. 2020). Homogeneity of variance was then tested using the betadisper and permutest functions to verify the effect of dispersion. To explore latitudinal patterns in our dataset, we focussed on a single habitat, the reef slope, and performed a Mantel test (ade4 R package; Dray and Dufour 2007) to test the relationship between community composition and the geographic distance between samples along the length of the reef. We also categorized the nine slope sites into Northern, Middle, and Southern sections, consisting of three sites each. To understand the differences in the eDNA communities recovered across the reef, we calculated the contributions of turnover (βjtu) and nestedness (βjne) components to the pairwise Jaccard’s dissimilarity index (βjac) using the betapart R package (Baselga & Orme 2012). A βjac index (beta diversity) of 0 indicates that species composition is identical between groups, whereas an index value of 1 indicates complete dissimilarity. The turnover component indicates species replacement between sites or groups, whereas nestedness indicates that the extent to which a group is a subset of another (Baselga and Orme 2012). To assess whether our unbalanced sampling effort produced a bias, we randomly selected five sites per reef zones to set an equal sampling effort of 25 samples per habitat and recomputed our diversity analyses a total of 50 times. The relative proportion of each phylum was calculated by adding up all ASVs (taxa) corresponding to the different phyla in relation to the total ASVs (taxa) recovered in each site. To detect differentially abundant taxa across habitats, we performed an Analysis of Compositions of Microbiomes with Bias Correction (ANCOM-BC) which accounts for the compositional nature of eDNA metabarcoding data (Mandal et al. 2015). We used the ancombc R package (Lin & Peddada 2020) to test for differences in abundance of metazoan taxa with a significance of p < 0.05 adjusted with the default Bonferroni correction. We evaluated how comprehensively eDNA surveyed the communities using species accumulation curves from the iNEXT package (Chao et al. 2014; Hsieh et al. 2020) and estimated the total richness of the system using the Chao2 estimator in SpadeR (Chao and Chiu 2014).
Results
Sequencing results and overall diversity
Metabarcoding of the rRNA 18S Universal eukaryote assay yielded 7,072,787 demultiplexed sequences. A total of 4,177,383 sequences remained after quality control and error filtering with the DADA2 pipeline. The average number of reads passing filters was 25,317 ( ± 11,693) per sample. The DADA2 pipeline generated 7751 ASVs, which were filtered down to 2778 ASVs by LULU. We removed ASVs with less than five total reads, resulting in a final dataset of 2061 ASVs. Following BLAST and LCA, a total of 603 eukaryotic ASVs were taxonomically identified (Fig. 2), representing approximately 29% of all ASVs recovered. Approximately one third of eukaryotic sequences were assigned to the kingdoms Protozoa and Chromista, including Bacillariophyta (diatoms), Apicomplexa, Radiolaria, Ciliophora (Fig. 2). However, for this study, we focused our analyses on metazoan taxa with the addition of red (Rhodophyta) and green (Chlorophyta) algae. The final taxonomic dataset included 401 taxa (ASVs) spanning 14 phyla, 35 classes, 84 orders, 112 families, 109 genera and 57 species detected in 118 seawater samples (two samples returned no taxonomic assignment).

Class-level dendrogram of all eukaryotic detections (603 taxa) recovered in the seawater samples at Ningaloo Reef
Across all samples, Arthropoda was the most represented group constituting 16.7% of all taxa identified. This was followed by Porifera (14.2%), Cnidaria (13%), Annelida (13%) and Mollusca (13%). Sequences belonging to Chordata (10.5%) were mostly derived from ascidian tunicates, whereas very few were assigned to fishes. Within Cnidaria, the reef-building coral genus Acropora, which is the most common genus of coral at Ningaloo, was widely detected across the reef. Some potential contaminants were detected in the field controls as well as across some of our seawater samples (12 metazoan ASVs). These hits were excluded from the analyses as they may have originated from the ship’s desalinated and UV-treated water which was used to rinse out the sampling bottles after the bleach sterilization step (Table S2).
Diversity signals across the reefscape
Levels of metazoan diversity in seawater samples collected from the lagoon were consistently greater than in samples collected from adjacent reef slope habitats. Lagoon alpha diversity was higher for both total ASVs and number of metazoan taxa identified (Fig. 3a, d). A total of 1683 ASVs were recovered in the lagoon, compared to 898 ASVs in the slope samples. A total of 520 ASVs (~ 25.2%) were shared between both habitats; 1163 ASVs were exclusively recovered in the lagoon, whereas 378 ASVs were only recovered on the slope (Fig. 3b). Metazoan alpha diversity was also higher in the lagoon (355 taxa) than on the slope (118 taxa). Of the 401 taxa detected, only 18% (72) were shared between reef zones. The remaining taxa had a unique signal corresponding to a specific habitat type, with 283 taxa only occurring in the lagoon and 46 taxa only found in the slope (Fig. 3e). As a result of this limited overlap, the level of eDNA assemblage dissimilarity between habitats was high (β-diversity = 0.8205), and taxonomic compositional differences were primarily due to a high rate of spatial turnover (0.5610), rather than nestedness (0.2595), indicating a pattern of species replacement when moving between habitats. Habitat-specific eDNA communities were apparent and clustering of samples based on reef zone was evident in the nMDS plots (of Jaccard dissimilarities) using both datasets (Fig. 3c, f). This was confirmed with the PERMANOVA which suggested significant differences in the eDNA communities detected among habitat types (ASV: R2 = 0.0988, p < 0.001, Taxa: R2 = 0.1167, p < 0.001). However, there was significant heterogeneity in dispersion (ASV: Pseudo-F = 22.649, p = 0.001, Taxa: Pseudo-F = 16.574, p = 0.001) which could impair the PERMANOVA results in an unbalanced design. While the PERMANOVA results should be interpreted with caution, the nMDS ordinations indicate strong divergence among eDNA communities across reef zones (Fig. 3c, f).

Boxplots of alpha diversity between habitats for the number of a ASVs and d metazoan taxa recovered. Venn diagrams showing the unique and shared b ASVs and e metazoan taxa recovered across habitats. Nonmetric multidimensional scaling (nMDS) of Jaccard dissimilarities with samples colour-coded by habitat type for the c ASV dataset and the f taxonomic dataset. Ordination stress values were c 0.1426 and f 0.2088
In addition to differences in eDNA communities across habitat types, we also detected a significant influence of geographic distance on community composition. The Mantel tests between the ecological (binary) and geographic (km) distances among samples revealed a significant positive correlation for both datasets (ASV: r = 0.3987, p = 0.001, Taxa: r = 0.3419, p = 0.001) (Fig. 4). Furthermore, PERMANOVA tests revealed significant differences in assemblages between the Northern, Middle, and Southern areas of the Ningaloo reef (ASV: R = 0.1657, p < 0.001, Taxa: R = 0.1418, p < 0.001). We did not find any evidence for heterogeneity of dispersion (ASV: Pseudo-F = 1.2301, p = 0.308, Taxa: Pseudo-F = 0.8896, p = 0.415). Levels of alpha diversity were similar across the reef slope samples collected from the Northern, Middle, and Southern sections of Ningaloo, with 497 ASVs (56 taxa), 507 ASVs (41 taxa) and 533 ASVs (64 taxa) recovered, respectively. Latitudinal metazoan community dissimilarity was high (β-diversity = 0.8314) and was almost entirely driven by spatial turnover (0.7943), indicating a pattern of species replacement when moving along the length of the reef.

Results of the Mantel test showing a significant positive correlation between ecological distance (binary) plotted against geographic distance (km) with regression lines for the A ASV dataset and B metazoan taxonomic dataset. (ASV: r = 0.3987, p = 0.001, Taxa: r = 0.3419, p = 0.001)
Community composition between reef habitats
The relative proportion of each phylum differed substantially in samples collected from the two reef zones (Fig. 5a), and there were consistent sets of organisms that were more frequently detected in each habitat. For example, annelid worms, molluscs, and cnidarians were well represented in the lagoon samples, and the phyla Sipuncula, Rhodophyta, Nematoda and Bryozoa were unique to the lagoon zone. In contrast, green algae, arthropods, and chordates were more frequently detected on the reef slope. Taxa such as echinoderms and other worms (Nemertea and Platyhelminthes) were detected in low numbers regardless of habitat type. Interestingly, reef slope sites showed wide variation in community composition among sites (Fig. 5a). This pattern seemed less evident in the lagoon samples, where the relative proportions of phyla were more constant across replicates and sites (Fig. 5a). We also found that at the taxonomic class level, only 51% (18) of classes were shared between habitats, with 15 classes found only in the lagoon samples (Fig. 5b). A total of 33 out of the 35 classes were found in the lagoon, whereas only 20 classes were found in the slope zone. This indicates that despite high levels of broadly distributed taxa, we also recovered localized eDNA signals associated solely in the lagoon samples. Overall, looking at the family level, 40.5% of all taxonomic families (n = 37) detected in the reef slope samples could be categorized as pelagic or free-swimming in nature, whereas 59.5% were benthic-associated organisms. In contrast, the taxonomic families (n = 102) detected in the lagoon samples were primarily (67.6%) benthic associated, whereas only 28.4% were categorized as pelagic, and 3.9% were parasitic organisms.

a Ridgeline plot showing the relative proportion of the 14 phyla recovered in the two habitats sampled. The relative proportion of each phylum was calculated by adding up all ASVs (taxa) corresponding to the different phyla in relation to the total ASVs (taxa) recovered in each site. b Heatmap of the presence and absence (P/A) of metazoan classes recovered with seawater eDNA. Classes are ordered alphabetically, and P/A was calculated at the site level
Key indicator taxa
Of the 401 total ASVs, 20 metazoan taxa were identified by ANCOM-BC as differentially abundant across reef zones (Fig. 6). The majority of these taxa (70%) were found to be more abundant in the lagoon samples, and these consisted primarily of habitat-forming benthic organisms such as sponges (order Haplosclerida), corals (orders Scleractinia and Anthoathecata), ascidians (order Stolidobranchia) and red algae (order Corallinales). Additional taxa also found to be of higher abundance included bivalves (order Mytiloida and Ostreida), polychaetes (order Terebellida) and copepods (order Decapoda). Inversely, there were six taxa that were more abundant in the slope samples, and these consisted of copepods (order Calanoida), free swimming tunicates (order Copelata) as well as sponges (order Clionaida) (Table S3).

Analysis of composition of microbiomes with bias correction (ANCOM-BC) identified 20 taxa that were differentially abundant across reef zones. Figure shows effect size (log fold change) and 95% confidence intervals, Bonferroni adjusted. Negative log fold change values indicate an increase in abundance in the Lagoon (pale blue), whereas positive log fold change values indicate an increase in abundance in the Slope (dark blue)
Sampling effort
Despite uncovering high biodiversity in our samples, our sampling effort did not completely capture the eukaryotic marine biodiversity of the Ningaloo region. ASV accumulation curves showed no plateauing of diversity across all sites, as well as within the reef slope and lagoon habitats, with estimates indicating more than 600 samples would be required to adequately characterise biodiversity (Fig. 7a). We captured approximately 46.6% of the total 4420 ASVs (SE 4010–4915) estimated by the Chao-2 index. Sample coverage curves across the lagoon sites indicated that only Stanley Pool approached sampling saturation (91.5%), although the sample coverage of the remaining sites was quite high (ranging from 82.1 to 85.8%), indicating near-sufficient sampling (Fig. 7b, Table S4). However, sample coverage across the slope sites was lower, ranging from 63.6% at Osprey to 87.6% at Point Edgar (Fig. 7c, Table S4). Our data suggest that to consistently reach over 80% in sample coverage, a minimum of 10 replicate 1 L samples need to be collected at each location to comprehensively census the gamma diversity of Ningaloo Reef.

Eukaryotic ASV accumulation curves across a all 1 L sample replicates (118), lagoon samples (74) and slope samples (44), as well as sample coverage estimates at the site-level for the b lagoon and c slope reef zones
Randomly selecting five sites per reef zone, resulting in an equal sampling effort of 25 samples per reef zone, we recomputed our analyses 50 times and found that patterns of diversity and community composition were consistent (Table S5). Levels of metazoan diversity in seawater samples collected from the lagoon remained consistently greater than in samples collected from adjacent reef slope habitats. On average, 267 taxa were detected across the 10 sites, with 222 taxa in the lagoon and 80 taxa in the slope. Only approximately 13.2% (± 3.1%) were shared across reef habitats. As a result of this limited overlap, the level of eDNA assemblage dissimilarity between habitats remained high (β-diversity ~ 0.8680 ± 0.0306), and taxonomic compositional differences were always primarily due to a high rate of spatial turnover (~ 0.7184 ± 0.0448) rather than nestedness (~ 0.1492 ± 0.0231).
Discussion
We used a Rapid Universal Metabarcoding Survey (RUMS) approach targeting the 18S gene region of marine eukaryotes to catalogue a total of 401 metazoan taxa spanning 14 phyla. Seawater eDNA signals were heterogeneous across two distinct reef habitat zones and revealed pronounced differences in the community profiles of reef habitats that can be periodically connected by tidal and other water currents. This study adds to growing evidence of the utility of eDNA for detecting fine-scale differences in community structure and provides novel insights into the spatial organization of eDNA within reef environments.
Community partitioning across the reefscape
eDNA metabarcoding offers the ability to detect localized signals of community composition corresponding to discrete marine habitat types (Jeunen et al. 2018; Oka et al. 2020; Ip et al. 2021). Most studies have compared eDNA signals across distinct and spatially separate environments such as coral reef, seagrass, mangroves, sandy shores, with few focusing on how more subtle signals can be structured across different reef zones (West et al. 2021). Our findings revealed that seawater samples collected from adjacent reef habitats returned distinct assemblages of metazoan diversity. The exposed shallow reef slope tends to be coral-dominated with relatively high cover, whereas the shallow lagoon consists largely of macroalgae meadows and sparse, patchy low-relief structures and coral bommies scattered across sand or rubble substrates (Cassata and Collins 2008; Kobryn et al. 2013; Vanderklift et al. 2020). As such, the lagoonal eDNA signal is expected to include macroalgae-associated organisms, which are unlikely to be represented in the outer reef slope samples. Furthermore, reef zones with different environmental gradients such as light levels, wave and temperature regimes can select for specific benthic assemblages that in turn attract a different suite of associated taxa (Thomson et al. 2020).
Beta diversity, the variance in species composition of biological assemblages among sites, can be partitioned into components of spatial turnover or nestedness (Baselga & Orme 2012). In this study, we observed a high β-diversity index of community dissimilarity (0.8205), which was primarily driven by the turnover component, indicating a pattern of species replacement when moving across habitat types. However, the nestedness component contributed a third of the dissimilarity index, which could indicate that the less diverse reef slope communities are a subset of the overall Ningaloo Reef communities recovered with eDNA. Indeed, few taxa were found to be unique to the reef slope samples (11.5%), whereas a large proportion of the diversity recovered was detected only in the lagoon samples (70.6%) or across both habitats (17.9%). Thus, the high eDNA-derived beta diversity observed at Ningaloo Reef was a result of both taxon replacement between habitats (the substitution of taxa in one site/habitat by different taxa in the other habitat), and taxon loss when moving from the lagoon to the reef slope (the less diverse assemblage resembling a subset of the richest one) (Baselga and Orme 2012). This signal was not biased by sample size as patterns of diversity and community composition described above remained the same after bootstrap resampling. Indeed, even with equal sampling effort, the lagoon samples were consistently more diverse than samples collected from the adjacent reef slope, and beta diversity remained high and largely driven by turnover. Furthermore, we detected a strong pattern of isolation by distance within our reef slope samples, as well as high community dissimilarity when comparing the Northern, Middle, and Southern sites, suggesting that communities are spatially partitioned across the length of the reef. Together, these results revealed cross-shore as well as latitudinal variation in metazoan communities in a dynamic, exposed and connected ecosystem, highlighting the ability of eDNA to provide localized diversity snapshots. It is however unclear whether these patterns hold over time, and accurate assessments of biodiversity rely on an understanding of species abundance distributions and how these change over time and space (Seymour et al. 2021). A temporal eDNA sampling approach would allow the identification of core resident species that are typically abundant, versus ephemeral species (Magurran and Henderson 2003).
Vertical dispersal of eDNA in the marine environment is thought to be limited, leading to distinct communities throughout the water column (Jeunen et al. 2020; Monuki et al. 2021). In this study, seawater was collected close to the surface in both habitats; however, the lagoon sites were shallower (< 6 m) than the reef slope sites (11–22 m). As a result, the slope-specific assemblages we recovered could be more representative of mid-water diversity, rather than benthic diversity which was probably well described in the lagoon samples. This could have potentially driven the divergence in alpha diversity we detected between reef zones. Indeed, our results reflected these patterns, and showed that worms (annelids, sipunculids, nematods) and benthic taxa were more dominant in the lagoon seawater samples. In contrast, free-swimming and pelagic species such as planktonic copepods and tunicates were well represented in the slope samples. Notable taxa missing from the slope samples included fishes, crinoids, and many types of worms. Sediment-dwelling worms can be harder to detect from seawater eDNA, and in our case were perhaps missed in the reef slope samples due to the larger distance between substrate and sampling depth (Koziol et al. 2018; Antich et al. 2020). Further studies investigating how eDNA signals differ throughout the water column are necessary to understand vertical partitioning in shallow coral reef habitats, as well as to inform on the best sampling methodologies to adequately capture comprehensive inventories of biodiversity.
eDNA as a monitoring tool for coral reef ecosystems
Traditionally, monitoring of coral reefs focuses on well-studied macrofauna such as corals and/or fishes which are used as surrogates for gamma diversity (Plaisance et al. 2011; Mellin et al. 2011). However, this approach ignores crucial components of reef-associated biota, such as cryptic species that live within the complex reef framework (Knowlton et al. 2010) or sediments. To remedy this issue, standardized 3D sampling devices called Autonomous Reef Monitoring Structures (ARMS) have been deployed globally; they enable the settlement of encrusting sessile species as well as mobile organisms that inhabit the stacks of plates, however they are long-term structures that require up to three years of deployment (Zimmerman and Martin 2004; Carvalho et al. 2019). Alternatively, eDNA metabarcoding can be used as a rapid biodiversity assessment tool to provide inventories of local species diversity. In this study, we used an eDNA RUMS approach and recovered 14 different metazoan phyla in a single survey conducted over five days. This highlights the potential of eDNA metabarcoding to characterize marine biodiversity across a broad taxonomic spectrum simultaneously, as well as capturing some of the reef cryptobiome that can be overlooked.
Limitations of the current approach
Despite the breadth of diversity recovered in this study using a single assay, our results only reflect a fraction of the total biodiversity of Ningaloo Reef. Indeed, a multi-assay approach has been shown to capture the most comprehensive overview of biodiversity (Stat et al. 2017; West et al. 2021). For example, a study at Coral Bay (Ningaloo Reef) calculated that the same 18S primer set used here detected only 44% of the total number of taxa recovered when combining 10 assays, highlighting the risk of severely underestimating the diversity of an area when only employing one universal assay (Stat et al. 2017). Our sampling recorded few fishes, and we were also unable to detect any megafauna, supporting the assertion that although universal markers provide great overall snapshots of biodiversity, their resolution within certain taxonomic groups can be poor (Stat et al. 2017). Indeed, fish eDNA studies have largely employed mitochondrial primers such as 12S or 16S because they allow for more specificity (Miya et al. 2015), whereas nuclear universal primers such as 18S often provide lower resolution but very wide taxonomic coverage (Deagle et al. 2014). For example, at Coral Bay, earlier work by Stat et al. (2017) identified only two operational taxonomic units (OTUs) corresponding to Actinopterygii fishes using 18S. For this reason, it is not unexpected that we also observed limited detectability of fishes. Additionally, DNA shedding rates vary not only among species but also across taxonomic groups, impacting the probability of detection of different taxa within an eDNA sample (Wood et al. 2020). Nonetheless, one of the key advantages of eDNA metabarcoding is that samples can be analysed again at a later date using a different suite of genetic markers, thus offering the possibility to retrospectively target specific taxa.
Inadequate sampling effort can critically influence the results obtained with eDNA metabarcoding (Juhel et al. 2020; Gold et al. 2021; Marwayana et al. 2021; West et al. 2021). ASV accumulation curves showed that our sampling effort was insufficient at capturing total estimated gamma diversity across Ningaloo Reef, and that overall, sample coverage was high but did not approach completeness. To achieve a more complete census, we sampled three sites within each lagoon location (n = 15), but still fell short of obtaining a complete picture of the estimated biodiversity for this reef zone. These results are typical of other eDNA metabarcoding surveys of species diversity in tropical ecosystems such as the Coral Triangle or Cocos Keeling Islands (Juhel et al. 2020; Marwayana et al. 2021; West et al. 2021). Since the majority of eDNA studies have been focussed on temperate marine ecosystems that typically harbour lower levels of species diversity (Port et al. 2016; Jeunen et al. 2018; Gold et al. 2021), it is likely that sampling protocols must be refined for highly diverse tropical ecosystems (Juhel et al. 2020; Marwayana et al. 2021; Stauffer et al. 2021). The tropical fringing reef ecosystem surveyed here will almost certainly contain assemblages of resident and transient species, and the transient component will complicate attempts to provide a comprehensive snapshot of diversity (Magurran and Henderson 2003). Transient, low-abundant species have low detectability, and their presence may even change over the timescale of an eDNA survey, making the complete sampling of gamma biodiversity very unlikely.
It can be beneficial to implement pilot studies to estimate optimal sampling intensity prior to undertaking large scale eDNA surveys, however that is not always possible. Another alternative to increased sampling (and higher costs) is to filter higher volumes of seawater since oligotrophic systems such as reefs do not usually pose problems related to the clogging of filters due to turbidity (Bessey et al. 2020; Marwayana et al. 2021). Given that replicated seawater samples can harbour distinct signals due to the patchiness of eDNA molecules, increasing sampling volumes will likely lead to better estimates of gamma diversity (Bessey et al. 2020; Stauffer et al. 2021).
Additionally, biodiversity estimates obtained from eDNA techniques are affected by the substrate selected for sampling and combining environmental sample types is recommended (Koziol et al. 2018). For example, sediment samples can yield higher overall biotic richness than seawater and are particularly efficient at detecting benthic taxa such as polychaete worms (Koziol et al. 2018). A recent study revealed that “ambient” seawater samples collected above the substrate contained different species assemblages than “crevice” seawater samples collected from within the reef matrix, and that an analysis focussed only on ambient seawater may not adequately characterize the presence of cryptic reef species such as chitinous or calcifying organisms (Nichols et al. 2021). For this reason, if the main aim of an eDNA survey is to maximize the biodiversity recovered from coral reefs, it will likely require sampling across multiple habitat types and/or reef zones, at multiple depths, and include a variety of substrate types.
Conclusion
Surveying coral reef biodiversity across a broad taxonomic spectrum over time will allow a better understanding of how reefs and their associated biota will react to changing impacts and conditions. We conducted a biodiversity survey using eDNA RUMS and found that despite high physical connectivity among adjacent habitats, metabarcoding of seawater eDNA could reveal strong differences in community composition. The implementation of eDNA metabarcoding into monitoring schemes, using either universal or multi-assay approaches, offers managers with a non-invasive, effective, and synoptic way to monitor biodiversity across habitats and trophic levels. With increasing anthropogenic impacts to threatened marine ecosystems, eDNA metabarcoding can provide a way to rapidly monitor post-disturbance effects to inform adaptive management responses.
Data availability
Raw MiSeq sequence data (fastq files), demultiplex indices file, metadata and binary ASV and taxonomic matrices are available at Zenodo.org (https://doi.org/10.5281/zenodo.5792468).
References
Antich A, Palacin C, Cebrian E, Golo R, Wangensteen OS, Turon X (2020) Marine biomonitoring with eDNA: Can metabarcoding of water samples cut it as a tool for surveying benthic communities? Mol Ecol 00:1–14. https://doi.org/10.1111/mec.15641
Ateweberhan M, Feary DA, Keshavmurthy S, Chen A, Schleyer MH, Sheppard CRC (2013) Climate change impacts on coral reefs: synergies with local effects, possibilities for acclimation, and management implications. Mar Pollut Bull 74:526–539. https://doi.org/10.1016/j.marpolbul.2013.06.011
Babcock RC, Thomson DP, Haywood MDE, Vanderklift MA, Pillans R, Rochester WA, Miller M, Speed CW, Shedrawi G, Field S, Evans R, Stoddart J, Hurley TJ, Thomson A, Gilmour J, Depczynski M (2021) Recurrent coral bleaching in north-western Australia and associated declines in coral cover. Mar Freshw Res 72:620–632. https://doi.org/10.1071/MF19378
Baker AC, Glynn PW, Riegl B (2008) Climate change and coral reef bleaching: an ecological assessment of long-term impacts, recovery trends and future outlook. Estuarine and Coastal Shelf Science 80:435–471. https://doi.org/10.1016/j.ecss.2008/09.003
Bakker J, Wangensteen OS, Baillie C, Buddo D, Chapman DD, Gallagher AJ, Guttridge TL, Hertler H, Mariana S (2019) Biodiversity assessment of tropical shelf eukaryotic communities via pelagic eDNA metabarcoding. Ecol Evol 9:14331–14355. https://doi.org/10.1002/ece3.5871
Baselga A, Orme CDL (2012) Betapart: An R package for the study of beta diversity. Methods Ecol Evol 3:808–812. https://doi.org/10.1111/j.2014-210X.2012.00224.x
Bessey C, Jarman SN, Berry O, Olsen YS, Bunce M, Simpson T, Power M, McLaughlin J, Edgar GJ, Keesing J (2020) Maximizing fish detection with eDNA metabarcoding. Environmental DNA 2:493–504. https://doi.org/10.1002/edn3.74
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13:581–583. https://doi.org/10.1038/nmeth.3869
Carvalho S, Aylagas E, Villalobos R, Kattan Y, Berumen M, Pearman JK (2019) Beyond the visual: using metabarcoding to characterize the hidden reef cryptobiome. Proc R Soc B. https://doi.org/10.1098/rspb.2018.2697
Cassata L, Collins LB (2008) Coral reef communities, habitats and substrates in and near sanctuary zones of Ningaloo Marine Park. J Coastal Res 24:139–151. https://doi.org/10.2112/05-0623.1
Chao A, Gotelli NJ, Hsieh TC, Sander EL, Ma KH, Colwell RK, Ellison AM (2014) Rarefaction and extrapolation with Hill numbers: a framework for sampling and estimation in species diversity studies. Ecological Monographs 84:45–67. https://doi.org/10.1890/13-0133.1
Chao A, Chiu CH (2014) Species richness: estimation and comparison (pp. 1–26). Wiley StatsRef: Statistics Reference Online
Deagle BE, Jarman SN, Coissac E, Pompanon F, Taberlet P (2014) DNA metabarcoding and the cytochrome c oxidase subunit 1 marker: not a perfect match. Biol Let. https://doi.org/10.1098/rsbl.2014.0562
Depczynski M, Gilmour J, Ridgway T, Barnes H, Heyward A, Holmes T, Moore J, Radford B, Thomson DP, Tinkler P (2013) Bleaching, coral mortality and subsequent survivorship on a West Australian fringing reef. Coral Reefs 32:233–238. https://doi.org/10.1007/s00338-012-0974-0
Descombes P, Wisz MS, Leprieur F, Parravicini V, Heine C, Olsen SM, Swingedouw D, Kulbicki M, Mouillot D, Pellissier L (2015) Forecasted coral reef decline in marine biodiversity hotspots under climate change. Glob Change Biol 21:2479–2487. https://doi.org/10.1111/gcb.12868
DiBattista JD, Reimer JD, Stat M, Masucci GD, Biondi P, De Brauwer M, Bunce M (2019) Digging for DNA at depth: rapid universal metabarcoding surveys (RUMS) as a tool to detect coral reef biodiversity across a depth gradient. Peer Journal. https://doi.org/10.7717/peerj.6379
Dray S, Dufour A-B (2007) Theade4 Package: Implementing the duality diagram for ecologists. J Stat Softw. https://doi.org/10.18637/jss.v022.i04
Eble JA, Daly-Engel TS, DiBattista JD, Koziol A, Gaither MR (2020) Chapter Two - Marine environmental DNA: Approaches, applications and opportunities. Adv Mar Biol 86:141–169. https://doi.org/10.1016/bs.amb.2020.01.001
Frøslev TG, Kjoller R, Bruun HH, Ejrnaes R, Brunbjerg AK, Pietroni C, Hansen AJ (2017) Algorithm for post-clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nat Commun 8:1188–1199. https://doi.org/10.1038/s41467-017-01312-x
Gelis ERE, Kamal MM, Subhan B, Bachtiar I, Sani LMI, Madduppa. (2021) Environmental biomonitoring of reef fish community structure with eDNA metabarcoding in the Coral Triangle. Environ Biol Fishes 8:887–903. https://doi.org/10.1007/s10641-021-01118-3
Gilmour J, Cook KL, Ryan NM, Puotinen ML, Green RH, Shedrawi G, Hobbs J-PA, Thomson DP, Babcock RC, Buckee J, Foster T, Richards ZT, Wilson SK, Barnes PB, Coutts TB, Radford BT, Piggott CH, Depczynski M, Evans SN, Schoepf V, Evans RD, Halford AR, Nutt CD, Bancroft KP, Heyward AJ, Oades D (2019) The state of Western Australia’s coral reefs. Coral Reefs 38:651–667. https://doi.org/10.1007/s00338-019-01795-8
Gold Z, Sprague J, Kushner DJ, Marin EZ, Barber PH (2021) eDNA metabarcoding as a biomonitoring tool for marine protected areas. PLoS ONE. https://doi.org/10.1371/journal.pone.0238557
Gong S, Jin X, Ren L, Tan Y, Xia X (2020) Unravelling heterogeneity of coral microbiome assemblages in tropical and subtropical corals in the South China Sea. Microorganisms. https://doi.org/10.3390/microorganisms8040604
Greenfield P (2017) Greenfield Hybrid Analysis Pipeline GHAP. Software Collection, CSIRO. https://doi.org/10.4225/08/59f98560eba25
Hsieh TC, Ma KH, Chao A (2020) iNEXT: iNterpolation and EXTrapolation for species diversity. R package version 2.0.20. http://chao.stat.nthu.edu.tw/wordpress/software-download/
Hughes TP, Anderson KD, Connolly SR, Heron SF, Kerry JT, Lough JM, Baird AH, Baum JK, Berumen ML, Bridge TC, Claar DC, Eakin MC, Gilmour JP, Graham NAJ, Harrison H, Hobbs J-PA, Hoey AS, Hoogenboom M, Lowe RJ, McCulloch MT, Pandolfi JM, Pratchett M, Schoepf V, Torda G, Wilson SK (2018) Spatial and temporal patterns of mass bleaching of corals in the Anthropocene. Science 359:80–83. https://doi.org/10.1126/science.aan8048
Ip YCA, Tay YC, Chang JJM, Ang HP, Tun KPP, Chou LM, Huang D, Meier R (2021) Seeking life in sedimented waters: Environmental DNA from diverse habitat types reveals ecologically significant species in a tropical marine environment. Environmental DNA 3:654–668. https://doi.org/10.1002/edn3.162
Jeunen G, Knapp M, Spencer HG, Lamare MD, Taylor HR, Stat M, Bunce M, Gemmell NJ (2018) Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Mol Ecol Resour 19:426–438. https://doi.org/10.1111/1755-0998.12982
Jeunen G, Lamare MD, Knapp M, Spencer HG, Taylor HR, Stat M, Bunce M, Gemmell NJ (2020) Water stratification in the marine biome restricts vertical environmental DNA (eDNA) signal dispersal. Environmental DNA 2:99–111. https://doi.org/10.1002/edn3.49
Johansson CL, Bellwood DR, Depczynski M (2010) Sea urchins, macroalgae and coral reef decline: a functional evaluation of an intact reef system, Ningaloo. Western Australia Marine Ecology Progress Series 414:65–74. https://doi.org/10.3354/MEPS08730
Jones GP, McCormick MI, Srinivasan M, Eagle JV (2004) Coral decline threatens fish biodiversity in marine reserves. Proc Natl Acad Sci 101:8251–8253. https://doi.org/10.1073/pnas.0401277101
Juhel J, Utama RS, Marques V, Vimono IB, Sugeha HY, Pouyaud L, Dejean T, Mouillot D, Hocdé R (2020) Accumulation curves of environmental DNA sequences sequences predict coastal fish diversity in the coral triangle. Proc R Soc B. https://doi.org/10.1098/rspb.2020.0248
Knowlton N, Brainard RE, Fischer R, Moews M, Plaisance L, Caley MJ (2010) Coral reef biodiversity. In: McIntyre AD (ed) Life in the World’s Oceans: Diversity, Distribution, and Abundance. Wiley-Blackwell, Oxford, pp 65–74
Kobryn HT, Wouters K, Beckley LE, Heege T (2013) Ningaloo Reef: Shallow marine habitats mapped using a hyperspectral sensor. PLoS ONE. https://doi.org/10.1371/journal.pone.0070105
Koziol A, Stat M, Simpson T, Jarman S, DiBattista JD, Harvey ES, Marnane M, McDonald J, Bunce M (2018) Environmental DNA metabarcoding studies are critically affected by substrate selection. Mol Ecol Resour 19:366–376. https://doi.org/10.1111/1755-0998.12971
Lin H, Peddada SD (2020) Analysis of composition of microbiomes with bias correction. Nat Commun. https://doi.org/10.1038/s41467-020-17041-7
Magurran AE, Henderson PA (2003) Explaining the excess of rare species in natural species abundance distributions. Nature 422:714–716. https://doi.org/10.1038/nature01547
Mandal S, Van Treuren W, White RA, Eggesbø M, Knight R, Peddada SD (2015) Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb Ecol Health Dis. https://doi.org/10.3402/mehd.v26.27663
Marwayana ON, Gold Z, Meyer CO, Barber PH (2021) Environmental DNA in a global biodiversity hotspot: Lessons from coral reef fish diversity across the Indonesian archipelago. Environmental DNA 00:1–17. https://doi.org/10.1002/edn3.257
Mellin C, Delean S, Caley J, Edgar G, Meekan M, Pitcher R, Przeslawski R, Williams A, Bradshaw C (2011) Effectiveness of biological surrogates for predicting patterns of marine biodiversity: A global meta-analysis. PLoS ONE. https://doi.org/10.1371/journal.pone.0020141
Miya M, Sato Y, Fukunaga T, Sado T, Poulsen JY, Sato K, Minamoto T, Yamamoto S, Yamanaka H, Araki H, Kondoh M, Iwasaki W (2015) MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. Royal Society Open Science. https://doi.org/10.1098/rsos.150088
Monuki K, Barber PH, Gold Z (2021) eDNA captures depth partitioning in a kelp forest ecosystem. PLoS ONE. https://doi.org/10.1371/journal.pone.0253104
Moore JAY, Bellchambers LM, Depczynski MR, Evans RD, Evans SN, Field SN, Friedman KJ, Gilmour JP, Holmes TH, Middlebrook R, Radford BT, Ridgway T, Shedrawi G, Taylor H, Thomson DP, Wilson SK (2012) Unprecedented Mass Bleaching and Loss of Coral across 12 degrees of Latitude in Western Australia in 2010–11. PLoS ONE 7:12–23. https://doi.org/10.1371/journal.pone.0051807
Mousavi-Derazmahalleh M, Stott A, Lines R, Peverley G, Nester G, Simpson T, Zawierta M, De La Pierre M, Bunce M, Christophersen CT (2021) eDNAFlow, an automated, reproducible and scalable workflow for analysis of environmental DNA sequences exploiting Nextflow and Singularity. Mol Ecol Resour 21:1697–1704. https://doi.org/10.1111/1755-0998.13356
Nguyen BN, Shen EW, Seemann J, Correa AMS, O’Donnell JL, Altieri AH, Knowlton N, Crandall KA, Egan SP, McMilan WO, Leray M (2020) Environmental DNA survey captures patterns of fish and invertebrate diversity across a tropical seascape. Sci Rep 10:1–14. https://doi.org/10.1038/s41598-020-63565-9
Nichols PK, Timmers M, Marko PB (2021) Hide ‘n seq: Direct versus indirect metabarcoding of coral reef cryptic communities. Environmental DNA 00:1–15. https://doi.org/10.1002/edn3.203
Oka S, Doi H, Miyamoto K, Hanahara N, Sado T, Miya M (2020) Environmental DNA metabarcoding for biodiversity monitoring of a highly diverse tropical fish community in a coral reef lagoon: Estimation of species richness and detection of habitat segregation. Environmental DNA 00:1–15. https://doi.org/10.1002/edn3.132
Oksanen JF, Blanchet G, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara B, Simpson GL, Solymos P, Stevens MHH, Szoecs E, Wagner H (2020) Vegan: Community Ecology Package. R package version 2.5–7. https://CRAN.R-project.org/package=vegan.
Oliver ECJ, Donat MG, Burrows MT, Moore PJ, Smale DA, Alexander LV, Benthuysen JA, Feng M, Gupta AS, Hobday AJ, Holbrook NJ, Perkins-Kirkpatrick SE, Scannell HA, Straub SC, Wernberg T (2018) Longer and more frequent marine heatwaves over the past century. Nat Commun. https://doi.org/10.1038/s41467-018-03732-9
Pearman JK, Anlauf H, Irigoien X, Carvalho S (2016) Please mind the gap- Visual census and cryptic biodiversity assessment at central Red Sea coral reefs. Mar Environ Res 118:20–30. https://doi.org/10.1016/j.marenvres.2016.04.011
Plaisance L, Caley MJ, Brainard RE, Knowlton N (2011) The diversity of coral reefs: What are we missing? PLoS ONE. https://doi.org/10.1371/journal.pone/0025026
Pochon X, Bott NJ, Smith KF, Wood SA (2013) Evaluating detection limits of next-generation sequencing for the surveillance and monitoring of international marine pests. PLoS ONE. https://doi.org/10.1371/journal.pone.0073935
Port JA, O’Donnell JL, Romero-Maraccini OC, Leary PR, Litvin SY, Nickols KJ, Yamahara KM, Kelly RP (2016) Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol Ecol 25:527–541. https://doi.org/10.1111/mec.13481
Pratchett MS, Hoey AS, Wilson SK, Messmer V, Graham NAJ (2011) Changes in biodiversity and functioning of reef fish assemblages following coral bleaching and coral loss. Diversity 3:424–452. https://doi.org/10.3390/d3030424
Rädecker N, Pogoreutz C, Voolstra CR, Wiedenmann J, Wild C (2015) Nitrogen cycling in coral: the key to understanding holobiont functioning? Trends Microbiol 23:490–497. https://doi.org/10.1016/j.tim.2015.03.008
Richardson LE, Graham NAJ, Hoey AS (2020) Coral species composition drives key ecosystem function on coral reefs. Proc R Soc B. https://doi.org/10.1098/rspb.2019.2214
Seymour M, Edwards FK, Cosby BJ, Bista I, Scarlett PM, Brailsford FL, Glanville HC, de Bruyn M, Carvalho GR, Creer S (2021) Environmental DNA provides higher resolution assessment of riverine biodiversity and ecosystem function via spatio-temporal nestedness and turnover partitioning. Communications Biology. https://doi.org/10.1038/s42003-021-02031-2
Stat M, Huggett MJ, Bernasconi R, DiBattista JD, Berry TE, Newman SJ, Harvey ES, Bunce M (2017) Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical environment. Sci Rep 7:12–23. https://doi.org/10.1038/s41598-017-12501-5
Stauffer S, Jucker M, Keggin T, Marques V, Andrello M, Bessudo S, Cheutin M, Borrero-Pérez GH, Richards E, Dejean T, Hocdé R, Juhel J, Ladino F, Letessier TB, Loiseau N, Maire E, Mouillot D, Martinezguerra MM, Manel S, Fernández AP, Valentini A, Velez L, Albouy C, Pellissier L, Waldock C (2021) How many replicates to accurately estimate fish biodiversity using environmental DNA on coral reefs? Ecol Evol 00:1–14. https://doi.org/10.1002/ece3.8150
Thomson DP, Babcock RC, Haywood MDE, Vanderklift MA, Pillans RD, Bessey C, Cresswell AK, Orr M, Boschetti F, Wilson SK (2020) Zone specific trends in coral cover, genera and growth-forms in the World-Heritage listed Ningaloo Reef. Mar Environ Res. https://doi.org/10.1016/j.marenvres.2020.105020
Vanderklift MA, Babcock RC, Barners PB, Cresswell AK, Feng M, Haywood MDE, Holmes TH, Lavery PS, Pillans RD, Smallwood CB, Thomson DP, Tucker AD, Waples K, Wilson SK (2020) The oceanography and marine ecology of Ningaloo, a World Heritage area. Oceanogr Mar Biol Annu Rev 58:143–178. https://doi.org/10.1201/9780429351495-4
West KM, Stat M, Harvey ES, Skepper CL, DiBattista JD, Richards ZT, Travers MJ, Newman SJ, Bunce M (2021) eDNA metabarcoding survey reveals fine-scale coral reef community variation across a remote, tropical island ecosystem. Mol Ecol 29:1069–1086. https://doi.org/10.1111/mec.15382
Wickham H (2016) ggplot2: Elegant graphics for data analysis. Springer-Verlag New York. https://ggplot2.tidyverse.org
Wood SA, Biessy L, Latchford JL, Zaiko A, von Ammon U, Audrezet F, Cristescu ME, Pochon X (2020) Release and degradation of environmental DNA and RNA in a marine system. Sci Total Environ. https://doi.org/10.1016/j.scitotenv.2019.135314
Zimmerman TL, Martin JW (2004) Artificial reef matrix structures (Arms) An inexpensive and effective method for collecting coral reef-associated invertebrates. Gulf and Caribbean Research. 16:59–64. https://doi.org/10.18785/grc.1601.08
Acknowledgements
We are grateful to the staff at TrEnD at Curtin University for their assistance with laboratory work and the crew of the Australian Institute of Marine Science RV Solander for their assistance in the field. We thank OOID Scientific for Fig. 1 illustration. We also acknowledge the Baiyungu, Thalanyji and Yinigurdira people, the traditional custodians of the land and waters on which field work at Nyinggulu (Ningaloo) was conducted and pay respects to their Elders, past, present, and emerging. Lastly, we thank the anonymous reviewers who kindly provided thoughtful and detailed feedback that resulted in an improved manuscript.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. This work was supported by the Australian Institute of Marine Science, with some funds provided by Santos Energy Ltd. Partial financial support was received from the Jock Clough Marine Foundation for laboratory analyses. The authors have no relevant financial or non-financial competing interests to disclose.
Author information
Authors and Affiliations
Contributions
LT and LD designed the study. LT and MM secured funding. LD, LT, AM, RL, TS, and JC contributed to the field work. LD conducted laboratory work with assistance from RL and TS. LD analysed the data and drafted the manuscript, with assistance from LT, SJ, and MM. All authors contributed to writing, reviewing, and editing of the final manuscript.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Topical Editor Steve Vollmer
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dugal, L., Thomas, L., Meenakshisundaram, A. et al. Distinct coral reef habitat communities characterized by environmental DNA metabarcoding. Coral Reefs 42, 17–30 (2023). https://doi.org/10.1007/s00338-022-02301-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-022-02301-3