
Abstract
Three species of giant barrel sponge are currently recognized in two distinct geographic regions, the tropical Atlantic and the Indo-Pacific. In this study, we used molecular techniques to study populations of giant barrel sponges across the globe and assessed whether the genetic structure of these populations agreed with current taxonomic consensus or, in contrast, whether there was evidence of cryptic species. Using molecular data, we assessed whether giant barrel sponges in each oceanic realm represented separate monophyletic lineages. Giant barrel sponges from 17 coral reef systems across the globe were sequenced for mitochondrial (partial CO1 and ATP6 genes) and nuclear (ATPsβ intron) DNA markers. In total, we obtained 395 combined sequences of the mitochondrial CO1 and ATP6 markers, which resulted in 17 different haplotypes. We compared a phylogenetic tree constructed from 285 alleles of the nuclear intron ATPsβ to the 17 mitochondrial haplotypes. Congruent patterns between mitochondrial and nuclear gene trees of giant barrel sponges provided evidence for the existence of multiple reproductively isolated species, particularly where they occurred in sympatry. The species complexes in the tropical Atlantic and the Indo-Pacific, however, do not form separate monophyletic lineages. This rules out the scenario that one species of giant barrel sponge developed into separate species complexes following geographic separation and instead suggests that multiple species of giant barrel sponges already existed prior to the physical separation of the Indo-Pacific and tropical Atlantic.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There has been much controversy over the processes driving evolution and speciation in marine environments (e.g., Mayr 1942; Rocha and Bowen 2008). Physical barriers are less obvious in seas and oceans than on land, and many marine organisms have long-range dispersal capabilities during early life stages. Taken together, these factors were believed to lead to fewer opportunities for allopatric speciation compared to terrestrial ecosystems (Palumbi 1997; Rocha and Bowen 2008). However, this is inconsistent with the high biodiversity found in coral reefs, which rivals numbers found in tropical rainforests (Reaka-Kudla et al. 1997). Coral reefs are currently among the most vulnerable of ecosystems (Bridge et al. 2013) due to the combined threat of climate change and anthropogenic stressors including pollution and overfishing (Hughes 1994; Pandolfi et al. 2005). It is, therefore, important to study and quantify the diversity of these systems and understand the evolutionary processes that have led to this diversity.
Marine speciation does not fundamentally differ from terrestrial speciation, but ecological partitions among populations are believed to be more important in the former, whereas geographic partitions are more important in the latter (Bowen et al. 2013). An increasing number of examples of non-allopatric speciation along ecological gradients (reviewed in Bowen et al. 2013) illustrate the evolutionary potential of tropical marine environments. Furthermore, numerous phylogenetic studies have provided evidence of cryptic species, i.e., species that are indistinguishable from congenerics in morphology and spatial distribution, but that are clearly differentiated genetically (Bickford et al. 2007). In contrast, certain species show strong genetic connectivity at a global scale (Horne et al. 2008; Reece et al. 2011) despite apparent morphological variation (Rocha et al. 2005).
Correct identification of species is a fundamental part of conservation and management, and misidentification of cryptic species may impair conservation efforts (Robinson et al. 2014). Genetic markers have become increasingly important tools to identify divergent cryptic species and have forced the rejection of the long-believed assumption of cosmopolitan distribution of certain species (Boury-Esnault et al. 1992; Knowlton 1993; Klautau et al. 1999). Molecular techniques have also helped to reconstruct the distributional patterns of invasive species, which have become major drivers of ecosystem change due to the increase in global shipping (Concepcion et al. 2010; Teske et al. 2011). Most studies that have focused on the distribution and evolution of marine species cover small spatial scales and become more useful when they are compared to more wide-ranging studies (Briggs and Bowen 2013; Cowman and Bellwood 2013a). A focus on wide-ranging studies within each marine phylum should therefore be a priority for the scientific community.
Sponges (Porifera) are an animal group with a relatively simple morphology and often pronounced morphological plasticity (Knowlton 2000). Hence, they can be notoriously difficult to identify to species or even to a higher taxonomic level due to the lack of reliable morphological markers (Knowlton 2000). They are considered the oldest multicellular animal lineage (van Soest et al. 2012), having evolved more than 500 million yr ago (Love et al. 2009; Maloof et al. 2010), and are widespread in many aquatic systems. On tropical reefs, sponge diversity and abundance can be higher than that of corals (Diaz and Rützler 2001). Unfortunately, this large and important animal group has long been understudied in coral reef ecology (Diaz and Rützler 2001). Most genetic studies of sponges have indicated the existence of cryptic species and refuted ocean-wide distributions of several taxa (Duran and Rützler 2006; Swierts et al. 2013; Bell et al. 2014; Knapp et al. 2015). However, these studies were done at small spatial scales, and, to the best of our knowledge, there has been no global phylogenetic study of any sponge taxon.
Giant barrel sponges (genus Xestospongia, family Petrosiidae, order Haplosclerida) are widely distributed throughout multiple tropical oceans. Giant barrel sponges are large and long-lived and have therefore been nicknamed ‘the redwoods of the reef’ (McMurray et al. 2008). These conspicuous sponges can measure up to a base diameter of more than 2.5 m (Nagelkerken et al. 2000) and can cover up to 9% of some reefs (Zea 1993); one specimen from Curaçao was estimated to be over 2300 yr old (Nagelkerken et al. 2000). Three species have been described, with the species delineation mainly based on geographic distributions. Xestospongia muta occurs in the tropical Atlantic, X. testudinaria in the Indo-Pacific from the Red Sea to Taiwan and X. bergquistia is thought to be confined to inshore environments in northern Australia where it lives in sympatry with X. testudinaria. Recent molecular studies have suggested that these species delineations are incorrect and that both X. muta and X. testudinaria consist of multiple sympatric species that apparently do not interbreed (Swierts et al. 2013; Bell et al. 2014). This has important implications for a number of published studies on the demography and population genetics of giant barrel sponges which assumed a single population of giant barrel sponge (López-Legentil and Pawlik 2009; McMurray et al. 2010; Richards et al. 2016).
The congruent identification of a phylogenetic lineage by multiple unlinked genetic loci indicates that it is genetically isolated from other such lineages, and thus qualifies as a species, because only in separate species will the coalescent histories of the different markers agree (Avise and Ball 1990; Coyne and Orr 2004; Padial et al. 2010). Nuclear DNA (nDNA) evolves independently from mitochondrial DNA (mtDNA); thus, congruent patterns across these markers support the existence of biological species (Goetze 2010; Padial et al. 2010). In sponges, mitochondrial variation is typically low (Wörheide et al. 2005), but previous studies of giant barrel sponges have shown that the combination of the adenosine triphosphate synthase subunit 6 gene (ATP6) with the I3-M11 partition of the cytochrome oxidase 1 gene (CO1) was informative (Rua et al. 2011; Swierts et al. 2013). The nuclear adenine triphosphate synthesis-β intron (ATPsβ) is very variable in giant barrel sponges, and because it is unlinked to the mtDNA, it serves as a good additional marker to identify potential species (Bentlage and Wörheide 2007; Swierts et al. 2013).
Molecular studies on giant barrel sponges using these mtDNA and nDNA markers have revealed some interesting results. For example, some haplotypes of the I3-M11 partition of the CO1 gene are shared between Indonesia (X. testudinaria) and Florida (X. muta); hence, two individuals from different ocean basins can be more closely related for this slowly evolving gene than two sympatric individuals on the same reef (Swierts et al. 2013; Setiawan et al. 2016). Giant barrel sponges from the tropical Atlantic and the Indo-Pacific sharing the same CO1 haplotype have both been related to an exterior morphology consisting of digitate structures (López-Legentil and Pawlik 2009; Swierts et al. 2013). These studies imply that the giant barrel sponge is a classic example of a tropical marine animal in which poor identification at the species level has led to an oversimplified taxonomic classification. Due to its conspicuousness, geographic range and available genetic markers, this group of sponges is suitable as a model for global sponge evolution and phylogeography. A better understanding of these species helps in our understanding of the evolutionary history of tropical marine species in general and marine sponges in particular, which is essential to our understanding of marine diversity.
In this study, we sequenced giant barrel sponges from reefs across the globe for a combination of the mtDNA genes ATP6 and CO1 and the nDNA intron ATPsβ. The first aim of this study was to assess how many species of giant barrel sponge are present globally and how they are distributed. Our second aim was to test whether the giant barrel sponges in the tropical Atlantic and the Indo-Pacific represent two monophyletic lineages. If this is the case, it would suggest that one species of giant barrel sponge in each ocean basin independently developed into different species and/or species complexes. However, if sponges do not form two distinct monophyletic groups in different ocean basins, it suggests that a species complex already existed prior to the ocean basins becoming separated. This information provides insight into genetic divergence among tropical reefs before physical barriers impeded gene flow between the Indo-Pacific and tropical Atlantic.
Materials and methods
Giant barrel sponges (Xestospongia spp.) were collected by SCUBA diving from 17 different locations (Table 1; Fig. 1). Sponge tissue for DNA extraction was immediately stored in absolute ethanol (98%) in a cool box. After 6–12 h, the ethanol was changed and samples were stored at −20 °C. Fifty-four sponge samples from Lembeh Island, Indonesia, were previously described in Swierts et al. (2013), but amplification and sequencing were repeated in this study to confirm haplotype assignment.

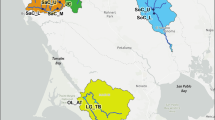
Location maps with haplotype frequencies of the mitochondrial DNA genes CO1 and ATP6 of giant barrel sponges. SE = Sint-Eustatius, the Netherlands; Cur = Santa Barbara, Curaçao; Sau = Jeddah, Saudi-Arabia; Tan = Dar es Salaam, Tanzania; May = Mayotte, France; Pat = Pattaya, Thailand; PQ = Phu Quoc, Vietnam; Koh = Koh Tao, Thailand; Tio = Tioman Island, Malaysia; Sin = St. John’s Island, Singapore, Jak = Jakarta Bay and Thousand Islands, Indonesia; Phu = Phuket, Thailand; Tai = Taiwan; HB = Halong Bay, Vietnam; Der = Derawan Islands, Indonesia; Lem = Lembeh Island, Indonesia; Spe = Spermonde Archipelago, Indonesia
DNA was extracted from sponge tissue using the DNeasy Blood and Tissue kit (Qiagen) following the manufacturer’s instructions. We sequenced 395 samples for a combination of the mitochondrial CO1 (Erpenbeck et al. 2002) and ATP6 genes (Rua et al. 2011).
For the CO1 gene, we used the primers C1-J2165 (5′-GAAGTTTATATTTTAATTTTACCDGG-3′) and C1-Npor2760 (5′-TCTAGGTAATCCAGCTAAACC-3′), which amplified a fragment of 544 base pairs (bp). Amplification was performed in a 25 µL total reaction volume with 15.5 µL sterile water, 5 µL dNTPs (2.5 mM), 2.5 µL coralload buffer (Qiagen), 0.4 µL of each primer (10 µM), 0.25 µL taq polymerase (Qiagen) and 1 µL DNA template (20 ng µL−1). For the ATP6 gene, we used the primers ATP6porF (5′-GTAGTCCAGGATAATTTAGG-3′) and ATP6porR (5′-GTTAATAGACAAAATACATAAGCCTG-3′), which amplified a product of 445 bp. Amplification was performed in a 25 µL total reaction volume with 14 µL sterile water, 5 µL dNTPs (2.5 mM), 2.5 µL coralload buffer (Qiagen), 1.5 µL BSA (Promega), 0.4 µL (10 µM) of each primer, 0.25 µL taq polymerase (Qiagen) and 1 µL DNA template (20 ng µL−1). For both genes, we used a PCR protocol that consisted of an initial denaturing step (95 °C for 5 min), followed by 35 cycles of denaturing (95 °C for 30 s), annealing (42 °C for 45 s) and extension (68 °C for 1.30 min), and a final extension step (72 °C for 10 min) executed in a T100 thermal cycler (Bio-Rad).
To test for congruent patterns at an independent genetic locus, the ATPsβ nuclear intron was amplified for a subset of 211 samples following Jarman et al. (2002). For this gene, we used the primers ATPSß-F (5′-ATGAGATGATCACATCAGGTG-3′) and ATPSß-R (5′-GGTTCGTTCATCTGTCC-3′), which amplified products in the range of 258–279 bp. Amplification was performed in a 25 µL total reaction volume with 14.55 µL sterile water, 4.2 Ll dNTPs (2.5 mM), 2.6 µL buffer (Qiagen), 1.6 Ll BSA (Promega) 0.4 µL of each primer (10 µM), 0.25 µL taq polymerase (Qiagen) and 1 µL DNA template (20 ng µL−1). The PCR protocol consisted of an initial denaturing step (95 °C for 5 min), followed by 35 cycles of denaturing (95 °C for 30 s), annealing (45 °C for 30 s) and extension (72 °C for 45 s), and a final extension step (72 °C for 4 min) executed in a T100 thermal cycler from Bio-Rad. All PCR products were sequenced in both directions by BaseClear, Leiden, the Netherlands or Macrogen Europe, Amsterdam, the Netherlands.
Sequences were checked using CodonCode Aligner version 3.7.1.2 (CodonCode Corporation). Double peaks were called when the height of the secondary peak was at least 60% of that of the primary peak in both the forward and reverse sequence reads. Samples that contained two nucleotide positions with double peaks were reconstructed using DnaSP v5.10.01 with the PHASE v2.1 algorithm (Stephens et al. 2001). Only reconstructed haplotypes with probabilities >0.9 were used for further analysis. Samples that contained many double peaks may have represented mixtures of multiple sequences and were therefore cloned using the pGEM-T Easy kit (Promega Corporation) or the TOPO-TA cloning kit (Thermo Fisher Scientific), following the manufacturers’ protocols. Primer sequences were trimmed of the final sequences, and alignments were obtained using ClustalW (Larkin et al. 2007) in Geneious v9.04 (Kearse et al. 2012) for both the combined mtDNA and single nDNA markers using the default software settings.
We made separate statistical parsimony networks for the combined mtDNA sequences (CO1 + ATP6) and the nDNA sequences with TCS v 1.21 (Clement et al. 2000). A maximum likelihood phylogenetic tree was constructed for the ATPsß-intron in Geneious using the PHYML plugin (Guindon et al. 2010) with the GTR model, which was the best fit model according to jModelTest2 (Guindon and Gascuel 2003; Darriba et al. 2012) based on the Akaike information criterion (Akaike 1974). The number of bootstrap replications was set at 1000. We also calculated Bayesian support values with MrBayes 3.2.6 (Huelsenbeck and Ronquist 2001; Ronquist and Huelsenbeck 2003). The analysis of every gene consisted of two independent runs of four Metropolis-coupled Markov chains, sampled at every 1,000th generation. Analyses were terminated after the chains converged significantly as indicated by an average standard deviation of split frequencies <0.01. Trees were visualized with FigTree v1.4.2 (Morariu et al. 2009).
Individuals were grouped based on a combination of mtDNA, nDNA and the geographic origin of the sample. Mean genetic distance was calculated between these groups for the mtDNA and nDNA genes in MEGA 7.0.21 (Kumar et al. 2016) using the Tamura–Nei model (Tamura and Nei 1993) with standard settings. We conducted an automated barcoding gap discovery (ABGD) analysis with standard settings to split our sequences into candidate species and compare those to our identified groups based on congruence between mtDNA, nDNA and geography (Puillandre et al. 2012).
Results
We obtained a total of 395 combined sequences of partial mitochondrial CO1 and ATP6 genes. In the final alignment of 989 base pairs, we found 13 variable sites: six were located in the CO1 gene and seven in the ATP6 gene, resulting in 17 different haplotypes (Table 2). Seven of the nine CO1-haplotypes (C1–C9) previously submitted to GenBank (López-Legentil and Pawlik 2009; Swierts et al. 2013; Setiawan et al. 2016) were present in this dataset. Re-analysis of the sample carrying the C3 haplotype from Lembeh Island showed that the sample was C2, and hence, this haplotype was wrongly identified (Swierts et al. 2013). Haplotype C7, described by Setiawan et al. (2016) from one sample from Tanzania, was not found in our dataset, and no new haplotypes were found for the CO1 gene. For the ATP6 gene, only three haplotypes were previously known (A1–A3; Swierts et al. 2013) and six new haplotypes were identified (A4–A9; GenBank accession numbers: KY381287–KY381292). Adding this gene to the CO1 gene expanded the number of haplotypes in our dataset from seven (C1, C2, C4–C6, C8, C9) to seventeen (Table 2).
As is common in sponges (Wörheide et al. 2005), mitochondrial variation was low (π = 0.0032). With one exception, connected haplotypes in the statistical parsimony network were differentiated by a single mutation (Fig. 2).

Haplotype network of the mitochondrial DNA genes CO1 and ATP6 of giant barrel sponges. Pie chart size is relative to the number of individuals with that haplotype. Colors indicate regions of origin. Lines connecting haplotypes represent one base substitution between two haplotypes; additional crossbars indicate an additional base substitution each. Green tropical Atlantic; red western Indian Ocean; yellow Red Sea; blue central Indo-Pacific
The tropical Atlantic haplotypes C2A5 and C9A5 are located at the opposite end of the network compared to the only other tropical Atlantic haplotype C8A2 and separated by 11 mutational steps. All 11 sequences from the Red Sea were identical and unique for the region (C5A7). In the western Indian Ocean, we found five different haplotypes in 21 sponges that were spread over the haplotype network; three of these haplotypes were only present in this region. Six haplotypes from the Indo-Pacific were represented by more than 25 individuals in our dataset, and the majority of these haplotypes were widespread and occurred at multiple sampling sites. Haplotype C5A2 was found in the central Indo-Pacific, but also in the tropical Atlantic. The four regions in which we sampled (central Indo-Pacific, tropical Atlantic, Red Sea, western Indian Ocean), which are geographically distant from one another, were characterized by different haplotype compositions, and all hosted unique haplotypes (Fig. 1).
We successfully amplified the nuclear intron ATPsß from 211 individuals. The nuclear intron ATPsß provided much more genetic variation (157 segregating sites; π = 0.0767) than the mitochondrial genes (13 segregating sites; π = 0.0032). A circular phylogenetic tree consisting of 285 alleles (137 homozygotes, 74 heterozygotes) was constructed for the nuclear intron and compared to the 17 different mitochondrial haplotypes (Fig. 3); a larger and more detailed rectangular phylogenetic tree is provided in the Electronic supplementary material, ESM, Fig. S1. All sequences were submitted to GenBank under accession numbers KY381293–KY381577. The nuclear sequences provided much more information than the mitochondrial markers, but were mostly phylogenetically congruent with the mtDNA. While not all branches in the nDNA tree were statistically supported and some mtDNA haplotypes were shared between regions, we could identify multiple groups that potentially operate as reproductively isolated populations. Sponges were assigned to a separate group when they possessed unique mtDNA haplotypes within one of the geographic regions and also formed a separate cluster of unique nuclear alleles (Fig. 3). Based on these criteria, the individuals from the central Indo-Pacific could be separated into three groups: group 1—haplotypes C1A1, C1A8 and C2A1; group 2—haplotypes C4A3 and C4A4; and group 3—haplotypes C5A2, C5A4 and C6A2. In the tropical Atlantic, the nuclear gene tree also contained three groups that were mostly congruent with mitochondrial haplotypes: group 7—haplotypes C2A5 and C9A5; group 8—haplotype C8A2; and group 9—haplotype C5A2. We found one group in the western Indian Ocean with mitochondrial haplotypes C2A8 and C2A9 (group 6), and one group in the Red Sea with mitochondrial haplotype C5A7 (group 5). The only individual with haplotype C5A6 was found in Taiwan; this individual had unique nuclear DNA and did not fit in any of the other three groups of the Indo-Pacific (group 4). Table 3 shows the mean genetic distances between these drafted groups. Mean genetic distance for the nDNA was considerably higher than for the mtDNA. An ABGD analysis on our nuclear data supported the groups 2, 3, 4, 5, 8 and 9 with recursive partitions at a prior maximal distance of 0.0046, while groups 1, 6 and 7 were not supported as separate groups (ESM S2). The statistical parsimony network constructed with TCS from our nuclear data resulted in seven unconnected statistical parsimony networks. Group 8 represented one network, group 3 represented three networks, and the remaining three networks consisted of the combinations of groups 2 and 9, groups 1, 6 and 7, and groups 4 and 5, respectively (ESM S3, S4).

Unrooted circular maximum likelihood tree of 285 alleles of the nuclear DNA gene ATPsβ. Values on branches indicate bootstrap support (only shown when >50) and Bayesian support value (only shown when >0.90). Dots on the branches indicate the number of individuals with that allele, and the colors of the dots indicate the haplotype of the individual for the mitochondrial CO1 and ATP6 haplotypes. Background colors represent geographic origin of the lineages. Abbreviations in the legend of the background colors indicate the current species consensus (XT, Xestospongia testudinaria; XM, Xestospongia muta)
The groups from the tropical Atlantic (7, 8, 9) and the Indo-Pacific (1, 2, 3) were not monophyletic per region for the nuclear marker, but rather were intertwined in a generally congruent pattern with the mtDNA (Fig. 3). They did not share any nuclear alleles, indicating that there has been no recent genetic exchange between giant barrel sponges from these areas. Also, heterozygotes were only found with both alleles within the same nuclear group providing further support that different groups are reproductively isolated (ESM Fig. S1). Note that not all groups are monophyletic in the nDNA tree. In particular, groups 1, 6 and 7 are ‘mixed’ in the tree, but they do not share any alleles. Also, some groups are only partly congruent, for example groups 4 and 5. Both groups show congruent patterns in the sense that they both host unique mtDNA haplotypes and unique nDNA alleles and are closely related to each other for both markers. However, there is an inconsistency in their placement relative to the other groups, since they are most closely related to group 8 in the nDNA phylogenetic tree (Fig. 3) but most closely related to group 3 in the haplotype network for the mtDNA (Fig. 2).
Discussion
Congruent patterns between mtDNA and nDNA markers of giant barrel sponges around the globe point to the existence of multiple genetically isolated taxa and support our hypothesis of the existence of additional species. The genetic differences between the nuclear clades are based on the variation of a single gene and the mitochondrial markers have low variation. Nevertheless, plotting mtDNA haplotypes to the nuclear phylogeny does not result in random distribution. All of the mtDNA haplotypes are confined to one nDNA group within a geographic region, suggesting biological species. Altogether, we identified at least eight potential giant barrel sponge species globally, yet the limitations of the markers make it impossible to exactly determine the number of species with the data presented in this study.
We found three potential giant barrel sponge species in the central Indo-Pacific (groups 1, 2, 3), three in the tropical Atlantic (groups 7, 8, 9), one in the western Indian Ocean (group 6) and one in the Red Sea (group 5). The lack of hybridization between these taxa, especially between those that are sympatric, indicates complete reproductive isolation. A ninth group (group 4) consisted of a single sample from Taiwan. Some genetic groups (e.g., groups 1, 6 and 7) were not statistically supported in the nuclear gene tree, but these could still represent (incipient) species because they occur in different parts of the world’s oceans and are thus geographically isolated. Certain other groups were statistically supported, which is, particularly in combination with their sympatric occurrence, a strong indication for speciation. It is important to note that some discrepancies exist in the results from our statistical analyses. Our own interpretation of nine groups differs from the results from the ABGD and TCS analyses, which both find only seven groups, but with partially different compositions. Also, the inconsistencies of the placement of certain groups relative to other groups illustrate that the phylogenetic relationships between the groups cannot be completely resolved with the combination of markers used in this study. The higher variation on the nDNA marker suggests that the evolutionary history of giant barrel sponges is better represented by this marker; however, additional genetic evidence and a thorough morphological analysis are required to delineate and describe distinct groups as species. Nevertheless, our data do indicate that the current taxonomic consensus with X. muta occurring in the tropical Atlantic and X. testudinaria in the Red Sea, western Indian Ocean and central Indo-Pacific, is incorrect.
Our results suggest the existence of three species in the tropical Atlantic, in line with previous suggestions based on a study of the sterol compositions of giant barrel sponges (Kerr et al. 1991). All CO1 haplotypes of samples from the tropical Atlantic in this analysis were previously described by López-Legentil and Pawlik (2009) from locations in the north (Florida) and west (Belize) of the tropical Atlantic and by de Bakker et al. (2016) from locations in the east (Saba Bank) of the tropical Atlantic. Therefore, these three groups seem to be distributed across the entire tropical Atlantic. Similarly, large geographic ranges can be observed for groups 1, 2 and 3 from the central Indo-Pacific, which have overlapping geographic ranges between locations more than 2000 km apart. Of particular interest is the finding that lineages in a given ocean basin were more closely related to lineages in another ocean basin than to lineages with which they co-occur. For instance, individuals from groups 3 and 9 can share the same mtDNA haplotype and are also closely related in the nuclear gene tree, but are found in the Indo-Pacific and tropical Atlantic, respectively. In other words, different species in each ocean basin do not form separate monophyletic clades. This suggests that distinct species of giant barrel sponges must have existed prior to the most recent physical separation of the tropical Atlantic and the Indo-Pacific.
At present, giant barrel sponges occur in the western Indo-Pacific (including the Red Sea and western Indian Ocean), the central Indo-Pacific and the tropical Atlantic. Giant barrel sponges have not been found in the eastern Pacific and the eastern tropical Atlantic. Hence, the sponges from the tropical Atlantic and the Indo-Pacific are separated by several barriers that have developed at various times throughout history (Cowman and Bellwood 2013b). The Tethys Seaway may have provided a potential migration route throughout the first half of the Cenozoic era between 60 and 30 million yr ago between the tropical Atlantic and the Indo-Pacific (Vermeij 2001; Harzhauser and Piller 2007). At that time, the western Tethys was the center of global marine biodiversity, but this subsequently shifted eastwards to its present location in the Indo-Australian archipelago (Renema et al. 2008). In the early Miocene, the African-Arabian Plate moved northwards to adjoin the Eurasian Plate (Aitchison et al. 2007), dividing the Tethys realm into a western and an eastern part: the ‘Terminal Tethyan Event’ (TTE). Previously, the closure of the Isthmus of Panama, approximately 3 million yr ago (Keigwin 1978, was suggested as the final geographic separation between giant barrel sponges from the tropical Atlantic and the Indo-Pacific (Montalvo and Hill 2011; Swierts et al. 2013). However, since giant barrel sponges do not occur east of New Caledonia, the scenario in which the tropical Atlantic and the Indo-Pacific locations became isolated due to the TTE should also be considered. If isolation followed the TTE, the lineages now living in sympatry must have evolved into different species before the TTE occurred. This could be a result of sympatric speciation within the Tethys Sea, or perhaps due to a series of speciation events from periodic geographic isolation as a result of changing sea levels (Haq et al. 1987).
Reproductive mechanisms can explain how different species of Xestospongia are maintained in sympatry. Sympatric populations of X. testudinaria and X. bergquistia spawn at different times of the year near Australia, possibly triggered by water temperature (Fromont and Bergquist 1994). This is comparable to corals of the Montastrea annularis species complex (van Veghel et al. 1996) in the tropical Atlantic, which are unlikely to interbreed due to a combination of temporal differences in spawning, sperm aging, gamete dispersal and dilution, and gametic incompatibility (Levitan et al. 2004). In the tropical Atlantic, X. muta has been observed to spawn and recruit twice a year, in spring and in late summer (Ritson-Williams et al. 2005; McMurray et al. 2008). However, it is possible that all giant barrel sponges spawn just once a year, and these different times in fact correspond to different mass spawning events of distinct species. Spawning events of giant barrel sponges have also been reported at different times of the year in the Indo-Pacific (Swierts et al. 2013; Röthing and Voolstra 2016).
Some of the previously assumed ‘morphological plasticity’ of giant barrel sponges might actually be morphological differentiation between species. For instance, sponges with haplotype C5 for the CO1 gene are associated with a digitate outer morphology in both the tropical Atlantic and the Indo-Pacific (López-Legentil and Pawlik 2009; Swierts et al. 2013). Other morphological characteristics might also differentiate between ‘cryptic’ species. For example, the spicules of X. muta have a broad size range (length 290–435 µm, width 8.3–23 µm) and vary between geographic locations (van Soest 1980). With possibly three species present in the tropical Atlantic, we have to consider that such a broad size range may be the result of lumping three different spicule size ranges and an uneven distribution of the various cryptic species among locations. Also in the tropical Atlantic, McMurray et al. (2014) found large differences in spongocoel morphology that were independent of sponge size and strongly influenced excurrent seawater velocity. Given that only three species have been described globally so far, scientific priority should be given to resolving this species complex further and differentiating between their potentially distinct ecological roles.
To our knowledge, the intertwined evolutionary history of tropical Atlantic and Indo-Pacific taxa we found for giant barrel sponges has never been found in other benthic reef animals. Usually, there is a general conformity between phylogeography and biogeography in marine animal groups, which suggests that geographic isolation is a starting point for divergences between species (Teske et al. 2011; Bowen et al. 2016). Most global marine species complexes are so-called ‘sibling species complexes’ that developed simultaneously in different ocean basins, potentially followed by cryptic invasions (Schwaninger 2008; Geller et al. 2010). The importance of geographic isolation, possibly related to sea currents, was suggested as a driving force in sponge speciation (DeBiasse et al. 2016). It may also be a driving force for giant barrel sponges, especially considering the groups that are confined to the Red Sea and the western Indian Ocean, which represent distinct biogeographic provinces (Briggs and Bowen 2012; Bowen et al. 2016). However, present-day ocean currents and geographic barriers cannot explain why giant barrel sponges in the tropical Atlantic and the Indo-Pacific do not form monophyletic lineages, and why in each ocean basin multiple genetically isolated lineages exist in sympatry. Trumpet fish represent a so-called ‘global ring species complex’, in which different lineages have come into contact after three to four million years of isolation and appear to be merging (Bowen et al. 2001). Four highly divergent lineages have been identified for vase tunicates; these four were originally limited to a certain geographic range, but two are now considered invasive with global distributions (Zhan et al. 2010). Sponge larvae, however, are generally considered to have low survival under environmental stress, and their transport in the ballast water of ships is unlikely and has not yet been reported (Klautau et al. 1999). Our data do not support any recent invasions of giant barrel sponges from one ocean basin to another and none of the candidate species has a global distribution. Instead, our results show a unique evolutionary history, suggesting intertwined species complexes in different ocean basins. This pattern may, however, be found in other tropical marine species, especially those with a long evolutionary history such as other globally distributed sponge groups. Resolving accurate species boundaries in such groups is important for the conservation of tropical marine ecosystems.
References
Aitchison JC, Ali JR, Davis AM (2007) When and where did India and Asia collide? J Geophys Res Solid Earth 112:B05423
Akaike H (1974) A new look at the statistical model identification. IEEE Trans Automat Contr 19:716–723
Avise JC, Ball RM (1990) Principles of genealogical concordance in species concepts and biological taxonomy. In: Futuyma D, Antonovics J (eds) Oxford surveys in evolutionary biology, vol 7., Oxford University PressOxford, UK, pp 45–67
Bell JJ, Smith D, Hannan D, Haris A, Jompa J, Thomas L (2014) Resilience to disturbance despite limited dispersal and self-recruitment in tropical barrel sponges: implications for conservation and management. PLoS One 9:e91635
Bentlage B, Wörheide G (2007) Low genetic structuring among Pericharax heteroraphis (Porifera: Calcarea) populations from the Great Barrier Reef (Australia), revealed by analysis of nrDNA and nuclear intron sequences. Coral Reefs 26:807–816
Bickford D, Lohman DJ, Sodhi NS, Ng PK, Meier R, Winker K, Ingram KK, Das I (2007) Cryptic species as a window on diversity and conservation. Trends Ecol Evol 22:148–155
Boury-Esnault N, Solé-Cava AM, Thorpe JP (1992) Genetic and cytological divergence between colour morphs of the Mediterranean sponge Oscarella lobularis Schmidt (Porifera, Demospongiae, Oscarellidae). Journal of Natural History 26:271–284
Bowen BW, Rocha LA, Toonen RJ, Karl SA (2013) The origins of tropical marine biodiversity. Trends Ecol Evol 28:359–366
Bowen BW, Bass AL, Rocha LA, Grant WS, Robertson DR (2001) Phylogeography of the trumpetfishes (Aulostomus): ring species complex on a global scale. Evolution 55:1029–1039
Bowen BW, Gaither MR, DiBattista JD, Iacchei M, Andrews KR, Grant WS, Toonen RJ, Briggs JC (2016) Comparative phylogeography of the ocean planet. Proc Natl Acad Sci U S A 113:7962–7969
Bridge TC, Hughes TP, Guinotte JM, Bongaerts P (2013) Call to protect all coral reefs. Nat Clim Chang 3:528–530
Briggs JC, Bowen BW (2012) A realignment of marine biogeographic provinces with particular reference to fish distributions. J Biogeogr 39:12–30
Briggs JC, Bowen BW (2013) Marine shelf habitat: biogeography and evolution. J Biogeogr 40:1023–1035
Clement M, Posada DCKA, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Concepcion GT, Kahng SE, Crepeau MW, Franklin EC, Coles SL, Toonen RJ (2010) Resolving natural ranges and marine invasions in a globally distributed octocoral (genus Carijoa). Mar Ecol Prog Ser 401:113–127
Cowman PF, Bellwood DR (2013a) The historical biogeography of coral reef fishes: global patterns of origination and dispersal. J Biogeogr 40:209–224
Cowman PF, Bellwood DR (2013b) Vicariance across major marine biogeographic barriers: temporal concordance and the relative intensity of hard versus soft barriers. Proc R Soc Lond B Biol Sci 280:20131541
Coyne JA, Orr HA (2004) Speciation. Sinauer Associates, Sunderland, MA
Darriba D, Taboada GL, Doallo R, Posada D (2012) jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772
de Bakker DM, Meesters EH, van Bleijswijk JD, Luttikhuizen PC, Breeuwer JAJ, Becking LE (2016) Population genetic structure, abundance, and health status of two dominant benthic species in the Saba Bank National Park, Caribbean Netherlands: Montastraea cavernosa and Xestospongia muta. PLoS One 11:e0155969
DeBiasse MB, Richards VP, Shivji MS, Hellberg ME (2016) Shared phylogeographical breaks in a Caribbean coral reef sponge and its invertebrate commensals. J Biogeogr 43:2136–2146
Diaz MC, Rützler K (2001) Sponges: an essential component of Caribbean coral reefs. Bull Mar Sci 69:535–546
Duran S, Rützler K (2006) Ecological speciation in a Caribbean marine sponge. Mol Phylogenet Evol 40:292–297
Erpenbeck D, Breeuwer JAJ, van der Velde H, van Soest R (2002) Unravelling host and symbiont phylogenies of halichondrid sponges (Demospongiae, Porifera) using a mitochondrial marker. Mar Biol 141:377–386
Fromont J, Bergquist PR (1994) Reproductive biology of three sponge species of the genus Xestospongia (Porifera: Demospongiae: Petrosida) from the Great Barrier Reef. Coral Reefs 13:119–126
Geller JB, Darling JA, Carlton JT (2010) Genetic perspectives on marine biological invasions. Ann Rev Mar Sci 2:367–393
Goetze E (2010) Species discovery in marine planktonic invertebrates through global molecular screening. Mol Ecol 19:952–967
Guindon S, Gascuel O (2003) A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst Biol 52:696–704
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Haq BU, Hardenbol J, Vail PR (1987) Chronology of fluctuating sea levels since the Triassic. Science 235:1156–1167
Harzhauser M, Piller WE (2007) Benchmark data of a changing sea—palaeogeography, palaeobiogeography and events in the Central Paratethys during the Miocene. Palaeogeogr Palaeoclimatol Palaeoecol 253:8–31
Horne JB, van Herwerden L, Choat JH, Robertson DR (2008) High population connectivity across the Indo-Pacific: congruent lack of phylogeographic structure in three reef fish congeners. Mol Phylogenet Evol 49:629–638
Huelsenbeck JP, Ronquist F (2001) MRBAYES: Bayesian inference of phylogeny. Bioinformatics 17:754–755
Hughes TP (1994) Catastrophes, phase shifts, and large-scale degradation of a Caribbean coral reef. Science 265:1547–1551
Jarman SN, Ward RD, Elliott NG (2002) Oligonucleotide primers for PCR amplification of coelomate introns. Mar Biotechnol 4:347–355
Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Mentjies P, Drummond A (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649
Keigwin LD (1978) Pliocene closing of the Isthmus of Panama, based on biostratigraphic evidence from nearby Pacific Ocean and Caribbean Sea cores. Geology 6:630–634
Kerr RG, Kerr SL, Djerassi C (1991) Biosynthetic studies of marine lipids. 26. Elucidation of the biosynthesis of mutasterol, a sponge sterol with a quaternary carbon in its side chain. J Org Chem 56:63–66
Klautau M, Russo CA, Lazoski C, Boury-Esnault N, Thorpe JP, Solé-Cava AM (1999) Does cosmopolitanism result from overconservative systematics? A case study using the marine sponge Chondrilla nucula. Evolution 1414–1422
Knapp IS, Forsman ZH, Williams GJ, Toonen RJ, Bell JJ (2015) Cryptic species obscure introduction pathway of the blue Caribbean sponge (Haliclona (Soestella) caerulea), (order: Haplosclerida) to Palmyra Atoll, Central Pacific. PeerJ 3:e1170
Knowlton N (1993) Sibling species in the sea. Annu Rev Ecol Syst 24:189–216
Knowlton N (2000) Molecular genetic analyses of species boundaries in the sea. Hydrobiologia 420:73–90
Kumar S, Stecher G, Tamura K (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol 2016:msw054
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, López R, Thompson JD, Gibson TJ, Higgins DG (2007) ClustalW and ClustalX version 2. Bioinformatics 23:2947–2948
Levitan DR, Fukami H, Jara J, Kline D, McGovern TM, McGhee KE, Swanson CA, Knowlton N (2004) Mechanisms of reproductive isolation among sympatric broadcast-spawning corals of the Montastraea annularis species complex. Evolution 58:308–323
López-Legentil S, Pawlik JR (2009) Genetic structure of the Caribbean giant barrel sponge Xestospongia muta using the I3-M11 partition of COI. Coral Reefs 28:157–165
Love GD, Grosjean E, Stalvies C, Fike DA, Grotzinger JP, Bradley AS, Kelly AE, Bhatia M, Meredith W, Snape CE, Bowring SA, Condon DJ, Summons RE (2009) Fossil steroids record the appearance of Demospongiae during the Cryogenian period. Nature 457:718–721
Maloof AC, Rose CV, Beach R, Samuels BM, Calmet CC, Erwin DH, Poirier GR, Yao N, Simons FJ (2010) Possible animal-body fossils in pre-Marinoan limestones from South Australia. Nat Geosci 3:653–659
Mayr E (1942) Systematics and the origin of species, from the viewpoint of a zoologist. Harvard University Press, Cambridge, MA, USA
McMurray SE, Blum JE, Pawlik JR (2008) Redwood of the reef: growth and age of the giant barrel sponge Xestospongia muta in the Florida Keys. Mar Biol 155:159–171
McMurray SE, Henkel TP, Pawlik JR (2010) Demographics of increasing populations of the giant barrel sponge Xestospongia muta in the Florida Keys. Ecology 91:560–570
McMurray S, Pawlik J, Finelli C (2014) Trait-mediated ecosystem impacts: how morphology and size affect pumping rates of the Caribbean giant barrel sponge. Aquat Biol 23:1–13
Montalvo NF, Hill RT (2011) Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl Environ Microbiol 77:7207–7216
Morariu VI, Srinivasan BV, Raykar VC, Duraiswami R, Davis LS (2009) Automatic online tuning for fast Gaussian summation. Advances in Neural Information Processing Systems 21:1113–1120
Nagelkerken I, Aerts L, Pors L (2000) Barrel sponge bows out. Reef Encounter 28:14–15
Padial JM, Miralles A, de la Riva I, Vences M (2010) The integrative future of taxonomy. Front Zool 7:16
Palumbi SR (1997) Molecular biogeography of the Pacific. Coral Reefs 16:S47–S52
Pandolfi JM, Jackson JBXC, Baron N, Bradbury RH (2005) Are US coral reefs on the slippery slope to slime? Science 307:1725–1726
Puillandre N, Lambert A, Brouillet S, Achaz G (2012) ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol Ecol 21:1864–1877
Reaka-Kudla ML, Wilson DE, Wilson EO (1997) Biodiversity II. Understanding and protecting our natural resources. Joseph Henry Press, Washington, USA, pp 83–108
Reece JS, Bowen BW, Larson A (2011) Long larval duration in moray eels (Muraenidae) ensures ocean-wide connectivity despite differences in adult niche breadth. Mar Ecol Prog Ser 437:269–277
Renema W, Bellwood DR, Braga JC, Bromfield K, Hall R, Johnson KG, Lunt P, Meyer CP, McMonagle LB, Morley RJ, O’Dea A, Todd JA, Wesselingh FP, Wilson MEJ, Pandolfi JM (2008) Hopping hotspots: global shifts in marine biodiversity. Science 321:654–657
Richards VP, Bernard AM, Feldheim KA, Shivji MS (2016) Patterns of population structure and dispersal in the long-lived “redwood” of the coral reef, the giant barrel sponge (Xestospongia muta). Coral Reefs 91:560–570
Ritson-Williams R, Becerro MA, Paul VJ (2005) Spawning of the giant barrel sponge Xestospongia muta in Belize. Coral Reefs 24:160
Robinson JP, White ER, Wiwchar LD, Claar DC, Suraci JP, Baum JK (2014) The limitations of diversity metrics in directing global marine conservation. Mar Policy 48:123–125
Rocha LA, Bowen BW (2008) Speciation in coral-reef fishes. J Fish Biol 72:1101–1121
Rocha LA, Robertson DR, Roman J, Bowen BW (2005) Ecological speciation in tropical reef fishes. Proc R Soc Lond B Biol Sci 272:573–579
Ronquist F, Huelsenbeck JP (2003) MRBAYES 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Röthing T, Voolstra CR (2016) Xestospongia testudinaria nighttime mass spawning observation in Indonesia. Galaxea 18:1–2
Rua CP, Zilberberg C, Solé-Cava AM (2011) New polymorphic mitochondrial markers for sponge phylogeography. J Mar Biol Assoc UK 91:1015–1022
Schwaninger HR (2008) Global mitochondrial DNA phylogeography and biogeographic history of the antitropically and longitudinally disjunct marine bryozoan Membranipora membranacea L. (Cheilostomata): another cryptic marine sibling species complex? Mol Phylogenet Evol 49:893–908
Setiawan E, de Voogd NJ, Swierts T, Hooper JN, Wörheide G, Erpenbeck D (2016) MtDNA diversity of the Indonesian giant barrel sponge Xestospongia testudinaria (Porifera: Haplosclerida)—implications from partial cytochrome oxidase 1 sequences. J Mar Biol Assoc UK 96:323–332
Stephens M, Smith N, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989
Swierts T, Peijnenburg KTCA, de Leeuw C, Cleary DFR, Hörnlein C, Setiawan E, Wörheide G, Erpenbeck D, de Voogd NJ (2013) Lock, stock and two different barrels: comparing the genetic composition of morphotypes of the Indo-Pacific sponge Xestospongia testudinaria. PLoS One 8:e74396
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Teske PR, Rius M, McQuaid CD, Styan CA, Piggott MP, Benhissoune S, Fuentes-Grünewald C, Walls K, Page M, Attard CRM, Cooke GM, McClusky CF, Banks SC, Barker NP, Cooke GM (2011) “Nested” cryptic diversity in a widespread marine ecosystem engineer: a challenge for detecting biological invasions. BMC Evol Biol 11:176
van Soest RWM (1980) Marine sponges from Curaçao and other Caribbean localities. part II. Haplosclerida. Studies on the fauna of Curaçao and other Caribbean islands 62:1–173
van Soest RWM, Boury-Esnault N, Vacelet J, Dohrmann M, Erpenbeck D, De Voogd NJ, Santodomingo N, Vanhoorne B, Kelly M, Hooper JN (2012) Global diversity of sponges (Porifera). PLoS One 7:e35105
van Veghel MLJ, Cleary DFR, Bak RPM (1996) Interspecific interactions and competitive ability of the polymorphic reef-building coral Montastrea annularis. Bull Mar Sci 58:792–803
Vermeij GJ (2001) Community assembly in the sea: geologic history of the living shore biota. In: Bertness M, Gaines S, Hay M (eds) Marine community ecology. Sinauer Press, Sunderland, MA, USA, pp 39–60
Wörheide G, Solé-Cava AM, Hooper JN (2005) Biodiversity, molecular ecology and phylogeography of marine sponges: patterns, implications and outlooks. Integr Comp Biol 45:377–385
Zea S (1993) Cover of sponges and other sessile organisms in rocky and coral reef habitats of Santa Marta, Colombian Caribbean Sea. Caribbean Journal of Science 29:75–88
Zhan A, Macisaac HJ, Cristescu ME (2010) Invasion genetics of the Ciona intestinalis species complex: from regional endemism to global homogeneity. Mol Ecol 19:4678–4694
Acknowledgements
We thank Friso Dekker, Maarten van Gemert, Christine Hörnlein, Yusheng Huang, Laurie van Reemst, Ee Crovetto, Ana-Rita Polonia, Rossana Freitas, Anne Bialecki, Mike Berumen, Jaaziel Garcia Hernandez, Sumaitt Putchakarn, Chad Scott, Nguyen Khac Bat, Swee Cheng Lim, Zarinah Waheed, Yosephine Tuti, Betsie Voetdijk, Peter Kuperus and Marcel Eurlings for logistic support, collecting and laboratory assistance. The feedback and constructive suggestions made during the review process are much appreciated and significantly improved this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Biology Editor Dr. Line K. Bay
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Swierts, T., Peijnenburg, K.T.C.A., de Leeuw, C.A. et al. Globally intertwined evolutionary history of giant barrel sponges. Coral Reefs 36, 933–945 (2017). https://doi.org/10.1007/s00338-017-1585-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-017-1585-6