
Abstract
A global reshaping of the immune responses occurs with ageing, indicated as immunosenescence, where mitochondria and mitochondrial metabolism play an important role. However, much less is known about the role of mitochondrial stress response in this reshaping and in particular of the molecules induced by such response, collectively indicated as mitokines. In this review, we summarize the current knowledge on the role of mitokines in modulating immune response and inflammation focusing on GDF15, FGF21 and humanin and their possible involvement in the chronic age-related low-grade inflammation dubbed inflammaging. Although many aspects of their biology are still controversial, available data suggest that these mitokines have an anti-inflammatory role and increase with age. Therefore, we hypothesize that they can be considered part of an adaptive and integrated immune-metabolic mechanism activated by mitochondrial dysfunction that acts within the framework of a larger anti-inflammatory network aimed at controlling both acute inflammation and inflammaging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
During ageing, an imbalance between inflammatory and specific immune responses occurs, leading to a decreased efficiency of these latter. It is known in fact that elderly people are more susceptible to infectious diseases and have a lower response to vaccination, with respect to young people. In particular, an increased inflammatory response blocks the activation of B and T cell responses [1,2,3,4,5,6]. This phenomenon, indicated as “immune paralysis” [7] or sometimes “immune activation” [8], can be considered one of the most striking clinical features of immunosenescence, and it is at least in part due to an excess activation of immune cells, mediated by pro-inflammatory cytokines [9,10,11,12,13,14]. B cells from both mice and humans are also impaired as a result of exposure to pro-inflammatory cytokines such as TNF-α [15, 16]. In fact, high levels of TNF-α induce a significant decrease in the capacity of B cells to produce protective antibodies [15, 16]. Other cell types like macrophages and NK cells undergo immune paralysis after a prolonged inflammation. To this regard, it has been reported that human monocytes display phagocytosis defects up to 6 months after a recovery from an inflammatory condition [17]. As far as NK cells, a decrease of NK cell number and functionality has been reported in experimental sepsis [18]. Moreover, it is also reported that chronic inflammation can drive the induction of myeloid-derived suppressor cells (MDSCs), which in turn create a microenvironment favourable to immune suppression [19,20,21]. To this regard, an almost universal phenomenon occurring during ageing is a state of chronic, low-grade, sterile inflammation that has been termed inflammaging [22]. Since many years, the role of energy metabolism in immune responses and inflammation is acknowledged (see below). However, the picture is much less clear as far as the role of mitochondrial stress response in these phenomena. In fact, it is known that a series of reactions are elicited in response to a mitochondrial dysfunction. In this short review, we will focus on the possible involvement of mitokines, i.e. soluble molecules produced in response to mitochondrial stress, in immunosenescence and inflammaging.
Inflammaging has long been considered a feature of immunosenescence, as it was thought that macrophages were the main responsible for the observed age-related increase of soluble inflammatory mediators [22]. Since then, the role of other cells and tissues as well as the contribution of other biological processes such as meta-flammation and cellular senescence has been recognized; however, the importance of immune cells in inflammaging is likely not negligible, considering in particular the capability of immune cells to recognize self-molecules such as uric acid crystals, heat shock proteins and mitochondrial components. These latter include cardiolipin, N-formyl peptides, TFAM transcription factor, mitochondrial DNA (mtDNA) and double-stranded RNA (dsRNA) [23, 24]. These self-molecules, when misplaced or altered in any way, can be sensed by macrophages through innate immunity receptors and can induce the production and release of pro- and anti-inflammatory mediators such as IL-6, TNF-α, IL-1β, RANTES and IL-1ra [25,26,27,28]. Collectively, these self-molecules are indicated as DAMPs (danger-associated molecular patterns), and their receptors are indicated as PRR (pattern recognition receptors). More recently, the pro-inflammatory role of at least some of those molecules has been extended also to non-immune cells, as they can be sensed also through widely expressed intracellular receptors, such as inflammasomes, mitochondrial antiviral signalling (MAVS) proteins, cGAS-Sting and toll-like receptor 9 (TLR 9), leading to a robust inflammatory and type I interferon response [24]. We have hypothesized that a possible cue for understanding inflammaging resides in the age-dependent increment in the production (or inefficient clearance) of these DAMPs, a phenomenon that we proposed to indicate as “garb-aging” [29]. As a solid example of garb-aging, we have reported that circulating levels of mtDNA increase with age and that people with highest levels of circulating mtDNA show higher concentrations of IL-6, TNF-a, RANTES and IL-1ra with respect to those with the lowest levels of mtDNA [30].
Other than sources of DAMPs, mitochondria are important modulator of immune responses also because of their role in energy metabolism, as it is known that T lymphocyte activation entails a shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis [31], though OXPHOS is still present and needed for the functional differentiation. In particular, it has been reported that succinate dehydrogenase is needed for T cell differentiation into Th1, while OXPHOS complex I, the malate-aspartate shuttle and the mitochondrial citrate export are required for proliferation [32]. Considering that one of the key features of ageing appears to be mitochondrial dysfunction, it is more than likely that such dysfunction can also impinge upon proper T cell function [23]. Accordingly, it has been recently reported that CD4+ T cells from elderly people are characterized by the presence of an elevated number of dysfunctional mitochondria engulfed into autophagosomes with respect to cells from young people, suggesting the presence of a defective mitochondrial turnover. These defective mitochondria may be the source of inflammatory stimuli, as mentioned above, and contribute to the impairment of immune defences in older people [33,34,35].
As far as B lymphocytes, it is reported that activation of naïve B lymphocytes leads to an increase in mitochondrial mass and number as well as expression of genes for TCA cycle and OXPHOS [36] and, on the other side, inhibition of OXPHOS (by oligomycin) or TCA (by glutamine depletion) blocks the activation of B cells and differentiation into plasmablasts, as well as proliferation and antibody class switching [36, 37]. These data suggest that also for B cells mitochondrial energy metabolism is important and that mitochondrial dysfunction can be at the basis of B cell functional impairment.
Finally, it is known that a main feature of the ageing bone marrow is a shift toward the production of myeloid cells at the expenses of lymphoid ones [38], and, consistently, it has been found that in a mouse model of mitochondrial impairment (UCP2 knockout, leading to an increased production of ROS), aged animals display an increased amount of monocytes and neutrophils and a decreased amount of B cells [39].
Mitochondrial stress response and immune responses
Despite the fact that, as mentioned in the previous paragraph, mitochondrial dysfunction can be a cause of immune response impairment, it has been proposed that a mild mitochondrial dysfunction could be beneficial for the cell as it could elicit an effective stress response [40]. This hypothesis was introduced a few years ago by Johnson and co-workers in a study conducted on Caenorhabditis elegans. They demonstrated that a mild disruption of mitochondrial electron transport chain (ETC) causes a transient DNA (nuclear and mitochondrial) damage leading to the activation of compensatory mechanisms that increase the lifespan in C. elegans [41]. A detailed discussion on the retrograde stress responses elicited by a mitochondrial stress, such as the mitochondrial unfolded protein response, is outside the scope of this short review, as many excellent studies have been published on this topic [40, 42, 43]. Rather, we will focus on the effects of some downstream products of this stress response on the immune function and on inflammation.
In their 2011 seminal paper, Dillin and co-workers demonstrated that the beneficial effect of a mild mitochondrial stress is not limited to the affected cell/tissue but also spreads to distal ones, thus conferring a global resistance and survival advantage to the whole organism [44]. In this paper, the authors presented evidence that an impairment of the electron transport chain localized in one tissue can be perceived in distal ones. In other words, the signalling of a localized mitochondrial perturbation can actually spread through the organism via soluble mediators that these authors indicated as “mitokines”. A mitokine should be a soluble molecule (protein, peptide or other) produced and secreted in response to a mitochondrial stress response and able to elicit an adaptive/compensatory response in distal cells even though not directly affected by the stressful event/stimulus. Since then, a number of molecules have been identified that fulfil this definition, including the neuronal peptide FLP2 in C. elegans [45], the fibroblast growth factor 21 (FGF21) [46,47,48] and the growth differentiation factor 15 (GDF15) [49, 50] and a series of mitochondrial DNA-encoded peptides that include humanin (HN) [51], MOTS-c [52, 53] and small humanin-like peptides (SHLPs) [54]. We have reported that some of them, namely, GDF15, FGF21 and HN, increase with age from young people to centenarians and are associated with worsened haematochemical parameters and, for nonagenarians and centenarians, with lower life expectancy [55]. Consistently, GDF15 has been reported to be the most upregulated protein in old age [56, 57] associated with many pathological conditions, such as type 2 diabetes (T2D), cardiovascular diseases (CVD), neurodegeneration and overall mortality [50, 58,59,60]. FGF21 is considered a pro-longevity hormone as it is able to modulate energy and lipid metabolism and extend animal lifespan [46, 47, 61,62,63]; however, it is also involved in accelerated ageing and premature death in Opa-1-deficient mice [64]. HN has been identified as a powerful anti-apoptotic factor with a cytoprotective role in many age-associated diseases such as Alzheimer’s disease (AD), T2D and CVD [65]. As a whole, these data support the idea of mitohormesis proposed by Ristow and Zarse [66]. According to this idea, the pro-longevity effects of some treatments like calorie restriction are ultimately due to an increased formation of ROS, which in turn activates a retrograde response of stress resistance culminating in lifespan extension. The observed age-related increase of mitokines could be then considered an attempt of mitohormesis aimed at increasing stress resistance [55, 67]; however, when the production of otherwise beneficial mitokines turns from acute to chronic (as it occurs with age), it becomes either no longer effective or even toxic [55]. In the next paragraphs, we will briefly summarize the present knowledge on the involvement of mitokines in the ageing of the immune system (immunosenescence), with particular regard to inflammaging.
Mitokines in immune responses
The role in immune response and inflammation of the mostly studied mitokines will be briefly described below, and, where available, data on ageing will be discussed.
GDF15
GDF15 was discovered over 20 years ago as a member of the transforming growth factor β (TGF-β) superfamily. The function of GDF15 is not still fully clear, since several findings propose opposite roles for this protein. While on one hand many studies suggest a protective/anti-inflammatory role for GDF15, on the other hand, many studies hypothesize a pathogenic/pro-inflammatory function.
Several studies in humans have reported that GDF15 activity increases under stress conditions in response to tissue insults. In healthy individuals, as well as young subjects, the circulating concentration of GDF15 is very low. Conversely, plasma GDF15 levels are higher in the elderly, in particular in the presence of pathological conditions, such as CVD, insulin resistance and T2D, neurodegeneration, renal chronic disease and cancer [50, 55], where it is supposed that GDF15 plays a protective role against different insults via PI3K–Akt, ERK1/2 and SMAD2/3 signalling pathways [68,69,70,71]. Recently, it has been shown that GDF15 decreases the expression of pro-inflammatory cytokines and prevents the activation of T cells in the liver of mice with fibrosis, while deficiency of GDF15 aggravates liver injury and fibrosis [72]. In agreement, Bootcov and co-workers found that the expression of GDF15 is induced in macrophages by IL-1β, IL-2, TNF-α and TGF-β and limits their activation, consequently blunting inflammation [73]. GDF15 is responsive to inflammation via p53 [74] and is necessary for tolerance to inflammation induced by viral or bacterial infections [75]. We have reported that GDF15 in humans is associated with increased number of total leukocytes and decreased number of lymphocytes [76]. This is also in agreement with a previous report showing an increase of GDF15 and a concomitant decrease in circulating CD4+ and CD8+ T cells in patients with COPD [77]. Therefore, all these findings suggest that GDF15 may have an anti-inflammatory role, although there are also studies suggesting the opposite. In particular, a study in mouse models indicates that the deletion of haematopoietic GDF15 reduces CCR2 expression and chemotaxis and improves plaque stability, with beneficial effects against atherosclerosis [78]. In agreement, another study in mice demonstrates that GDF15 is involved in the progression of atherosclerosis by regulating apoptotic cell death and IL-6 inflammatory response [79]. The hypothesis that GDF15 may play detrimental role is also supported by many studies on the effects of GDF15 on cachexia. Higher circulating GDF15 levels in fact were found to be associated with the development of cachexia in both animal models and human patients [80,81,82]. In particular, gain- and loss-of-function experiments in mouse models suggest that GDF15 is a mediator of cancer cachexia [81]. In addition, GDF15 is also considered a marker of all-cause mortality [50, 83], and in agreement in our previous study, we found that, among mitokines, GDF15 is the most associated with mortality in old age [55]. Therefore, the role of GDF15 is more complex than expected and possibly double-sided. In a condition of mild or transient stress (or when a pathology is in an early stage), the increase in circulating GDF15 levels could be protective; on the contrary, when the stress is elevated and chronic (or when a pathology is in an advanced stage), the continuous/chronic release of GDF15 may become detrimental [55, 84]. It is also possible that, very simply, in chronic situations, the detrimental effects that caused GDF15 expression overcome GDF15’s beneficial ones. Interestingly, GDF15 appears the most upregulated protein during the ageing process, as mentioned above [56, 57]; therefore, its importance is probably higher than expected and plays a crucial role in the interconnection between metabolism, inflammation and immune response in old age.
Given the large number of actions associated with GDF15, a still unresolved question is how GDF15 acts and by what type of receptors. To date, the only confirmed GDF15 receptor is the GDNF receptor family member GFRAL, acting with its co-receptor RET. However, several studies in mice, as well as in nonhuman primates, reported that GFRAL expression is present only in hindbrain neurons but not in other peripheral tissues [85, 86]; therefore, it is not yet clear how GDF15 can act directly on peripheral tissues in the absence of any known receptors. It is then possible that still not recognized receptors exist other than GFRAL or that GDF15 may exerts its effects through other molecules. Actually, previous studies showed that GDF15 might act on different cell types through TGF-β and its receptors, as GDF15 is involved in the regulation of TGF-β/Smad signalling pathway [74, 87,88,89,90]. However, these findings must be interpreted with caution, as it has been reported that commercial preparations of recombinant GDF15 can be contaminated with variable levels of TGF-β [91].
HN
HN plays a cytoprotective role against oxidative stress, apoptosis and inflammatory response [65]. A role for HN in immune responses has been, in fact, demonstrated in several studies, suggesting that HN has a role in the attenuation of inflammation and it has therefore to be considered an anti-inflammatory mediator [92,93,94,95]. In particular, the neuroprotective role of HN has been at least in part attributed to the capability of HN to interact with the gp130 subunit of the IL-6 receptor, leading to a decreased in vitro production of pro-inflammatory cytokines, such as IL-6, IL-1β and TNFα [92, 94]. HN attenuates inflammation and macrophages infiltration, as well as apoptosis, also in the early stage of kidney disease in ApoE-deficient mice [93]. Moreover, it is possible that the anti-inflammatory activity is partly mediated by the well-known anti-apoptotic effects of HN, as it can dampen the production of pro-inflammatory, apoptosis-related DAMPs. To mention only few examples of the anti-apoptotic activities of HN, it has been demonstrated that HN physically interacts with IGFBP-3, a pro-apoptotic protein [96]. In particular, IGFBP-3 expression is very elevated in brain affected by AD suggesting a role for IGFBP-3 in cell death [97]. Moreover, it has been demonstrated that HN interacts with the apoptosis-inducing protein Bax, creating a HN-Bax complex. In this way, HN retains Bax in the cytoplasm and prevents the translocation of Bax to mitochondria, thus protecting from apoptosis [98]. This activity is important also for inflammation, as the activation of Bax/Bak complexes leads not only to the release of pro-apoptotic molecules but also of mtDNA and dsRNA that can activate a pro-inflammatory response via MAVS, cGAS-Sting, inflammasomes and NF-κB [24]. Consistently with the idea that HN has not only cytoprotective but also anti-inflammatory roles, it has been found that HN has protective roles not only in AD [94] but also in T2D, CVD and atherosclerosis [95, 99,100,101], which are all considered inflammaging-related diseases [102, 103]. Moreover, very recently it has been reported that HN, and probably other mitochondria-derived peptides, improves not only health status but also lifespan [104]. In particular, Yen and co-workers found that the overexpression of HN increases lifespan in C. elegans, while middle-aged mice treated with the potent humanin analogue HNG showed an improvement in health parameters [104]. Literature data are still controversial as far as the age-related changes in HN circulating levels. At variance with some studies that showed a decrease of HN with ageing in both mice and humans, we have found in humans an increase of HN with age. To further support this finding, we have also observed that higher levels of HN and GDF15 are present in people with accelerated ageing such as Down Syndrome (DS) persons, with respect to their siblings of similar age [55]. In agreement with our finding, a recent work by Salemi et al. [105] shows a significant upregulation of HN in fibroblasts from DS persons compared with non-trisomic siblings [105]. As a whole, it is tempting to speculate that these increases in HN concentrations can be considered an attempt of the tissue/organism to counteract the detrimental effects of inflammation/inflammaging.
FGF21
FGF21 is an important regulatory protein of energy metabolism and inflammatory processes. Several studies of gain- and loss-of-function indicate that FGF21 is a key metabolic mediator to improve the compromised mitochondrial function and reduce inflammation and apoptosis in several organs [106,107,108]. An in vitro study in LPS-stimulated murine macrophagic cells (RAW 264.7) demonstrated that a pre-incubation with FGF21 reduces the expression of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6, increases the level of IL-10 and inhibits the activation of the nuclear factor-κB (NF-κB) [109]. This finding was then confirmed and further investigated by many other studies. For example, a recent study from Gao et al. [110] demonstrated that FGF21 suppressed inflammation and apoptosis caused by LPS stimulation via inhibition of TLR4/MYD88/NF-κB signalling pathway in both Balb/c mice and BEAS-2B or THP-1 cells [110]. Moreover, in a rat model of atherosclerosis, the upregulation of FGF21 affected inflammation and oxidative stress by increasing the expression of Nrf2-ARE (nuclear factor erythroid 2-related factor 2-antioxidant response elements) signalling-related proteins [111], which are involved in cellular antioxidant and anti-inflammatory pathways, as well as in the protection of mitochondria.
In addition to its anti-inflammatory role, FGF21 seems to play also an important protective role against thymus ageing by delaying age-related thymic involution in mice [112]. In particular, the overexpression of FGF21 reduces the generation of ectopic lipids and inflammation, while its loss-of-function in middle-aged mice accelerates thymic involution. Therefore, it seems that FGF21 can be considered a key regulator of the immune function able to counteract at least in part the immune dysfunction occurring with ageing. In agreement with these positive roles in immune function, FGF21 has a pro-longevity activity in mice [93] and is considered a marker of successful ageing [113]. However, the precise role of FGF21 in ageing is still highly questioned. In fact, it is reported that FGF21 could be responsible for the accelerated ageing phenotype observed in Opa-1 KO mice [64], and consistently, in our previous study, we have found that circulating FGF21 levels increase with age in subjects without evident pathologies and are particularly higher in centenarians. Moreover, we have found that FGF21 is positively associated with worsened parameters, such as higher insulinemia and HOMA-IR, and is inversely correlated with survival in oldest subjects [55]. Therefore, whether the role of FGF21 in ageing is beneficial or detrimental is still to be clarified. It has to be considered that, similarly to GDF15, an acute or chronic production of FGF21 may have different or even opposite biological meanings. Therefore, it could be speculated that an acute increment of FGF21 is good for health and lifespan, while a chronically elevated level of circulating FGF21 could be detrimental or, more likely, could be interpreted as an attempt of the organism to counteract an overwhelming stress.
Other mitokines
Much less is known regarding the role in immune responses/inflammation of mitokines such as FLP2, MOTS-c and SHLPs.
FLP2 neuropeptide is important for the regulation of mitochondrial stress response in the nervous system of C. elegans. FLP2 is released by neurons during mitochondrial dysfunction and transmits to distal tissues a signal for the induction of UPRmt [45]. However, its overexpression does not extend worms’ lifespan [45]. No data are available regarding the possible involvement of FLP2 in the ageing process, as well as inflammation or immune response. However, since many studies indicate that UPRmt affects inflammatory responses [114,115,116,117], it is possible that, at least indirectly, FLP2 can have a role as well.
MOTS-c is a peptide of 16 amino acids discovered few years ago by Lee et al. [53]. MOTS-c is expressed in several tissues, such as skeletal muscle and adipose tissue, but it is also present at circulating level, both in mice and humans [53, 118]. Studies suggested that MOTS-c is a key regulator of cellular metabolism and inflammatory processes [53, 119]. It has been shown that MOTS-c counteracts inflammation by reducing the levels of pro-inflammatory cytokines, such as IL-6, IL-1β and TNFα, and increasing those of the anti-inflammatory cytokine IL-10 [53, 120]. In particular, MOTS-c decreases the bacterial load in mice with sepsis by enhancing the bactericidal capacity of macrophages and thus improving the survival of mice [120]. Moreover, MOTS-c suppresses inflammation also by controlling NF-κB and STAT1 pathway [121]. As far ageing, it seems that MOTS-c circulating levels decrease with ageing [53, 122].
SHLPs are a class of peptides (from 1 to 6) expressed in several tissues and detectable at plasma level [54]. To date little is known about their precise role; however, in vitro studies demonstrated that they have a number of biological effects. In particular, SHLP2 and SHLP3 increase cell viability and reduce apoptosis, while SHLP6 significantly enhances apoptosis [54]. Similar to HN, SHLP2 and SHLP3 play a critical role in metabolism, apoptosis and inflammation [54]. The mechanism by which SHLPs regulate the expression of metabolic and inflammatory markers remains still unclear; however, data from Cobb et al. suggest that SHLP2 and SHLP3 regulate the levels of leptin, while SHLP3 regulates also those of IL-6 and MCP-1 [54]. In addition, SHLP2 treatment protects against Aβ1–42-induced cell death and thus can counteract AD [54]. Moreover, SHLP2 circulating levels decline with age. Altogether, these data suggest that SHLP2 and SHLP3 have potential beneficial effects and can counteract (at least some) age-related diseases [54, 123, 124].
Conclusions and perspectives
Even if the role of mitochondrial stress response in modulating the immune responses is only partially understood, available data point out to an anti-inflammatory role of the most studied mitokines. As summarized in the previous paragraphs, GDF15, FGF21 and HN have been proposed as anti-inflammatory molecules in many experimental conditions, in both humans and animal models. As their blood levels appear to increase with age (as many pro-inflammatory mediators do), it is possible that they are part of an integrated, molecular, immune-metabolic machinery/network able to respond to (among others) mitochondrial stress, whose role is to set up a coordinated, systemic anti-inflammatory response in both acute and chronic conditions. In this latter case, the mitokine response could be considered part of an “anti-inflammaging” process whose existence was proposed some years ago as a consequence of inflammaging [124]. In fact, the capability to downregulate both acute and chronic inflammatory responses is crucial to maintain homeostasis in young subjects and to avoid/postpone age-related diseases (CVD, T2D, neuro-inflammation and cancer, among others) in the elderly. Many other anti-inflammatory molecules are known, including resolvins, maresins, adiponectin, IL-10, TGF-β, etc. Some of them are particularly elevated in centenarians [125,126,127,128], suggesting that these exceptional people owe their longevity, among others, to a successful balancing between pro- and anti-inflammatory mediators. Now, the discovery that other molecules endowed with anti-inflammatory and immunomodulatory activity increase with age not only in centenarians but also in elderly people suggests that a large, comprehensive anti-inflammaging network is physiologically put in place likely as a general attempt of the ageing organism to cope with the progressive increase of chronic inflammation (inflammaging) and its deleterious effects. However, ultimately this (beneficial) response can be insufficient to counteract the detrimental accumulation with age of molecular insults, particularly in those old people affected by major are-related pathologies. Thus, the increased blood levels of mitokines such as GDF15, FGF21 and HN become correlated with altered haematochemical parameters and lower life expectancy [55]. A simplified version of this hypothesis is presented in Fig. 1. The key concept expressed in the figure is the balancing between pro- and anti-inflammatory stimuli that are tightly interconnected via feedback loops to produce a net result of detrimental or successful adaptation to a chronic stress. The other key concept is that the source of the chronic stress is not necessarily an external noxa, but it can be the inescapable, progressive process of deterioration of crucial organelles such as mitochondria. Thus, all the systems aimed at sensing and responding to mitochondrial dysfunction occupy the centre of the stage for effective adaptation/remodelling of the different cell types and organs involved, leading eventually to either successful or unsuccessful ageing. As far as immune responses other than inflammation, the precise role of mitochondrial stress response and mitokines is less clear, and more studies are needed to better clarify this topic, in particular in the framework of immunosenescence and of the crosstalk between the immune system and other organs such as the liver and adipose tissue. As other types of stresses, like ER stress, can also elicit the production of some mitokines, future studies should disentangle the precise role of mitochondrial dysfunction with respect to other stresses in modulating the immune responses via mitokine production. Moreover, studies aimed at better clarifying the direct responsiveness of mitokines to inflammatory or immunological stimuli would be also desirable. However, should the hypothesis that mitokines are part of an attempt to modulate acute and chronic inflammatory reactions be confirmed, a reappraisal of the biological meaning of their association with morbidity (age-related diseases) and mortality, is urgently needed, also considering their possible use (positive modulation of their expression) as therapeutic tools and targets.

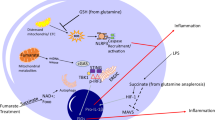
Schematic representation of the role of mitochondria in healthy ageing or unsuccessful ageing and the onset of age-related diseases (ARDs). In healthy ageing, stress stimulates the mitochondrial unfolded protein response (UPRmt) and the production of mitokines (HN, FGF21, GDF15) that act to inhibit production and activity of inflammatory cytokines (including IL-1β, IL-18, IL-6, type I IFN, TNF-α) through yet not clarified mechanisms and thus preserve a balance between inflammatory and specific immune responses; in unsuccessful ageing, an imbalance between inflammatory and specific immune responses occurs, with a high production of reactive oxygen species (ROS) and danger-associated molecular patterns (DAMPs) leading to an increase of inflammatory cytokines that contribute to the onset of ARDs. GDF15, FGF21 and HN can be part of an immune-metabolic machinery/network activated by both acute and chronic stressors impinging on mitochondria aimed at modulating inflammaging. Many other anti-inflammatory molecules are known, including IL-10, resolvins, maresins, etc
References
Czesnikiewicz-Guzik M, Lee WW, Cui D, Hiruma Y, Lamar DL, Yang ZZ, Ouslander JG, Weyand CM, Goronzy JJ (2008) T cell subset-specific susceptibility to aging. Clin Immunol 127:107–118. https://doi.org/10.1016/j.clim.2007.12.002
Cain D, Kondo M, Chen H, Kelsoe G (2009) Effects of acute and chronic inflammation on B-cell development and differentiation. J Invest Dermatol 129:266–277. https://doi.org/10.1038/jid.2008.286
Condotta SA, Rai D, James BR, Griffith TS, Badovinac VP (2013) Sustained and incomplete recovery of naive CD8+ T cell precursors after sepsis contributes to impaired CD8+ T cell responses to infection. J Immunol 190:1991–2000. https://doi.org/10.4049/jimmunol.1202379
Shen-Orr SS, Furman D, Kidd BA, Hadad F, Lovelace P, Huang YW et al (2016) Defective signaling in the JAK-STAT pathway tracks with chronic inflammation and cardiovascular risk in aging humans. Cell Syst 3:374–384.e4. https://doi.org/10.1016/j.cels.2016.09.009
Beyer M, Abdullah Z, Chemnitz JM, Maisel D, Sander J, Lehmann C, Thabet Y, Shinde PV, Schmidleithner L, Köhne M, Trebicka J, Schierwagen R, Hofmann A, Popov A, Lang KS, Oxenius A, Buch T, Kurts C, Heikenwalder M, Fätkenheuer G, Lang PA, Hartmann P, Knolle PA, Schultze JL (2016) Tumor-necrosis factor impairs CD4(+) T cell-mediated immunological control in chronic viral infection. Nat Immunol 17:593–603. https://doi.org/10.1038/ni.3399
Hagen M, Derudder E (2020) Inflammation and the alteration of B-cell physiology in aging. Gerontology 66:105–113. https://doi.org/10.1159/000501963
Fulop T, Dupuis G, Baehl S, Le Page A, Bourgade K, Frost E et al (2016) From inflamm-aging to immune-paralysis: a slippery slope during aging for immune-adaptation. Biogerontology 17:147–157
Frasca D, Ferracci F, Diaz A, Romero M, Lechner S, Blomberg BB (2016) Obesity decreases B cell responses in young and elderly individuals. Obesity (Silver Spring) 24:615–625. https://doi.org/10.1002/oby.21383
Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G (2008) Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun Ageing 5:6. https://doi.org/10.1186/1742-4933-5-6
McElhaney JE, Effros RB (2009) Immunosenescence: what does it mean to health outcomes in older adults? Curr Opin Immunol 21:418–424. https://doi.org/10.1016/j.coi.2009.05.023
Haralambieva IH, Painter SD, Kennedy RB, Ovsyannikova IG, Lambert ND, Goergen KM, Oberg AL, Poland GA (2015) The impact of immunosenescence on humoral immune response variation after influenza A/H1N1 vaccination in older subjects. PLoSOne 10:e0122282. https://doi.org/10.1371/journal.pone.0122282
Briceño O, Lissina A, Wanke K, Afonso G, von Braun A, Ragon K, Miquel T, Gostick E, Papagno L, Stiasny K, Price DA, Mallone R, Sauce D, Karrer U, Appay V (2016) Reduced naïve CD8(+) T-cell priming efficacy in elderly adults. Aging Cell 15:14–21. https://doi.org/10.1111/acel.12384
Kennedy RB, Simon WL, Gibson MJ, Goergen KM, Grill DE, Oberg AL, Poland GA (2016) The composition of immune cells serves as a predictor of adaptive immunity in a cohort of 50- to 74-year-old adults. Immunology 148:266–275. https://doi.org/10.1111/imm.12599
Parmigiani A, Alcaide ML, Freguja R, Pallikkuth S, Frasca D, Fischl MA, Pahwa S (2013) Impaired antibody response to influenza vaccine in HIV-infected and uninfected aging women is associated with immune activation and inflammation. PLoS One 8:e79816. https://doi.org/10.1371/journal.pone.0079816
Frasca D, Romero M, Diaz A, Alter-Wolf S, Ratliff M, Landin AM, Riley RL, Blomberg BB (2012) A molecular mechanism for TNF-alpha-mediated downregulation of B cell responses. J Immunol 188:279–286. https://doi.org/10.4049/jimmunol.1003964
Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB (2014) High TNF-α levels in resting B cells negatively correlate with their response. Exp Gerontol 54:116–122. https://doi.org/10.1016/j.exger.2014.01.004
Roquilly A, Jacqueline C, Davieau M, Mollé A, Sadek A, Fourgeux C, Rooze P, Broquet A, Misme-Aucouturier B, Chaumette T, Vourc’h M, Cinotti R, Marec N, Gauttier V, McWilliam HEG, Altare F, Poschmann J, Villadangos JA, Asehnoune K (2020) Alveolar macrophages are epigenetically altered after inflammation, leading to long-term lung immunoparalysis. Nat Immunol 21:636–648. https://doi.org/10.1038/s41590-020-0673-x
Jensen IJ, Winborn CS, Fosdick MG, Shao P, Tremblay MM, Shan Q, Tripathy SK, Snyder CM, Xue HH, Griffith TS, Houtman JC, Badovinac VP (2018) Polymicrobial sepsis influences NK-cell-mediated immunity by diminishing NK-cell-intrinsic receptor-mediated effector responses to viral ligands or infections. PLoS Pathog 14:e1007405. https://doi.org/10.1371/journal.ppat.1007405
Song X, Krelin Y, Dvorkin T, Bjorkdahl O, Segal S, Dinarello CA, Voronov E, Apte RN (2005) CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1β-secreting cells. J Immunol 175:8200–8208. https://doi.org/10.4049/jimmunol.175.12.8200
Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S (2007) Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 67:4507–4513. https://doi.org/10.1158/0008-5472.CAN-06-4174
Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67:10019–10026. https://doi.org/10.1158/0008-5472.CAN-07-2354
Franceschi C, Bonafè M, Valensin S, Olivieri F, De Luca M, Ottaviani E et al (2000) Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci 908:244–254. Review. https://doi.org/10.1111/j.1749-6632.2000.tb06651.x
McGuire PJ (2019) Mitochondrial dysfunction and the aging immune system. Biology (Basel) 8:E26. https://doi.org/10.3390/biology8020026
Riley JS, Tait SWG (2020) Mitochondrial DNA in inflammation and immunity. EMBO Rep:e49799. https://doi.org/10.15252/embr.201949799
Laskin DL (2009) Macrophages and inflammatory mediators in chemical toxicity: a battle of forces. Chem Res Toxicol 22:1376–1385. https://doi.org/10.1021/tx900086v
Davis BK, Wen H, Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735. https://doi.org/10.1146/annurev-immunol-031210-101405
Arango Duque G, Descoteaux A (2014) Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol 5:491. https://doi.org/10.3389/fimmu.2014.00491
Turner MD, Nedjai B, Hurst T, Pennington DJ (2014) Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 1843:2563–2582. https://doi.org/10.1016/j.bbamcr.2014.05.014
Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S (2017) Inflammaging and 'Garb-aging'. Trends Endocrinol Metab 28:199–212. https://doi.org/10.1016/j.tem.2016.09.005
Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D et al (2014) Circulating mitochondrial DNA increases with age and is a familiar trait: implications for "inflamm-aging". Eur J Immunol 44:1552–1562. https://doi.org/10.1002/eji.201343921
Tarasenko T, Pacheco SE, Koenig MK, Gomez-Rodriguez J, Kapnick SM, Diaz F et al (2017) Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab 25:1256–1268 e1257. https://doi.org/10.1016/j.cmet.2017.05.007
Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R et al (2019) Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571:403–407. https://doi.org/10.1038/s41586-019-1311-3
Bektas A, Schurman SH, Gonzalez-Freire M, Dunn CA, Singh AK, Macian F et al (2019) Age-associated changes in human CD4(+) T cells point to mitochondrial dysfunction consequent to impaired autophagy. Aging (Albany NY) 11:9234–9263. https://doi.org/10.18632/aging.102438
Pereira BI, Devine OP, Vukmanovic-Stejic M, Chambers ES, Subramanian P, Patel N, Virasami A, Sebire NJ, Kinsler V, Valdovinos A, LeSaux CJ, Passos JF, Antoniou A, Rustin MHA, Campisi J, Akbar AN (2019) Senescent cells evade immune clearance via HLA-E-mediated NK and CD8+ T cell inhibition. Nat Commun 10:2387. https://doi.org/10.1038/s41467-019-10335-5
Callender LA, Carroll EC, Bober EA, Akbar AN, Solito E, Henson SM (2020) Mitochondrial mass governs the extent of human T cell senescence. Aging Cell 19:e13067. https://doi.org/10.1111/acel.13067
Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA (2018) Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience 5:99–109. https://doi.org/10.1016/j.isci.2018.07.005
Heyse S, Connolly T, Chiles T (2015) The regulation and role of L-glutamine in B-cell activation. FASEB J 29:888.17
Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL (2005) Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A 102:9194–9199. https://doi.org/10.1073/pnas.0503280102
Kretzschmar C, Roolf C, Timmer K, Sekora A, Knübel G, Escobar HM et al (2016) Uncoupling protein 2 deficiency results in higher neutrophil counts and lower B-cell counts during aging in mice. Exp Hematol 44:1085–1091.e2. https://doi.org/10.1016/j.exphem.2016.08.003
Rose G, Santoro A, Salvioli S (2017) Mitochondria and mitochondria-induced signalling molecules as longevity determinants. Mech Ageing Dev 165(Pt B):115–128. https://doi.org/10.1016/j.mad.2016.12.002
Rea SL, Ventura N, Johnson TE (2007) Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol 5:e259
Yao RQ, Ren C, Xia ZF, Yao YM (2020) Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy 12:1–17. https://doi.org/10.1080/15548627.2020.1725377
Ji T, Zhang X, Xin Z, Xu B, Jin Z, Wu J, Hu W, Yang Y (2020) Does perturbation in the mitochondrial protein folding pave the way for neurodegeneration diseases? Ageing Res Rev 57:100997. https://doi.org/10.1016/j.arr.2019.100997
Durieux J, Wolff S, Dillin A (2011) The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144:79–91. https://doi.org/10.1016/j.cell.2010.12.016
Shao LW, Niu R, Liu Y (2016) Neuropeptide signals cell non-autonomous mitochondrial unfolded protein response. Cell Res 26:1182–1196. https://doi.org/10.1038/cr.2016.118
Kim KH, Jeong YT, Kim SH, Jung HS, Park KS, Lee HY, Lee MS (2013a) Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem Biophys Res Commun 440:76–81. https://doi.org/10.1016/j.bbrc.2013.09.026
Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, Ko TH, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, Lee MS (2013b) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19:83–92. https://doi.org/10.1038/nm.3014
Davis RL, Liang C, Edema-Hildebrand F, Riley C, Needham M, Sue CM (2013) Fibroblast growth factor 21 is a sensitive biomarker of mitochondrial disease. Neurology. 81:1819–1826. https://doi.org/10.1212/01.wnl.0000436068.43384.ef
Yatsuga S, Fujita Y, Ishii A, Fukumoto Y, Arahata H, Kakuma T, Kojima T, Ito M, Tanaka M, Saiki R, Koga Y (2015) Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann Neurol 78:814–823. https://doi.org/10.1002/ana.24506
Fujita Y, Taniguchi Y, Shinkai S, Tanaka M, Ito M (2016) Secreted growth differentiation factor 15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr Gerontol Int 1:17–29. https://doi.org/10.1111/ggi.12724
Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, Nishimoto I (2001) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci U S A 98(11):6336–6341
Zarse K, Ristow M (2015) A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell Metab 21:355–356. https://doi.org/10.1016/j.cmet.2015.02.013
Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, Cohen P (2015) The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab 21:443–454. https://doi.org/10.1016/j.cmet.2015.02.009
Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK et al (2016) Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 8:796–809. https://doi.org/10.18632/aging.100943
Conte M, Ostan R, Fabbri C, Santoro A, Guidarelli G, Vitale G, Mari D, Sevini F, Capri M, Sandri M, Monti D, Franceschi C, Salvioli S (2019) Human aging and longevity are characterized by high levels of mitokines. J Gerontol A Biol Sci Med Sci 74:600–607. https://doi.org/10.1093/gerona/gly153
Tanaka T, Biancotto A, Moaddel R, Moore AZ, Gonzalez-Freire M, Aon MA, Candia J, Zhang P, Cheung F, Fantoni G, CHI consortium, Semba RD, Ferrucci L (2018) Plasma proteomic signature of age in healthy humans. Aging Cell 17:e12799. https://doi.org/10.1111/acel.12799
Lehallier B, Gate D, Schaum N, Nanasi T, Lee SE, Yousef H, Moran Losada P, Berdnik D, Keller A, Verghese J, Sathyan S, Franceschi C, Milman S, Barzilai N, Wyss-Coray T (2019) Undulating changes in human plasma proteome profiles across the lifespan. Nat Med 25:1843–1850. https://doi.org/10.1038/s41591-019-0673-2
Corre J, Hébraud B, Bourin P (2013) Concise review: growth differentiation factor 15 in pathology: a clinical role? Stem Cells Transl Med 2:946–952. https://doi.org/10.5966/sctm.2013-0055
Adela R, Banerjee SK (2015) GDF-15 as a target and biomarker for diabetes and cardiovascular diseases: a translational prospective. J Diabetes Res 2015:490842–490814. https://doi.org/10.1155/2015/490842
Fujita Y, Ito M, Kojima T, Yatsuga S, Koga Y, Tanaka M (2015) GDF15 is a novel biomarker to evaluate efficacy of pyruvate therapy for mitochondrial diseases. Mitochondrion 20:34–42. https://doi.org/10.1016/j.mito.2014.10.006
Liu JJ, Foo JP, Liu S, Lim SC (2015) The role of fibroblast growth factor 21 in diabetes and its complications: a review from clinical perspective. Diabetes Res Clin Pract 108:382–389. https://doi.org/10.1016/j.diabres.2015.02.032
Salminen A, Kaarniranta K, Kauppinen A (2017) Regulation of longevity by FGF21: interaction between energy metabolism and stress responses. Ageing Res Rev 37:79–93. https://doi.org/10.1016/j.arr.2017.05.004
Xie T, Leung PS (2017) Fibroblast growth factor 21: a regulator of metabolic disease and health span. Am J Physiol Endocrinol Metab 313:E292–E302. https://doi.org/10.1152/ajpendo.00101.2017
Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G et al (2017) Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 25:1374–1389.e6. https://doi.org/10.1016/j.cmet.2017.04.021
Thummasorn S, Shinlapawittayatorn K, Khamseekaew J, Jaiwongkam T, Chattipakorn SC, Chattipakorn N (2018) Humanin directly protects cardiac mitochondria against dysfunction initiated by oxidative stress by decreasing complex I activity. Mitochondrion 38:31–40. https://doi.org/10.1016/j.mito.2017.08.001
Ristow M, Zarse K (2010) How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 45:410–418. https://doi.org/10.1016/j.exger.2010.03.014
Klaus S, Ost M (2020) Mitochondrial uncoupling and longevity - a role for mitokines? Exp Gerontol 130:110796. https://doi.org/10.1016/j.exger.2019.110796
Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, Hewett TE, Breit SN, Molkentin JD (2006) GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res 98:342–350
Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, Bolomini-Vittori M, Korf-Klingebiel M, Napp LC, Hansen B, Kanwischer A, Bavendiek U, Beutel G, Hapke M, Sauer MG, Laudanna C, Hogg N, Vestweber D, Wollert KC (2011) GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med 17:581–588
Kalli M, Minia A, Pliaka V, Fotis C, Alexopoulos LG, Stylianopoulos T (2019) Solid stress-induced migration is mediated by GDF15 through Akt pathway activation in pancreatic cancer cells. Sci Rep 9:978. https://doi.org/10.1038/s41598-018-37425-6
Tarfiei GA, Shadboorestan A, Montazeri H, Rahmanian N, Tavosi G, Ghahremani MH (2019) GDF15 induced apoptosis and cytotoxicity in A549 cells depends on TGFBR2 expression. Cell Biochem Funct 37:320–330. https://doi.org/10.1002/cbf.3391
Chung HK, Kim JT, Kim HW, Kwon M, Kim SY, Shong M, Kim KS, Yi HS (2017) GDF15 deficiency exacerbates chronic alcohol- and carbon tetrachloride-induced liver injury. Sci Rep 7:17238
Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN (1997) MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A 94:11514–11519
Tan M, Wang Y, Guan K, Sun Y (2000) PTGF-beta, a type beta transforming growth factor (TGF-beta) superfamily member, is a p53 target gene that inhibits tumor cell growth via TGF-beta signaling pathway. Proc Natl Acad Sci U S A 97:109–114. https://doi.org/10.1073/pnas.97.1.109
Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM et al (2019) GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell 178:1231–1244.e11
Conte M, Martucci M, Mosconi G, Chiariello A, Cappuccilli M, Totti V, Santoro A, Franceschi C, Salvioli S (2020) GDF15 plasma level is inversely associated with level of physical activity and correlates with markers of inflammation and muscle weakness. Front Immunol 11:915. https://doi.org/10.3389/fimmu.2020.00915
Freeman CM, Martinez CH, Todt JC, Martinez FJ, Han MK, Thompson DL et al (2015) Acute exacerbations of chronic obstructive pulmonary disease are associated with decreased CD4+ & CD8+ T cells and increased growth & differentiation factor-15 (GDF-15) in peripheral blood. Respir Res 16:94. https://doi.org/10.1186/s12931-015-0251-1
de Jager SC, Bermúdez B, Bot I, Koenen RR, Bot M, Kavelaars A et al (2011) Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J Exp Med 208:217–225. https://doi.org/10.1084/jem.20100370
Bonaterra GA, Zügel S, Thogersen J, Walter SA, Haberkorn U, Strelau J, Kinscherf R (2012) Growth differentiation factor-15 deficiency inhibits atherosclerosis progression by regulating interleukin-6-dependent inflammatory response to vascular injury. J Am Heart Assoc 1:e002550. https://doi.org/10.1161/JAHA.112.002550
Breit SN, Johnen H, Cook AD, Tsai VW, Mohammad MG, Kuffner T et al (2011) The TGF-β superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors 29:187–195. https://doi.org/10.3109/08977194.2011.607137
Lerner L, Tao J, Liu Q, Nicoletti R, Feng B, Krieger B, Mazsa E, Siddiquee Z, Wang R, Huang L, Shen L, Lin J, Vigano A, Chiu MI, Weng Z, Winston W, Weiler S, Gyuris J (2016) MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J Cachexia Sarcopenia Muscle 7:467–482. https://doi.org/10.1002/jcsm.12077
Tsai VW, Brown DA, Breit SN (2018) Targeting the divergent TGFβ superfamily cytokine MIC-1/GDF15 for therapy of anorexia/cachexia syndromes. Curr Opin Support Palliat Care 12:404–409. https://doi.org/10.1097/SPC.0000000000000384
Wiklund FE, Bennet AM, Magnusson PK, Eriksson UK, Lindmark F, Wu L et al (2010) Macrophage inhibitory cytokine-1 (MIC-1/GDF15): a new marker of all-cause mortality. Aging Cell 9:1057–1064. https://doi.org/10.1111/j.1474-9726.2010.00629.x
Wollert KC, Kempf T, Wallentin L (2017) Growth differentiation factor 15 as a biomarker in cardiovascular disease. Clin Chem 63:140–151. https://doi.org/10.1373/clinchem.2016.255174
Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjær SB, Wu X, Huang T, Hultman K, Paulsen SJ, Wang J, Bugge A, Frantzen JB, Nørgaard P, Jeppesen JF, Yang Z, Secher A, Chen H, Li X, John LM, Shan B, He Z, Gao X, Su J, Hansen KT, Yang W, Jørgensen SB (2017) GFRAL is the receptor for GDF15 and is required for the anti-obesity effects of the ligand. Nat Med 23:1158–1166. https://doi.org/10.1038/nm.4394
Mullican SE, Lin-Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, Beck SC, South VJ, Dinh TQ, Cash-Mason TD, Cavanaugh CR, Nelson S, Huang C, Hunter MJ, Rangwala SM (2017) GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med 23:1150–1157. https://doi.org/10.1038/nm.4392
Lee DH, Yang Y, Lee SJ, Kim K, Koo TH, Shin SM, Song KS, Lee YH, Kim YJ, Lee JJ, Choi I, Lee JH (2003) Macrophage inhibitory cytokine-1 induces the invasiveness of gastric cancer cells by up-regulating the urokinase-type plasminogen activator system. Cancer Res 63:4648–4655
Codó P, Weller M, Kaulich K, Schraivogel D, Silginer M, Reifenberger G et al (2016) Control of glioma cell migration and invasiveness by GDF-15. Oncotarget 7:7732–7746. https://doi.org/10.18632/oncotarget.6816
Min KW, Liggett JL, Silva G, Wang R, Shen R, Eling TE et al (2016) NAG-1/GDF15 accumulates in the nucleus and modulates transcriptional regulation of the Smad pathway. Oncogene 35:377–388. https://doi.org/10.1038/onc.2015.95
Lu Y, Ma J, Li Y, Huang J, Zhang S, Yin Z, Ren J, Huang K, Wu G, Yang K, Xu S (2017) CDP138 silencing inhibits TGF-β/Smad signaling to impair radioresistance and metastasis via GDF15 in lung cancer. Cell Death Dis 8:e3036. https://doi.org/10.1038/cddis.2017.434
Olsen OE, Skjærvik A, Størdal BF, Sundan A, Holien T (2017) TGF-β contamination of purified recombinant GDF15. PLoS One 12:e0187349. https://doi.org/10.1371/journal.pone.0187349
Hashimoto Y, Kurita M, Aiso S, Nishimoto I, Matsuoka M (2009) Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX-1/gp130. Mol Biol Cell 20:2864–2873. https://doi.org/10.1091/mbc.E09-02-0168
Zhang X, Urbieta-Caceres VH, Eirin A, Bell CC, Crane JA, Tang H, Jordan KL, Oh YK, Zhu XY, Korsmo MJ, Bachar AR, Cohen P, Lerman A, Lerman LO (2012) Humanin prevents intra-renal microvascular remodeling and inflammation in hypercholesterolemic ApoE deficient mice. Life Sci 91:199–206. https://doi.org/10.1016/j.lfs.2012.07.010
Zhao ST, Zhao L, Li JH (2013) Neuroprotective peptide humanin inhibits inflammatory response in astrocytes induced by lipopolysaccharide. Neurochem Res 38:581–588. https://doi.org/10.1007/s11064-012-0951-6
Gottardo MF, Jaita G, Magri ML, Zárate S, Ayala MM, Ferraris J et al (2014) Antiapoptotic factor humanin is expressed in normal and tumoral pituitary cells and protects them from TNF-α-induced apoptosis. PLoS One 9:e111548. https://doi.org/10.1371/jpuornal.pone.0111548
Ikonen M, Liu B, Hashimoto Y, Ma L, Lee KW, Niikura T, Nishimoto I, Cohen P (2003) Interaction between the Alzheimer's survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc Natl Acad Sci U S A 100:13042–13047
Rensink AA, Gellekink H, Otte-Höller I, ten Donkelaar HJ, de Waal RM, Verbeek MM, Kremer B (2002) Expression of the cytokine leukemia inhibitory factor and pro-apoptotic insulin-like growth factor binding protein-3 in Alzheimer's disease. Acta Neuropathol 104:525–533
Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC, Reed JC (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423:456–461
Zacharias DG, Kim SG, Massat AE, Bachar AR, Oh YK, Herrmann J, Rodriguez-Porcel M, Cohen P, Lerman LO, Lerman A (2012) Humanin, a cytoprotective peptide, is expressed in carotid atherosclerotic [corrected] plaques in humans. PLoS One 7:e31065. https://doi.org/10.1371/journal.pone.0031065
Kuliawat R, Klein L, Gong Z, Nicoletta-Gentile M, Nemkal A, Cui L, Bastie C, Su K, Huffman D, Surana M, Barzilai N, Fleischer N, Muzumdar R (2013) Potent humanin analog increases glucose-stimulated insulin secretion through enhanced metabolism in the β cell. FASEB J 27:4890–4898. https://doi.org/10.1096/fj.13-231092
Thummasorn S, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N (2017) High-dose humanin analogue applied during ischemia exerts cardioprotection against ischemia/reperfusion injury by reducing mitochondrial dysfunction. Cardiovasc Ther:35. https://doi.org/10.1111/1755-5922.12289
Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A (2018) (2018a) Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14(10):576–590. https://doi.org/10.1038/s41574-018-0059-4
Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, Grignolio A, Monti D, Capri M, Salvioli S (2018b) The continuum of aging and age-related diseases: common mechanisms but different rates. Front Med (Lausanne) 5:61. https://doi.org/10.3389/fmed.2018.00061
Yen K, Mehta HH, Kim SJ, Lue Y, Hoang J, Guerrero N et al (2020) The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging (Albany NY) 12:10.18632/aging.103534
Salemi M, Ridolfo F, Salluzzo MG, Cannarrella R, Giambirtone M, Caniglia S, Tirolo C, Ferri R, Romano C (2020) Humanin gene expression in fibroblast of Down syndrome subjects. Int J Med Sci 17:320–324. https://doi.org/10.7150/ijms.39145
Ji K, Zheng J, Lv J, Xu J, Ji X, Luo YB, Li W, Zhao Y, Yan C (2015) Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR-YY1-PGC1α pathway. Free Radic Biol Med 84:161–170. https://doi.org/10.1016/j.freeradbiomed.2015.03.020
Sa-Nguanmoo P, Tanajak P, Kerdphoo S, Satjaritanun P, Wang X, Liang G et al (2016) FGF21 improves cognition by restored synaptic plasticity, dendritic spine density, brain mitochondrial function and cell apoptosis in obese-insulin resistant male rats. Horm Behav 85:86–95. https://doi.org/10.1016/j.yhbeh.2016.08.006
Restelli LM, Oettinghaus B, Halliday M, Agca C, Licci M, Sironi L, Savoia C, Hench J, Tolnay M, Neutzner A, Schmidt A, Eckert A, Mallucci G, Scorrano L, Frank S (2018) Neuronal mitochondrial dysfunction activates the integrated stress response to induce fibroblast growth factor 21. Cell Rep 2018;24(6):1407–1414. doi: https://doi.org/10.1016/j.celrep.2018.07.023
Yu Y, He J, Li S, Song L, Guo X, Yao W, Zou D, Gao X, Liu Y, Bai F, Ren G, Li D (2016) Fibroblast growth factor 21 (FGF21) inhibits macrophage-mediated inflammation by activating Nrf2 and suppressing the NF-κB signaling pathway. Int Immunopharmacol 38:144–152. https://doi.org/10.1016/j.intimp.2016.05.026
Gao J, Liu Q, Li J, Hu C, Zhao W, Ma W, Yao M, Xing L (2020) Fibroblast growth factor 21 dependent TLR4/MYD88/NF-κB signaling activation is involved in lipopolysaccharide-induced acute lung injury. Int Immunopharmacol 80:106219. https://doi.org/10.1016/j.intimp.2020.106219
Jia H, Cheng J, Zhou Q, Peng J, Pan Y, Han H (2018) Fibroblast growth factor 21 attenuates inflammation and oxidative stress in atherosclerotic rat via enhancing the Nrf1-ARE signaling pathway. Int J Clin Exp Pathol 11:1308–1317
Youm YH, Horvath TL, Mangelsdorf DJ, Kliewer SA, Dixit VD (2016) Prolongevity hormone FGF21 protects against immune senescence by delaying age-related thymic involution. Proc Natl Acad Sci U S A 113:1026–1031. https://doi.org/10.1073/pnas.1514511113
Sanchis-Gomar F, Pareja-Galeano H, Santos-Lozano A, Garatachea N, Fiuza-Luces C, Venturini L, Ricevuti G, Lucia A, Emanuele E (2015) A preliminary candidate approach identifies the combination of chemerin, fetuin-A, and fibroblast growth factors 19 and 21 as a potential biomarker panel of successful aging. Age (Dordr) 37:9776. https://doi.org/10.1007/s11357-015-9776-y
Rath E, Berger E, Messlik A, Nunes T, Liu B, Kim SC, Hoogenraad N, Sans M, Sartor RB, Haller D (2012) Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61:1269–1278. https://doi.org/10.1136/gutjnl-2011-300767
Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM (2014) Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516:414–417. https://doi.org/10.1038/nature13818
Lin YF, Haynes CM (2016) Metabolism and the UPR(mt). Mol Cell 61:677–682. https://doi.org/10.1016/j.molcel.2016.02.004
Martinez BA, Petersen DA, Gaeta AL, Stanley SP, Caldwell GA, Caldwell KA (2017) Dysregulation of the mitochondrial unfolded protein response induces non-apoptotic dopaminergic neurodegeneration in C. elegans models of Parkinson's disease. J Neurosci 37:11085–11100. https://doi.org/10.1523/JNEUROSCI.1294-17.2017
Lu H, Tang S, Xue C, Liu Y, Wang J, Zhang W et al (2019) Mitochondrial-derived peptide MOTS-c increases adipose thermogenic activation to promote cold adaptation. Int J Mol Sci 20:E2456. https://doi.org/10.3390/ijms20102456
Kim SJ, Mehta HH, Wan J, Kuehnemann C, Chen J, Hu JF et al (2018) Mitochondrial peptides modulate mitochondrial function during cellular senescence. Aging (Albany NY) 10(6):1239–1256. https://doi.org/10.18632/aging.101463
Zhai D, Ye Z, Jiang Y, Xu C, Ruan B, Yang Y, Lei X, Xiang A, Lu H, Zhu Z, Yan Z, Wei D, Li Q, Wang L, Lu Z (2017) MOTS-c peptide increases survival and decreases bacterial load in mice infected with MRSA. Mol Immunol 92:151–160. https://doi.org/10.1016/j.molimm.2017.10.017
Yan Z, Zhu S, Wang H, Wang L, Du T, Ye Z et al (2019) MOTS-c inhibits osteolysis in the mouse calvaria by affecting osteocyte-osteoclast crosstalk and inhibiting inflammation. Pharmacol Res 147:104381. https://doi.org/10.1016/j.phrs.2019.104381
Fuku N, Pareja-Galeano H, Zempo H, Alis R, Arai Y, Lucia A, Hirose N (2015) The mitochondrial-derived peptide MOTS-c: a player in exceptional longevity? Aging Cell 14:921–923. https://doi.org/10.1111/acel.12389
Nashine S, Cohen P, Nesburn AB, Kuppermann BD, Kenney MC (2018) Characterizing the protective effects of SHLP2, a mitochondrial-derived peptide, in macular degeneration. Sci Rep 8:15175. https://doi.org/10.1038/s41598-018-33290-5
Mehta HH, Xiao J, Ramirez R, Miller B, Kim SJ, Cohen P, Yen K (2019) Metabolomic profile of diet induced obesity mice in response to humanin and small humanin-like peptide 2 treatment. Metabolomics 15:88. https://doi.org/10.1007/s11306-019-1549-7
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Ageing Dev 128:92–105
Lio D, Scola L, Crivello A, Colonna-Romano G, Candore G, Bonafè M, Cavallone L, Franceschi C, Caruso C (2002) Gender-specific association between −1082 IL-10 promoter polymorphism and longevity. Genes Immun 3:30–33
Bucci L, Ostan R, Cevenini E, Pini E, Scurti M, Vitale G, Mari D, Caruso C, Sansoni P, Fanelli F, Pasquali R, Gueresi P, Franceschi C, Monti D (2016) Centenarians' offspring as a model of healthy aging: a reappraisal of the data on Italian subjects and a comprehensive overview. Aging (Albany NY) 8:510–519
Storci G, De Carolis S, Papi A, Bacalini MG, Gensous N, Marasco E et al (2019) Genomic stability, anti-inflammatory phenotype, and up-regulation of the RNAseH2 in cells from centenarians. Cell Death Differ 26:1845–1858. https://doi.org/10.1038/s41418-018-0255-8
Funding
Open access funding provided by Alma Mater Studiorum - Università di Bologna within the CRUI-CARE Agreement. The study was partially supported by the Roberto and Cornelia Pallotti Legacy for Cancer Research to S.S., Russian Federation mega grant “DPM-AGEING” (grant 074-02-2018-330) on Digitalized and Personalized Medicine of Healthy Ageing, 2018–2021, at the Lobachevsky State University of Nizhny Novgorod to C.F.
Author information
Authors and Affiliations
Contributions
Maria Conte and Stefano Salvioli: concept and writing of the manuscript. Morena Martucci and Antonio Chiariello: literature search and manuscript revision. Claudio Franceschi: critical discussion. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
This article is a contribution to the special issue on: Immunosenescence: New Biomedical Perspectives - Guest Editors: Claudio Franceschi, Aurelia Santoro and Miriam Capri
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Conte, M., Martucci, M., Chiariello, A. et al. Mitochondria, immunosenescence and inflammaging: a role for mitokines?. Semin Immunopathol 42, 607–617 (2020). https://doi.org/10.1007/s00281-020-00813-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-020-00813-0