
Abstract
Understanding genetic structure and diversity among remnant populations of rare species can inform conservation and recovery actions. We used a population genetic framework to spatially delineate gene pools and estimate gene flow and effective population sizes for the endangered California Freshwater Shrimp Syncaris pacifica. Tissues of 101 individuals were collected from 11 sites in 5 watersheds, using non-lethal tissue sampling. Single Nucleotide Polymorphism markers were developed de novo using ddRAD-seq methods, resulting in 433 unlinked loci scored with high confidence and low missing data. We found evidence for strong genetic structure across the species range. Two hierarchical levels of significant differentiation were observed: (i) five clusters (regional gene pools, FST = 0.38–0.75) isolated by low gene flow were associated with watershed limits and (ii) modest local structure among tributaries within a watershed that are not connected through direct downstream flow (local gene pools, FST = 0.06–0.10). Sampling sites connected with direct upstream-to-downstream water flow were not differentiated. Our analyses suggest that regional watersheds are isolated from one another, with very limited (possibly no) gene flow over recent generations. This isolation is paired with small effective population sizes across regional gene pools (Ne = 62.4–147.1). Genetic diversity was variable across sites and watersheds (He = 0.09–0.22). Those with the highest diversity may have been refugia and are now potential sources of genetic diversity for other populations. These findings highlight which portions of the species range may be most vulnerable to future habitat fragmentation and provide management consideration for maintaining local effective population sizes and genetic connectivity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat loss and fragmentation pose severe threats to biodiversity and are regarded as a major concern for endangered species. The ways in which they affect population structure and dynamics of a species depend on many factors, including fragment size and connectivity, the species’ dispersal strategy, and habitat suitability for survival and reproduction (Frankham et al. 2017). Populations that remain large and maintain high connectivity will generally lose genetic diversity more slowly than smaller and more isolated populations. Thus, smaller, isolated populations with limited gene flow may be at higher risk of extinction because genetic diversity is a central component of long-term resilience in the face of environmental change (Lewis 2006; Delaney et al. 2010; Callens et al. 2011; Frankham et al. 2017).
Most freshwater systems are highly vulnerable to population isolation due to both historical and recent anthropogenic landscape features. In networks of streams and rivers, physical settings such as elevation, drainage configuration, unidirectional water flow, limited lateral connectivity and extreme hydrological events (ex. droughts, floods) all influence population size and gene flow (Bohonak and Jenkins 2003; Crispo et al. 2006; Díez-del-Molino et al. 2016, 2018; Beer et al. 2019; Whelan et al. 2019). Human activities such as agriculture, urbanization, and dams also tend to reduce genetic connectivity among populations (Horreo et al. 2011; Roberts et al. 2013; Beer et al. 2019; Richmond et al. 2018). For these reasons, freshwater systems tend to have a higher density of threatened species than marine and terrestrial systems (Dudgeon et al. 2006; Strayer and Dudgeon 2010).
Larval development and dispersal strategies vary among freshwater animal taxa. This variation is associated with species geographic range and regional environmental features that dictate connectivity (Bohonak 1999; Bohonak and Jenkins 2003; Page and Hughes 2007). In freshwater shrimps, the family Atyidae has three distinct life cycles and associated dispersal capabilities: (1) extended larval development, in which females produce hundreds of eggs, larvae progress through 9 to 12 freshwater and saltwater stages, and juveniles migrate upstream (Hancock 1998; Bauer 2011a; Herrera-Correal et al. 2013; Yatsuya et al. 2013); (2) abbreviated larval development where species have larger eggs and a short freshwater planktonic larval phase (Hancock 1998; Bauer 2013; Herrera-Correal et al. 2013; Yatsuya et al. 2013); and (3) direct development into non-planktonic juveniles, where the entire life cycle is spent in a relatively small portion of the stream or river (Hancock 1998; Bauer 2013; Herrera-Correal et al. 2013; Yatsuya et al. 2013).
The California Freshwater Shrimp (Syncaris pacifica, Holmes 1894) is an endangered species listed both Federally (in 1988) and under the California Endangered Species Act (in 1980). It is a stream-dwelling crustacean with direct development and the only remaining species in the genus (following the extinction of S. pasadenae; Hedgpeth 1968). It is a low dispersal species endemic to less than 20 perennial streams in Sonoma, Napa and Marin County in Northern California where its habitat is naturally fragmented and has been further divided by urbanization and agricultural activities (U.S. Fish and Wildlife Service 1998). Phylogenetic analysis of atyid shrimp suggests that S. pacifica belongs to a relictual lineage from the Jurassic era that has persisted across their distribution range for more than 200 million years (Von Rintelen et al. 2012).
Features of the California Freshwater Shrimp life cycle and demography have been described previously (Eng 1981; Martin and Wicksten 2004; U.S. Fish and Wildlife Service 2011). In addition to direct development, S. pacifica is sexually dimorphic. Females tend to be larger than males when they reach maturity during their second summer (35–50 mm vs. 30–35 mm; Eng 1981). Sexual dimorphism is commonly associated with direct development and larger egg size in atyid shrimp (Bauer 2004, 2011b; Christodoulou and Anastasiadou 2017). Gravid females carrying ≈ 50–120 eggs are found during the fall and eggs hatch in May-June. The small juveniles can grow quickly, reaching ≈ 19 mm before the onset of winter stream temperatures. Although growth during the winter months is slow, juveniles reach ≈ 31 mm in body length by the end of the first year. S. pacifica usually mature in the second year and the lifespan is cited as approximately three years (Eng 1981).
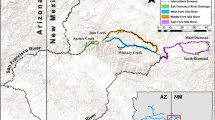
Current ecological data suggest that the California Freshwater Shrimp persists in small population fragments (e.g., 129–464 per km in Olema Creek; U.S. Fish and Wildlife Service 1998; Fong and Reichmuth 2019). In response to these low population numbers, the 1998 U.S. Fish and Wildlife Service recovery plan divided the species into four management units based on the drainage: (1) tributary streams to the lower Russian River, (2) coastal streams flowing directly into the Pacific Ocean, (3) streams draining into Tomales Bay, and 4) streams flowing into north San Pablo Bay (see Fig. 1). The primary goals of the recovery plan include delisting the species when populations are sufficiently abundant and suitable habitat is secured and managed. The recovery plan and the 2011 five-year evaluation review of the species include prioritization of genetic studies to help define and thereby aid in the preservation of remnant genetic variability within each drainage unit (U.S. Fish and Wildlife Service 1998, 2011).
Empirical analyses are needed to address these recommendations. To date, the available information on this species consists largely of presence surveys and local density estimates. These surveys provide a baseline of current distributions and census population sizes but do not provide information on conservation units and the levels of connectivity among them. Furthermore, count data do not translate well into effective population size or underlying genetic diversity. In this study, we used genomic RAD-seq data to delineate contemporary gene pools (based solely on genic similarity among individuals) in the California Freshwater Shrimp and estimate contemporary gene flow and effective population sizes. Our results significantly advance the data available for conservation and management of this species across its entire range.
Methods

Sampling locations of the California Freshwater Shrimp (Syncaris pacifica), in northern California. Circles represent sampling sites, and the watersheds are highlighted with colors. Samples were collected hierarchically based on watershed, followed by tributary streams within watershed and within stream sites separated with direct flow. Map sources include NHD Waterbodies and Flowlines (USGS 2019), and the ESRI Community baseman layer (ArcGIS, OSM)
Sample collection and DNA extraction
Syncaris pacifica occurs in perennial streams north of San Francisco Bay and south of the Russian River basin in low elevation areas (maximum 125 m) of Sonoma, Napa, and Marin Counties. The drainage system of this species’ range can be divided into streams that flow to: Tomales Bay (e.g., Lagunitas and Olema Creek), San Pablo Bay (Sonoma Creek), the Pacific Ocean (e.g., Salmon Creek), and the Russian River (e.g., Jonive Creek and Green Valley Creek; U.S. Fish and Wildlife Service 1998).
A total of 101 samples were collected between October 2019 – July 2021, using a D-frame net, across the known species distribution range, from 11 stream segments in all 5 watersheds (see Fig. 1). Two or three abdominal appendages were non-invasively taken from 6 to 11 individuals per sampling site, preserved in cold 95% ethanol on site, and stored at -20 °C freezer before laboratory processing. DNA was extracted using a modified protocol with the DNeasy blood and tissue extraction kit (Qiagen, Valencia, CA). The DNA concentration of extractions was measured using a Qubit 3.0 fluorometer (Thermo Fisher Scientific).
ddRAD-seq library preparation and sequencing
Extracted DNA (10 ng/ul) was sent to the Cornell Genomics Facility for double digest RAD-seq library preparation following Peterson et al. (2012), using a combination of the SbfI-high fidelity and MspI restriction enzymes, and 5–7 bp barcodes. Single-end sequencing was performed in an Illumina NextSeq500 platform for a total read length of 150 bp.
SNP discovery and genotyping
Raw sequences were adapter-trimmed, de-multiplexed, filtered, and processed using programs wrapped in the Stacks pipeline (Catchen et al. 2013). To minimize bias in the final data set, guidelines (Paris et al. 2017; Rochette and Catchen 2017) suggest running the pipeline several times with different parameter combinations. Throughout this process, we sought to maximize accuracy (e.g., reducing presumed paralogous loci and low-confidence genetic data) while maintaining a high number of loci, sampling sites, and individuals per sampling site. We set the minimum number of raw reads required to form a stack to 3 (parameter m in ustacks), while different values were tested for the maximum mismatches between alleles (heterozygous) to form a locus (parameter M in ustacks) and the number of mismatches allowed between sample loci when building the alleles catalog (parameter n in cstacks). Parameter values are generally selected to yield a high number of polymorphic loci found in 80% (parameter r80 in populations) or more of the individuals within a sampling site. We retained only loci present in 80% of the samples and only alleles with an overall minor allele frequency of 0.02 or higher (--min-maf parameter in populations).
The filtered data set consisted of 101 samples from 11 sampling sites in five watersheds (Green Valley, Salmon, Sonoma, Point Reyes, and Napa) genotyped at 736 loci (95,606 total base pairs, 1723 polymorphic sites). After completing the Stacks pipeline, this was reduced to 433 variant sites, with an average of 8% missing data per individual (range 2–24%).
The data generated in Stacks were exported in two formats for downstream analyses: (A) “data set A”: consisting of full haplotype data (short sequences) for the haplotype-based F-statistics, and (B) “data set B”: consisting of only the first SNP per locus, for all other analyses since they do not accommodate linkage among polymorphic sites within a short sequence.
Local genetic diversity
Using Stacks, we calculated four genetic diversity indices for each sampling site using data set A: the number of alleles found only in a specific sampling site (private alleles), observed heterozygosity (Ho), expected heterozygosity (He), nucleotide diversity (π), and the inbreeding coefficient (FIS).
Regional gene pools
We inferred gene pools via four approaches that use different aspects of the genomic data set and require different assumptions. First, we calculated genetic divergence (as FST) for data set A between all pairs of sampling sites. Each unique haplotype (short sequence read) was treated as a categorically different allele, whether they differed by one SNP or more than one. Statistical significance for these pairwise statistics was determined using nonparametric tests for all polymorphic loci jointly in Arlequin 3.0 (Excoffier et al. 2005). We ran 10,100 permutations and used a Bonferroni-corrected significance of p = 0.0009 to account for multiple testing.
Second, genetic differences were visualized in a typical “isolation by distance” plot (Slatkin 1993), where genetic similarity or genetic difference between pairs of sampling sites is plotted against the pairwise geographical distance. A typical expectation is that genetic similarity will decrease as the distance between sampling sites increases. We did this analysis using the similarity metric Slatkin’s M and data set A. Although the scatterplot contains 55 points, each is a pairwise contrast between 11 independent sampling sites, and these sites only represent 5 regional gene pools (see Results). Therefore, we treated this analysis only as a data visualization, with no statistical test performed due to a small number of independent gene pools.
Third, a model-free statistical approach was used to cluster individuals into putative gene pools. A discriminant analysis of principal component (DAPC) was performed using the R package Adegenet version 2.0.2 (Jombart 2008; R Core Team 2021) on the total data set. This method is similar to a principal component analysis (PCA) but consists of several distinct phases:
-
a.
PCA was used to reduce the complete data set B (433 nucleotide sites) to 30 principal components to obtain an uncorrelated genetic variance matrix;
-
b.
This matrix was analyzed using discriminant analysis (DA) to separate individuals into clusters using K-means clustering. The function find.cluster was used to test the most likely value of K in a range of 1 to 11 (the total number of sampling sites), using a Bayesian Information Criterion. Analysis did not include geographic information and results were based solely on genetic information;
-
c.
We then concluded that the five watersheds (Table 1) represent regional gene pools. We then determined whether additional hierarchical population (localized gene pools, model K = 1 to 3) structure is statistically detectable within each watershed (except Napa River which had one sampling site). This seemed likely, since lotic (flowing) freshwater systems inherently nest streams within watersheds, and sampling sites within streams.
Finally, we implemented the Bayesian clustering algorithm of STRUCTURE (Pritchard et al. 2000). This is the most widely used method for objectively clustering individuals into gene pools, under the assumption of random mating within gene pools. For this study, “clusters” from STRUCTURE were interpreted as gene pools. The workflow was as follows (Evanno et al. 2005; Janes et al. 2017; Cullingham et al. 2020):
-
a.
STRUCTURE was run using data set B with the admixture model, correlated allele frequencies, and with clusters ranging from K = 1 to 11, with n = 10 replicates for each K. Individual sample results were plotted using STRUCTURE PLOT version 2.0 (Ramasamy et al. 2014);
-
b.
Posterior probability for the eleven models was calculated using mean ln Pr(X|K) and Bayes’ rule following Pritchard et al. (2000). We judged the samples to represent a single gene pool if the K = 1 model had the highest posterior probability, K = 1 was not contradicted by other analyses, and there was a reasonable biological interpretation;
-
c.
When the Pritchard method indicated that K ≠ 1, the Δ K method proposed by Evanno et al. (2005) was used to determine K. (The Δ K method is generally regarded as more accurate than the Pritchard method for K > 1). The CLUMPAK web server (Kopelman et al. 2015) was used for these calculations. We concluded that the Δ K method represented the true number of gene pools if its results were not contradicted by further analyses, and there was a reasonable biological interpretation. STRUCTURE was run hierarchically similar to the DAPC analysis.
Gene flow
Because we found that watersheds correspond with regional genetic clusters, we estimated contemporary levels of gene flow between watersheds using BayesAss v.3, adjusted for multiple SNP markers (Wilson and Rannala 2003; Mussmann et al. 2019). This program estimates migration rates between each gene pool pair, with accuracy expected for migration rates less than 0.01 and FST higher than 0.1 (Faubet et al. 2007; Samarasin et al. 2017). We used data set B for this analysis. Three different seeds were performed for each MCMC run. A burn-in of one million steps was discarded, and the following four million iterations sampled every five thousand steps were used calculate the posterior probability distribution. Mixing parameters were set to (-a 0.3 -f 0.3 -m 0.3), after running different parameters in a test trial, as recommended by Rannala (2015). Convergence was accessed using the program TRACER (Rambaut et al. 2018) by plotting the parameter values against the MCMC and using (rule of thumb) effective sample size greater than 200.
Effective population size
For each watershed, we estimated contemporary effective population size (Ne) using data set B and the linkage disequilibrium method implemented in NeEstimator version 2.1 (Do et al. 2014). Results reported here are for a minimum allele threshold of 0.02 and include a 95% confidence interval from jackknifing over individuals.
Results
Local genetic diversity
Observed heterozygosity (Ho) and expected heterozygosity (He) varied widely across sampling sites and watersheds, despite similar sample sizes (Table 1). Genetic diversity was much higher in the Green Valley watershed (He = 0.15–0.18, π = 0.16–0.19) than other areas (He = 0.07–0.10, π = 0.11–0.15). The distribution of private alleles also varied substantially across sampling sites. For example, the Napa River site had 28 private alleles that were found in no other samples, while sites in the Salmon Creek and Sonoma Creek watersheds each had 0 or 1 private alleles. Local inbreeding coefficients were very low (FIS = 0.02–0.05; Table 1) because Ho and He were nearly identical within a site.
Regional gene pools
There was consensus among the genetic divergence analyses that the five watersheds define regional gene pools. FST = 0.34–0.72 between sites in different watersheds, and FST ≤ 0.12 among sites within a watershed (Table 2). Our limited sampling also suggests that differentiation patterns are hierarchical. FST = 0.07–0.11 among different tributaries within a watershed, and FST ≈ 0 between sites in the same stream (regardless of geographical distance). Pairwise FST values were significantly greater than 0 (after Bonferroni correction for multiple tests) for all pairs of sampling sites not located in the same stream. We also observed that comparisons involving the Green Valley Creek watershed tended to have lower FST than others. The overall conclusion of hierarchical population structure was highlighted in the isolation by distance plot (Fig. 2).
The DAPC concluded that K = 5 is the most likely number of clusters. The first four discriminant function axes explained 81% of the overall variance. Each cluster consisted of individuals from the same watershed, and large distances separated the five watersheds in principal component space (Fig. 3A). This aligned with the FST and isolation by distance results. Within each watershed, individuals did not obviously segregate by sampling sites in principal component space (Fig. 3A inserts).
The four watersheds other than Napa River were reanalyzed separately using DAPC for models of K = 1, 2, 3. In three watersheds (Lagunitas Creek, Salmon Creek, Sonoma Creek), no substructure was observed in the clustering results. However, a model with K = 2 was favored for Green Valley watershed, with Jonive Creek clearly separated from the two Green Valley Creek sites. The data support two localized gene pools in separate tributaries (Fig. 3B).
The STRUCTURE clustering algorithm also supported the general conclusion that this species exists in regional gene pools that correspond to watersheds. There was less clarity regarding the optimal value of K. The ln Pr(X|K) criterion suggested posterior probabilities > 0 for all models K = 5 and higher, with K = 6 favored (pp > 0.99). However, the Δ K method favored K = 2 clusters, with a secondary peak at K = 4 and third peak at K = 5. We discounted the K = 2 model as too simplistic and in contradiction with the other analyses, and further analyzed the K = 4, 5, 6 models for common features (see Fig. 4A for K = 5).
-
Lagunitas Creek watershed: in all three models (K = 4, 5, 6), all shrimp had nearly 100% ancestry from a cluster (gene pool) unique to that watershed.
-
Salmon Creek watershed: in all three models, all shrimp had nearly 100% ancestry from a cluster (gene pool) unique to that watershed.
-
Sonoma Creek and Napa River watersheds: in the K = 5 and K = 6 models, the Sonoma Creek and Napa River samples had near 100% ancestry from a gene pool that was unique to their watershed. These two watersheds comprised a single gene pool in the K = 4 model.
-
Green Valley Creek watershed: the three sites from the Green Valley watershed corresponded to a single gene pool in the K = 4 and K = 5 models. In the K = 6 model, ancestry in this watershed was represented by two clusters (gene pools). JC represented a single gene pool with near-100% ancestry for all individuals, while each GV_U and GV_L shrimp had approximately 50% of their ancestry in each cluster. This would imply that every GV_U and GV_L shrimp is the hybrid offspring of parents from two very distinct gene pools. We judged that interpretation to be biologically unreasonable, since GV (Green Valley Creek) is a small tributary with no upstream bifurcations.
The four watersheds other than Napa River were reanalyzed separately using STRUCTURE for models of K = 1, 2, 3. Within all four watersheds, posterior probabilities using ln Pr(X|K) suggested K > 1 clusters, and the Δ K method favored the K = 2 model. The K = 2 result for Green Valley watershed separated Jonive Creek from the two Green Valley Creek sites, as two localized gene pools in separate tributaries (Fig. 4B).
Unlike Green Valley watershed, the K = 2 models for the other watersheds did not separate individuals by tributary or sampling site. Instead, each resulting model ascribed near-identical patterns of mixed ancestry to all individuals within the same watershed: 60 − 90% ancestry from a single dominant gene pool, and the remainder from a minor gene pool. Because this could not be explained by stream flow patterns and known upstream populations, we judged these models to be methodological artifacts. Thus, we accepted the simpler STRUCTURE model of K = 1 (no localized gene pools) within Lagunitas Creek watershed, within Salmon Creek watershed, and within Sonoma Creek watershed. This matches the within-watershed DAPC conclusions of K = 2 localized gene pools only within the Green Valley Creek watershed.

Isolation by distance plot showing the relationship between genetic similarity (Slatkin’s M) between pairs of sampling sites, and geographic distance (Euclidean)

Discriminant analysis of principal components (DAPC) plot. Individuals are labeled by sampling site, although location information was not used in the analysis. A): DAPC results for all individuals (smallest points, plotted against the first two DAPC axes). Magnified rectangles show the sampling site for each individual. B): DAPC results for the Green Valley Creek watershed in a separate analysis. Smoothed density curves for the optimal K = 2 model are plotted on the first DAPC axis

Gene pools inferred from the Bayesian clustering analysis in STRUCTURE. A: Analysis of the entire data set for the K = 5 model. Each column in the stacked bar chart represents one individual. Colors indicating the proportion of an individual’s genomic ancestry attributed to each of five gene pools. B: STRUCTURE results for the Green Valley Creek watershed in a separate analysis (K = 2 model)
Gene flow
The tracer inspections of MCMC were all stationary and change in BayesAss parameters did not result in difference of migration rate estimates. We found very low contemporary migration rates between the five watersheds (approximately the past three generations for this analysis). Depending on the watershed pair and directionality, 95% confidence intervals ranged from m = (0.00, 0.03) to m = (0.00, 0.08).
Effective population size
Effective population size (Ne) estimates were low, but with large confidence intervals: Lagunitas Creek Ne = 84.7 (95% CI = 30.3, ∞), Green Valley Creek Ne = 62.4 (95% CI = 35.9, 177.7) and Sonoma Creek Ne = 147.1 (56.2, ∞). The estimation method failed to converge for Salmon Creek watershed and Napa River watershed, which happens when the sampling error is less than theoretically expected.
Discussion
Effective management of threatened and endangered species is greatly enhanced by an understanding of how landscape features promote or restrict gene flow, and whether the species exists in gene pools with discrete boundaries. In this study we used RAD-seq data to delineate gene pools and estimate gene flow and effective population size in the endangered California Freshwater Shrimp, that persists in a highly fragmented landscape. Our main findings indicate that (i) gene pools boundaries correlate with watersheds, (ii) these gene pools are isolated with low to no gene flow, and (iii) the gene pools have small effective population sizes. These findings have important implications for evolutionary potential and conservation of this species. The high population structure suggests that the gene pools should be managed separately, and low effective population sizes imply these gene pools may have reduced capacity to adapt to a changing environment.
Local genetic diversity
Diversity metrics (e.g., He) varied among sampling sites, but were generally lower compared to a previous study on a confamilial shrimp species using similar markers (Rahman et al. 2018) sampled in Eastern Australia across 15 km distance. However, the Green Valley watershed displayed exceptionally high genetic diversity consistent with the results from Rahman et al. (2018). The higher genetic diversity in Green Valley watershed is somewhat intriguing and we hypothesize that it could result from either (i) this watershed may have been a refugium during glaciation periods and maintains more ancestral polymorphisms than recently colonized watersheds, and/or (ii) the Green Valley watershed may have a larger current effective population size. The latter interpretation seems unlikely, due to a lower estimate of recent effective population sizes compared to other sites. Future analyses aimed at decoupling these two hypotheses, could include simulation analysis with Approximate Bayesian Computation, calibrated on current genetic diversity estimates with larger sample sizes.
Regional gene pools and gene flow
Two hierarchical levels of structure among populations of the Freshwater Shrimp were observed. The higher level contained five clusters (regional gene pools) corresponding to watersheds, with very restricted gene flow among them. More locally, sites in different tributary streams that are not directly connected with water flow still achieve moderate gene flow. We observed consistently high genetic similarity between sites that have direct upstream-to-downstream water flow, so that these should be treated as single gene pools.
The highly structured populations were consistently associated with watershed boundaries, suggesting that watershed and stream network configuration creates natural barriers to dispersal among regional gene pools in directly developing species such as the California Freshwater Shrimp. This pattern is expected for a species with a life history which: (1) lacks either a terrestrial dispersal phase (as found in most freshwater invertebrates) or a planktonic larval stage (found in many other shrimp species) that would facilitate dispersal, (2) reproduces during the summer when rain is minimal and water flow is limited, and (3) lives in a naturally fragmented habitat exacerbated by anthropogenic activities. Although genetic studies of other freshwater atyid shrimp are rare, Page and Hughes (2007) conducted a multispecies phylogeographic analysis of closely related freshwater shrimp in Australia and found that direct development species were highly divergent within a regional scale (< 80 km).
The watersheds can be considered “islands” of genetic divergence for the California Freshwater Shrimp. Within the “islands,” gene flow is constrained by unidirectional flow. This can be seen within the Lagunitas watershed, where Lagunitas creek is genetically divergent from its tributary Olema Creek, and in Green Valley watershed where the Green Valley Creek is divergent from its tributary Jonive Creek (Table 2). Moreover, increased stream distance is associated with increased genetic differentiation within tributary streams and among watersheds. Within streams, no genetic differentiation was observed, suggesting high dispersal and gene flow between sites. For example, in the Salmon Creek watershed, sites separated by > 10 km did not show significant genetic differentiation.
We found very limited to no contemporary gene flow between pairs of regional gene pools. These low migration rates are likely to represent true gene flow estimates because they are concordant with the results from the STRUCTURE and FST analysis, which show clear subdivision between regional gene pools.
The lack of genetic difference on sites connected through direct flow within a stream are more likely to be a true pattern and not an artifact of the limited sample sizes. Even though sample sizes were modest for this study, FST results were significantly different from zero for similar sample sizes in tributary streams, suggesting that the number of individuals and the SNPs used were enough to recover the patterns of population structure in this species.
Effective population size
Effective population sizes were very small in each watershed. Census population sizes are generally low in this species (< 1000 per kilometer of stream distance: USGS 1998; Fong and Reichmuth 2019) and in animals, effective population sizes can be 50% (or even an order of magnitude) lower than the census size (Frankham 1995; Luikart et al. 2010). For the California Freshwater Shrimp, surveys have suggested that about 20% of individuals are breeders and the sex ratio may be biased towards females (Fong and Reichmuth 2019). Thus, our analyses match the predicted effect of genetic drift on small and isolated populations.
Current models suggest that populations with Ne ≤ 50–100 are vulnerable to inbreeding depression, while those with Ne ≤ 500–1000 are less likely to maintain sufficient evolutionary potential (Franklin and Frankham 1998; Frankham et al. 2014). The lower bound confidence intervals reported for the California Freshwater Shrimp fall below the lower threshold of population viability, highlighting the vulnerability of the species. However, given the higher, and in two cases, undefined, upper bound of these confidence intervals, these estimates could be refined with increased sample size and number of loci to help yield precise parameters estimates.
The observed patterns of divergence and effective population size in the California Freshwater Shrimp highlight the vulnerability of small and isolated populations to genetic drift – leading to reduced (neutral and adaptive) genetic diversity. Because the effect of natural selection is overwhelmed by genetic drift in such populations, the potential to adapt to rapid climate change is likely limited. Additionally, this loss of genetic diversity can result in inbreeding and associated inbreeding depression by unmasking the expression of rare deleterious recessive alleles (Frankham et al. 2017). Our results also highlight how species with life history traits similar to that of the California Freshwater Shrimp - direct development, low dispersal rate and reproduction during low stream flow are particularly vulnerable to population structure, small population sizes and thus the effect of genetic drift.
Implications for management
Understanding the level of population differentiation is important for informing management of endangered species. This is the first study to examine population genetic structure and diversity across remaining populations of S. pacifica. Results contribute baseline information as to whether populations of the California Freshwater Shrimp form distinct groups or comprise a metapopulation. We found that populations of S. pacifica from different watersheds comprise distinct groups, each experiencing independent evolutionary and demographic processes. Two levels of significant differentiation associated with landscape configuration were observed: watersheds, which are isolated by no recent gene flow, and within watersheds, a moderate level of gene flow between tributary streams. These isolated watersheds consist of effective population sizes that are small, even compared to previous census data. Indeed, while sampling we noticed that juveniles were approximately 80% of the sampled individuals, indicating that few breeding adults were present and contributing to each site. Our results support current management units (watersheds) and goals that focus on increasing the population size of each management unit in S. pacifica (U.S. Fish and Wildlife Service 1998). Possible actions could include artificially increasing gene flow within watersheds or creating juvenile nurseries for growth and restocking in the wild, but evidence of highly isolated regional gene pools supports limiting exchange to geographically close sites and testing for local adaptations; breeding experiments could be beneficial before implementation. The FST results presented here can help identify gene pools that have less differentiation and are therefore better candidates for exchangeability. Finally, including standardized and robust monitoring of genetic diversity can help measure the progress and success of the management actions and trends in populations.
Data availability
Raw RADseq data have been submitted to the Sequence Read Archive (BioProject ID: PRJNA1091710); Data matrices and associated parameter files have been submitted to Dryad (https://doi.org/10.5061/dryad.41ns1rnnp).
References
Bauer RT (2004) Remarkable shrimps: adaptations and natural history of the carideans. University of Oklahoma
Bauer RT (2011a) Amphidromy and migrations of freshwater shrimps. I. Costs, benefits, evolutionary origins, and an unusual case of amphidromy. In: New frontiers in crustacean biology 145–156. Brill
Bauer RT (2011b) Amphidromy and migrations of freshwater shrimps. ii. delivery of hatching larvae to the sea, return juvenile upstream migration, and human impacts. In: New frontiers in crustacean biology 157–168. Brill
Bauer RT (2013) Amphidromy in shrimps: a life cycle between rivers and the sea. Lat Am J Aquat Res 41:633–650. https://doi.org/10.3856/vol41-issue4-fulltext-2
Beer SD, Bartron ML, Argent DG, Kimmel WG (2019) Genetic assessment reveals population fragmentation and inbreeding in populations of brook trout in the Laurel Hill of Pennsylvania. Trans Am Fish Soc 148:620–635. https://doi.org/10.1002/tafs.10153
Bohonak AJ (1999) Dispersal, gene flow, and population structure. The Quarterly review of biology 74:21–45. University of Chicago Press
Bohonak AJ, Jenkins DG (2003) Ecological and evolutionary significance of dispersal by freshwater invertebrates. Ecol Lett 6:783–796. https://doi.org/10.1046/j.1461-0248.2003.00486.x
Callens TO, Galbusera P, Matthysen E, Durand EY, Githiru M, Huyghe JR, Lens LU (2011) Genetic signature of population fragmentation varies with mobility in seven bird species of a fragmented Kenyan cloud forest. Mol Ecol 20:1829–1844. https://doi.org/10.1111/j.1365-294X.2011.05028.x
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Christodoulou M, Anastasiadou C (2017) Sexual dimorphism in the shrimp genus Atyaephyra (Caridea: Atyidae): the case study of Atyaephyra Thyamisensis. J Crustac Biol 37:588–601. https://doi.org/10.1093/jcbiol/rux062
Crispo E, Bentzen P, Reznick DN, Kinnison MT, Hendry AP (2006) The relative influence of natural selection and geography on gene flow in guppies. Mol Ecol 15:49–62. https://doi.org/10.1111/j.1365-294X.2005.02764.x
Cullingham CI, Miller JM, Peery RM, Dupuis JR, Malenfant RM, Gorrell JC, Janes JK (2020) Confidently identifying the correct K value using the ∆K method: when does K = 2? Mol Ecol 29:862–869. https://doi.org/10.1111/mec.15374
Delaney KS, Riley SPD, Fisher RN (2010) A rapid, strong, and convergent genetic response to urban habitat fragmentation in four divergent and widespread vertebrates. PLoS ONE 5:e12767. https://doi.org/10.1371/journal.pone.0012767
Díez-del-Molino D, Araguas RM, Vera M, Vidal O, Sanz N, García-Marín JL (2016) Temporal genetic dynamics among mosquitofish (Gambusia holbrooki) populations in invaded watersheds. Biol Invasions 18:841–855. https://doi.org/10.1007/s10530-016-1055-z
Díez-del-Molino D, García-Berthou E, Araguas RM, Alcaraz C, Vidal O, Sanz N, García-Marín JL (2018) Effects of water pollution and river fragmentation on population genetic structure of invasive mosquitofish. Sci Total Environ 637:1372–1382. https://doi.org/10.1016/j.scitotenv.2018.05.003
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (ne) from genetic data. Mol Ecol Resour 14:209–214. https://doi.org/10.1111/1755-0998.12157
Dudgeon D, Arthington AH, Gessner MO, Kawabata ZI, Knowler DJ, Lévêque C, Naiman RJ, Prieur-Richard AH, Soto D, Stiassny ML, Sullivan CA (2006) Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev Camb Philos Soc 81:163–182. https://doi.org/10.1017/S1464793105006950
Eng LL (1981) Distribution, life history, and status of the California Freshwater Shrimp, Syncaris pacifica (Holmes). California Department of Fish and Game. Inland Fisheries Endangered Species Program Special Publication 81 – 1
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:117693430500100. https://doi.org/10.1177/117693430500100003
Faubet P, Waples RS, Gaggiotti OE (2007) Evaluating the performance of a multilocus bayesian method for the estimation of migration rates. Mol Ecol 16:1149–1166. https://doi.org/10.1111/j.1365-294X.2007.03218.x
Fong D, Reichmuth M (2019) Lagunitas Creek watershed distributional survey for California freshwater shrimp (Syncaris pacifica) in Golden Gate National Recreation Area and Point Reyes National Seashore. 2018 Annual Status Report to the U.S. National Park Service. March 2019
Frankham R (1995) Effective population size/adult population size ratios in wildlife: a review. Genet Res 66:95–107. https://doi.org/10.1017/S0016672308009695
Frankham R, Bradshaw CJ, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol Conserv 170:56–63. https://doi.org/10.1016/j.biocon.2013.12.036
Frankham R, Ballou JD, Ralls K, Eldridge M, Dudash MR, Fenster CB, Lacy RC, Sunnucks P (2017) Genetic management of fragmented animal and plant populations. Oxford University Press
Franklin IR, Frankham R (1998) How large must populations be to retain evolutionary potential? Anim Conserv Forum 1(1):69–70. https://doi.org/10.1111/j.1469-1795.1998.tb00228.x
Hancock MA (1998) The relationship between egg size and embryonic and larval development in the freshwater shrimp. Freshw Biol 39:715–723. https://doi.org/10.1046/j.1365-2427.1998.00323.x
Hedgpeth JW (1968) The atyid shrimp of the genus Syncaris in California. Int Revue Der Gesamten Hydrobiol 53:511–524
Herrera-Correal J, Mossolin EC, Wehrtmann IS, Mantelatto FL (2013) Reproductive aspects of the caridean shrimp Atya scabra (Leach, 1815) (Decapoda: Atyidae) in São Sebastião Island, southwestern Atlantic, Brazil. Lat Am J Aquat Res 41:676–684. https://doi.org/10.3856/vol41-issue4-fulltext-4
Horreo JL, Martinez JL, Ayllon F, Pola IG, Monteoliva JA, Heland M, Garcia-Vazquez EV (2011) Impact of habitat fragmentation on the genetics of populations in dendritic landscapes. Freshw Biol 56:2567–2579. https://doi.org/10.1111/j.1365-2427.2011.02682.x
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The K = 2 conundrum. Mol Ecol 26:3594–3602. https://doi.org/10.1111/mec.14187
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour 15:1179–1191. https://doi.org/10.1111/1755-0998.12387
Lewis SL (2006) Tropical forests and the changing earth system. Philos Trans R Soc Lond B Biol Sci 361:195–210. https://doi.org/10.1098/rstb.2005.1711
Luikart G, Ryman N, Tallmon DA, Schwartz MK, Allendorf FW (2010) Estimation of census and effective population sizes: the increasing usefulness of DNA-based approaches. Conserv Genet 11:355–373. https://doi.org/10.1007/s10592-010-0050-7
Martin JW, Wicksten MK (2004) Review and redescription of the freshwater atyid shrimp genus Syncaris Holmes, 1900, in California. J Crustac Biol 24:447–462. https://doi.org/10.1651/c-2451
Mussmann SM, Douglas MR, Chafin TK, Douglas ME (2019) BA3-SNPs: contemporary migration reconfigured in BayesAss for next-generation sequence data. Methods Ecol Evol 10:1808–1813. https://doi.org/10.1111/2041-210X.13252
Page TJ, Hughes JM (2007) Radically different scales of phylogeographic structuring within cryptic species of freshwater shrimp (Actyidae: Caridina). Limnol Oceanogr 52:1055–1066. https://doi.org/10.4319/lo.2007.52.3.1055
Paris JR, Stevens JR, Catchen JM (2017) Lost in parameter space: a road map for stacks. Methods Ecol Evol 8:1360–1373. https://doi.org/10.1111/2041-210X.12775
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 31:e37135. https://doi.org/10.1371/journal.pone.0037135
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
R Core Team (2021) R: a language and environment for statistical computing. R Foundation for Statistical Computation, Vienna, Austria
Rahman S, Schmidt D, Hughes J (2018) De novo SNP discovery and strong genetic structuring between upstream and downstream populations of Paratya Australiensis Kemp, 1917 (Decapoda: Caridea: Atyidae). J Crustac Biol 38:166–172. https://doi.org/10.1093/jcbiol/rux117
Ramasamy RK, Ramasamy S, Bindroo BB, Naik VG (2014) STRUCTURE PLOT: a program for drawing elegant STRUCTURE bar plots in user friendly interface. SpringerPlus 3:1–3
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Rannala B (2015) BayesAss Edition 3.0 user’s Manual. University of California Davis
Richmond JQ, Backlin AR, Galst-Cavalcante C, O’Brien JW, Fisher RN (2018) Loss of dendritic connectivity in southern California’s urban riverscape facilitates decline of an endemic freshwater fish. Mol Ecol 27:369–386. https://doi.org/10.1111/mec.14445
Roberts JH, Angermeier PL, Hallerman EM (2013) Distance, dams and drift: what structures populations of an endangered, benthic stream fish? Freshw Biol 10:2050–2064. https://doi.org/10.1111/fwb.12190
Rochette NC, Catchen JM (2017) Deriving genotypes from RAD-seq short-read data using Stacks. Nat Protoc 12:2640–2659. https://doi.org/10.1038/nprot.2017.123
Samarasin P, Shuter BJ, Wright SI, Rodd FH (2017) The problem of estimating recent genetic connectivity in a changing world. Conserv Biol 31:126–135. https://doi.org/10.1111/cobi.12765
Slatkin M (1993) Isolation by distance in equilibrium and non-equilibrium populations. Evol 47:264–279. https://doi.org/10.1111/j.1558-5646.1993.tb01215.x
Strayer DL, Dudgeon D (2010) Freshwater biodiversity conservation: recent progress and future challenges. J North Am Benthol Soc 29:344–358. https://doi.org/10.1899/08-171.1
U.S. Fish and Wildlife Service (1998) California Freshwater Shrimp (Syncaris pacifica Holmes) Recovery Plan. Portland. Oregon 94
U.S. Fish and Wildlife Service (2011) California Freshwater Shrimp (Syncaris pacifica) 5-Year review. Summary and Evaluation
von Rintelen K, Page TJ, Cai Y, Roe K, Stelbrink B, Kuhajda BR, Iliffe TM, Hughes J, von Rintelen T (2012) Drawn to the dark side: a molecular phylogeny of freshwater shrimps (Crustacea: Decapoda: Caridea: Atyidae) reveals frequent cave invasions and challenges current taxonomic hypotheses. Mol Phylogenet Evol 63:82–96. https://doi.org/10.1016/j.ympev.2011.12.015
Whelan NV, Galaska MP, Sipley BN, Weber JM, Johnson PD, Halanych KM, Helms BS (2019) Riverscape genetic variation, migration patterns, and morphological variation of the threatened round Rocksnail, Leptoxis ampla. Mol Ecol 28:1593–1610. https://doi.org/10.1111/mec.15032
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191. https://doi.org/10.1093/genetics/163.3.1177
Yatsuya M, Ueno M, Yamashita Y (2013) Life history of the amphidromous shrimp Caridina leucosticta (Decapoda: Caridea: Atyidae) in the Isazu River, Japan. J Crustac Biol 33:488–502. https://doi.org/10.1163/1937240x-00002113
Acknowledgements
This work was funded by a Cooperative Agreement between San Diego State University and U.S. Geological Survey (G19AC00105). U.S. Fish and Wildlife Services approved this project under Biological Opinion 08ESMF00-2019-F-2482. Sampling within National Park Service lands was conducted under Permit PORE-2019-SCI-0035, and elsewhere under a California Endangered Species Act Memorandum of Understanding. We thank Mike Reichmuth, Mariska Obedzinski, Steve Lee, Dave Cook, and Larry Serpa for logistic support on sampling trips. We also thank Julia Smith, Anna Mitelberg and Steve Bogdanowicz for laboratory and sequencing assistance, Paul Maier for bioinformatics support, Elise Watson for GIS support, and Michael Hellberg, Brian Halstead and two anonymous reviewers for comments on the manuscript. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Author information
Authors and Affiliations
Contributions
AJB, AGV and RNF conceptualized and designed the research. DF, AMA and AJB coordinated fieldwork. Laboratory work was led by AGV and AMA. AMA conducted the analysis and wrote the manuscript with supervision of AJB. All the authors edited and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ada, A.M., Vandergast, A.G., Fisher, R.N. et al. Conservation genetics of the endangered California Freshwater Shrimp (Syncaris pacifica): watershed and stream networks define gene pool boundaries. Conserv Genet (2024). https://doi.org/10.1007/s10592-024-01621-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10592-024-01621-x