
Abstract
Background
Although T cells, especially CD8+, have been implicated in chronic obstructive pulmonary disease (COPD) pathogenesis, their role during acute exacerbations (AE-COPD) is uncertain.
Methods
We recruited subjects with COPD and a history of previous AE-COPD and studied them quarterly to collect blood and spontaneously expectorated sputum while stable. During exacerbations (defined by a change in symptoms plus physician diagnosis and altered medications), we collected blood and sputum before administering antibiotics or steroids. We used flow cytometry to identify leukocytes in peripheral blood, plus Luminex® analysis or ELISA to determine levels of inflammatory biomarkers in serum and sputum supernatants.
Results
Of 33 enrolled subjects, 13 participated in multiple stable visits and had ≥1 AE-COPD visit, yielding 18 events with paired data. Flow cytometric analyses of peripheral blood demonstrated decreased CD4+ and CD8+ T cells during AE-COPD (both absolute and as a percentage of all leukocytes) and significantly increased granulocytes, all of which correlated significantly with serum C-reactive protein (CRP) concentrations. No change was observed in other leukocyte populations during AE-COPD, although the percentage of BDCA-1+ dendritic cells expressing the activation markers CD40 and CD86 increased. During AE-COPD, sICAM-1, sVCAM-1, IL-10, IL-15 and GDF-15 increased in serum, while in sputum supernatants, CRP and TIMP-2 increased and TIMP-1 decreased.
Conclusions
The decrease in CD4+ and CD8+ T cells (but not other lymphocyte subsets) in peripheral blood during AE-COPD may indicate T cell extravasation into inflammatory sites or organized lymphoid tissues. GDF-15, a sensitive marker of cardiopulmonary stress that in other settings independently predicts reduced long-term survival, is acutely increased in AE-COPD. These results extend the concept that AE-COPD are systemic inflammatory events to which adaptive immune mechanisms contribute.
Trial registration
NCT00281216, ClinicalTrials.gov.
Similar content being viewed by others


Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive, debilitating and highly prevalent condition that is believed to result from an abnormal response to inhaled oxidants and which is characterized by lung destruction, airways remodeling, and mucus hypersecretion. COPD is punctuated by episodic worsening in respiratory function and symptoms, termed acute exacerbations of COPD (AE-COPD). These events are associated with accelerated lung function decline, reduced quality of life [1, 2], plus increased mortality, which is appreciable for events requiring hospitalization and which persists for at least five years [3, 4]. AE-COPD also drive the majority of the economic burden of this common disease [5]. Preventing these expensive and potentially lethal events should be a major public health goal.
Despite this great impact of AE-COPD, understanding of the responsible pathogenic mechanisms remains incomplete. There is consensus that a plurality of AE-COPD are associated with isolation of respiratory microbes, with some contribution from environmental triggers [6–8], and some events lacking evidence of inflammation, currently of incompletely understood etiology [9]. However, the acute beneficial role of systemic steroid therapy suggests that overly-exuberant inflammation, rather than simply presence of microbes, might determine symptomatology in most AE-COPD. Hence, continued investigation of inflammatory mechanisms during AE-COPD remains important.
We have hypothesized that AE-COPD would be associated with systemic evidence of inappropriate immune activation, especially of the adaptive immune system [10]. Lung T cells, especially CD8+ T cells, have been implicated in COPD pathogenesis [11–13], but data on their role in AE-COPD are limited. During AE-COPD, numbers of lymphocytes increase in both sputum and airway biopsies [14–16] and a CD8+ type-2-mediated immune reaction has been identified in sputum [17]. However, in the peripheral blood, a practical sample for monitoring systemic responses, there are few data during AE-COPD on the activation status and phenotype of immune cells or on their correlation with circulating biomarkers. The lack of universally-accepted animal models of AE-COPD necessitates use of human samples to address this question.
To that end, we designed a prospective cohort study to investigate subjects during the stable state and at onset of AE-COPD. Peripheral blood was used for both immunophenotyping of various leukocytes populations and to analyze 41 analytes in serum. Sputum supernatants were used to analyze 36 analytes. Our results provide novel data supporting AE-COPD as systemic inflammatory events to which T cells contribute, and which even when not requiring hospitalization, are associated with pulmonary vascular stress.
Materials & methods
Study design
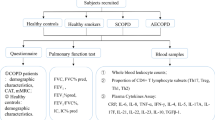
This observational longitudinal cohort study aimed to collect biomarkers of AE-COPD, for paired, within-subject comparison with samples collected at enrollment during clinical stability. Accordingly, we recruited participants at high risk of repeated AE-COPD who agreed to return to the study sites during suspected exacerbations. Individual subjects were followed for up to 3 years.
Ethics, consent and permissions
Subjects were recruited both from Pulmonary and General Medicine clinics at VA Ann Arbor Healthcare System (VAAAHS) and the University of Michigan Health System (UMHS) and by use of additional media (flyers, UMHS website for clinical research volunteers). Studies and consent procedures were performed in accordance with the Declaration of Helsinki and were approved by each site’s Institutional Review Board. Written consent to participate was obtained before any study procedures. All participants understood the purpose of the study and consented to publication of individual data. The study period was from December 2006 until March 2010.
Subject population & participation
Subjects were enrolled while clinically stable ≥6 weeks post AE-COPD or other use of antibiotics. Inclusion criteria were a diagnosis of COPD by GOLD guidelines [18]; a post-bronchodilator forced expiratory volume in one second (FEV1) of ≤ 70 % predicted; cigarette smoking exposure ≥ 20 pack years; 40 years ≤ age ≤ 80 years; daily productive cough ≥3 months annually for two consecutive years; and at least one AE-COPD requiring medical attention and a medication change (addition of oral steroids, antibiotics or both) annually for the previous three years.
Exclusion criteria included unstable heart disease; illnesses anticipated to result in death within two years; prednisone > 20 mg per day; asthma, cystic fibrosis, clinically significant bronchiectasis, lung cancer, or other inflammatory or fibrotic lung diseases.
Participants underwent spirometry, prospectively-collected questionnaires, clinical evaluation by a pulmonologist, posterior-anterior and lateral chest radiographs and collection of peripheral blood and spontaneously expectorated sputum at enrollment and during unscheduled visits when they sought medical attention for presumptive AE-COPD. During scheduled follow-up visits when participants were clinically stable, study coordinators performed identical data collection and evaluation (omitting radiographs).
AE-COPD was defined as a change above baseline in respiratory symptoms (increased dyspnea, cough, and sputum volume or purulence) plus addition by a study physician of antibiotics, oral steroids or both, based on review of history, physical examination and chest radiographs to exclude pneumonia. A diagnosis of AE-COPD was made prior to collection of samples (so study physicians had no knowledge of biomarker results, including serum CRP) and of the Breathlessness, Cough, and Sputum Scale questionnaire. Only if the unscheduled visit was determined clinically to be an AE-COPD meriting treatment using oral steroids, antibiotics or both, were peripheral blood and spontaneously expectorated sputum specimens collected, which occurred before initiating any new medications.
Sample collection & processing
Peripheral blood was collected into two Vacutainer blood collection tubes (Becton Dickinson, Franklin Lakes, NJ). One containing heparin was used for whole blood flow cytometric analyses; the other without additives was allowed to clot for 30 min at room temperature before centrifugation and then serum was collected into cryovials and stored at −80 °C until analysis.
Spontaneously expectorated sputum was immediately transferred to the laboratory, on ice, for processing. Dense, non-liquid portions of sputum were selected and weighed. Sputolysin® (EMD Millipore, Billerica, MA) was prepared by dissolving a 1 mL vial in 9 mL distilled water. The prepared Sputolysin was added at 2x the weight of the sputum and incubated in a 37 °C waterbath with vortexing every 5 min until the sample became viscous. An equal volume of PBS was added and the mixture was filtered through a 48 μm nylon gauze mesh filter, centrifuged and the supernatant was stored at −80 °C until analysis.
Flow cytometry
Whole blood was dispersed into tubes (100 μL each) containing antibodies and incubated for 25 min, protected from light, with shaking. BD FACS™ Lysing Solution (Becton Dickinson) was used to lyse erythrocytes according to manufacturer’s instructions. We used monoclonal antibodies from a variety of vendors as indicated in Additional file 1: Supplemental Materials & Methods. We assessed viability in all experiments using a Live/Dead Fixable Near-IR Dead Cell Stain Kit for 633 nm excitation (Invitrogen, Carlsbad, CA). Immediately after staining, cells were fixed by addition of staining buffer containing 2 % paraformaldehyde; tubes were then stored at 4 °C in a rack wrapped in aluminum foil until analyzed.
Staining was analyzed on a LSR II flow cytometer (BD Bioscience, San Jose, CA), equipped with four lasers as described in detail [19]. Data were collected using FACSDiva software (BD Biosciences) with automatic compensation and were analyzed using FlowJo software (Tree Star, Ashland, OR). We collected at least 10,000 viable CD45+ events per sample in each experiment. Details of our gating strategy are provided in Additional file 1: Supplemental Materials & Methods, and have recently been published together with representative raw data [20].
Serum & sputum protein measurements
Serum and sputum samples were stored at −80 °C until all could be analyzed simultaneously. We used two methodologies to analyze protein concentrations. The majority of analytes were measured using a Luminex 200 system® (Luminex Corporation, Austin, TX), according to the manufacturer’s instructions. We used multiplex bead sets from a variety of vendors as indicated in Additional file 1: Supplemental Materials & Methods. For four analytes (GDF-15, IL-18, IL-23p19 and IFN-β), multiplex beads were not available; instead, ELISAs were used according to manufacturers’ instructions.
Statistical analyses
We used GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA) on a Macintosh Quad-Core Intel Xeon computer running OS 10.8.3 (Apple; Cupertino, CA). We used Wilcoxon matched-pairs signed rank test to look for differences between stable and exacerbation visits. A two-tailed p value of < 0.05 was considered to indicate significance. Spearman nonparametric correlation coefficient was used.
Results
Patient enrollment and completion
We enrolled 33 COPD subjects; six completed only the baseline visit and were excluded. Another 14 subjects completed multiple visits in the stable state; however, 13 did not complete an AE-COPD visit, despite an interval self-reported AE-COPD, and were excluded. A single subject did not have any AE-COPD. The remaining 13 subjects participated in multiple stable visits and had ≥1 AE-COPD visit, yielding 18 total events with paired stable and AE-COPD data. Age, sex, smoking history, spirometry, number of AE-COPD, and inhaled corticosteroid (ICS) use for each of the 13 subjects are shown (Table 1). All events were treated successfully in the outpatient setting.
Among the 18 captured AE-COPD events, 14 occurred 2–13 months after the preceding stable visit (5.3 ± 3.2 months, mean ± SD). However, in the other four, the AE-COPD occurred within one week after a scheduled stable visit. Given the possibility that data were not collected during a period of true stability, in these analyses we instead used paired data from a subsequent scheduled stable visit 2–5 months after the AE-COPD.
Exacerbations resulted in increased symptom scores and elevated CRP levels
All decisions to treat and hence to collect data at an AE-COPD visit were made by clinicians blinded to data from study questionnaires or biomarkers. However, as an independent confirmation that subjects were experiencing an actual AE-COPD, we retrospectively evaluated the severity of respiratory symptoms using the Breathlessness, Cough, and Sputum Scale (BCSS) [21]. The BCSS is based on a three-item questionnaire rating breathlessness, cough and sputum on a scale of 0 (no symptoms) to 4 (severe symptoms), which are summed. BCSS total score significantly increased during AE-COPD (Fig. 1a), from a median of 3.5 during stable visits to 8.0 during AE-COPD.

AE-COPD confirmed by increases in BCSS total score and serum CRP levels. AE-COPD events were identified by physician diagnosis (after exclusion of pneumonia by chest radiograph) plus the clinical decision to prescribe oral steroids or antibiotics. Although these decisions were blinded to biomarker results, the diagnosis of AE-COPD was retrospectively confirmed by highly significant increases in (a) BCSS total scores and (b) serum CRP levels. Open circles represent individual subjects; mean ± SEM of the grouped data are shown by the single red circles. The Wilcoxon matched-pairs signed rank test was used to determine significant differences between visits
A study published shortly before the launch of ours, which assessed the ability of 36 different biomarkers to confirm the presence of AE-COPD and to predict their severity, found CRP to be the most selective analyte [22]. Serum CRP levels in our subjects, which were measured by the clinical laboratories, were also significantly increased during AE-COPD (Fig. 1b) (stable, 0.58 ± 0.40 mg/dL vs. AE-COPD, 6.4 ± 7.5 mg/dL; mean ± SD). In our study, BCSS correlated with CRP levels (r = 0.43, p = 0.011). Collectively, these findings provide additional evidence that these events were valid AE-COPD.
Peripheral blood CD4+ and CD8+ T cells decreased during AE-COPD
CD4+ T cells and CD8+ T cells significantly decreased in the blood during AE-COPD, relative to the paired stable visit (Fig. 2a-d). This change occurred both as a percent of all CD45+ cells (CD4+ T cells: stable, 17.4 ± 1.7 % of CD45+ leukocytes vs. during AE-COPD, 10.0 ± 4.2 %; mean ± SEM; p = 0.002) (CD8+ T cells: stable, 10.4 ± 2.2 % vs. AE-COPD, 5.9 ± 1.0 %; p = 0.02) and in absolute numbers. By Spearman correlation analyses, CD4+ and CD8+ T cells correlated significantly and inversely with serum CRP, both relatively (percentage CD4+ T cells: r = −0.74, p < 0.0001; percentage CD8+ T cells: r = −0.46, p = 0.005) and as absolute cell numbers (CD4+ T cells: r = −0.60, p = 0.0001; CD8+ T cells: r = −0.41, p = 0.013).

Peripheral blood CD4+ and CD8+ T cells decreased during AE-COPD while CD15+ granulocytes increased. Peripheral blood was collected during stable visits and during AE-COPD, prior to medication (n = 18 paired samples). Cells were stained and analyzed by flow cytometry. Results are shown for the individual cell type as a percentage of all CD45+ leukocytes (a, c, e) and as absolute cell numbers per μL (b, d, f). A, B. CD4+ T cells. C, D. CD8+ T cells. E, F. CD15+ cells; note difference in scale of panels E & F. Open circles represent individual subjects; mean ± SEM of the grouped data are shown by the single red circles. The Wilcoxon matched-pairs signed rank test was used to determine significant differences between visits
We did not see changes during AE-COPD in the distribution in peripheral blood of B cells, NK cells or any of the three types of DCs, in either relative or absolute terms. However, CD15+ granulocytes increased significantly (Fig. 2e-f), both as a percent of CD45+ cells (stable, 54.5 ± 4.3 % of CD45+ cells vs. AE-COPD, 68.9 ± 3.3 %) and in absolute numbers. CD15+ cells correlated directly with serum CRP (percentage CD15+ cells, r = 0.59, p = 0.0003; absolute CD15+ cells, r = 0.49, p = 0.003).
Because the flow cytometric staining used in this study had not been designed to distinguish neutrophils from eosinophils, we retrospectively examined data from the differential cell counts performed by the clinical laboratories. There were no significant changes in eosinophils during AE-COPD in our subjects (stable, 2.59 ± 2.0 % vs. AE-COPD, 1.41 ± 0.83 %; mean ± SEM, n = 15 events; p = 0.170). By contrast, results of the clinical differential counts confirmed that the change in granulocytes we observed was due to an increase in neutrophils (stable, 70.71 ± 5.37 % vs. AE-COPD, 79.04 ± 8.29 %; p = 0.013). We found no change in numbers of platelets (stable, 266.6 ± 47.2 × 103/μL vs. AE-COPD, 258.4 ± 54.4 × 103/μL; p = 0.33).
Data from representative individuals demonstrated a recurring pattern, in which AE-COPD were marked by decreased percentages of both CD4+ and CD8+ T cells in peripheral blood, followed by a rebound in those cell types at the next stable visit (Fig. 3). Interestingly, in some subjects who had a scheduled visit shortly before a clinically-defined exacerbation (Fig. 3a, b), the decline in cell frequency could actually be observed shortly before the AE-COPD. This finding suggests that immune activation might precede symptoms, but a larger study will be needed to test that possibility. In contrast, the sole subject with no reported AE-COPD events did not display a change in T cell frequency (Fig. 3e).

Representative graphs from individual subjects tracking percentages of CD4+ and CD8+ T cells during stable and exacerbation visits. Peripheral blood was collected during stable visits and during AE-COPD, prior to medication. Cells were stained and analyzed by flow cytometry. Panels a-e each depicted the data for a single subject; CD4+ T cells (upper panels, blue) and CD8+ T cells (lower panels, red) shown as a percentage of all CD45+ leukocytes. a, b. Two subjects with multiple AE-COPD; c, d. two subjects with a single AE-COPD; and e, subject with no AE-COPD. Stable visits are shown by open circles and AE-COPD are shown in solid circles labeled with “Ex”
Activation markers CD40 and CD86 are increased on BDCA-1 DCs during AE-COPD
We also analyzed the surface expression of receptors specific to the various leukocyte populations. On both CD4+ and CD8+ T cells, we analyzed CD69, CD25, CD27, CD62L and IL-18R, which collectively contribute to defining T cell activation history. There was no difference in the percentage of T cells expressing these receptors between stable and AE-COPD visits, nor were there any significant changes in B cells or NK cells (data not shown). On the three DC population, we analyzed the activation and maturation markers CD40, CD80, CD83 and CD86. Despite an absence of change in the numbers or frequency of DCs, we found that the percentage of mDC1 that expressed CD40 and CD86 increased significantly during AE-COPD (Fig. 4). By contrast, expression of CD80 and CD83 did not change on mDC1, nor were there any significant changes in receptor expression on mDC2 or pDC (not shown).

CD40 and CD86 increased on BDCA-1+ DCs from peripheral blood during AE-COPD. Peripheral blood was collected during stable visits and during AE-COPD, prior to medication (n = 10 paired samples). Cells were stained and analyzed by flow cytometry. After gating on BDCA-1+ DCs, the percent of cells expressing (a) CD40 and (b) CD86 was determined. Open circles represent individual subjects; mean ± SEM of the grouped data are shown by the single red circles. The Wilcoxon matched-pairs signed rank test was used to determine significant differences between visits
Increased production of specific analytes in the serum during AE-COPD
We analyzed serum for 41 different analytes using a combination of Luminex® and ELISA assays (Table 2). In addition to CRP, considered above, five analytes increased significantly during AE-COPD: soluble intracellular adhesion molecule (sICAM)-1, soluble vascular cell adhesion molecule (sVCAM)-1, IL-10, IL-15, and growth and differentiation factor 15 (GDF-15) (Fig. 5a-e). Serum IL-15 measurements during stable visits were at or near the limit of detection, however IL-15 levels were significantly increased in all but one of the AE-COPD events. Interestingly, CCL11 showed a trend to decrease during AE-COPD (Fig. 5f), which did not attain statistical significance (stable, 79.7 ± 41.1 pg/mL vs. AE-COPD, 67.8 ± 56.7 pg/mL; mean ± SD; p = 0.052).

Soluble adhesion molecules and inflammatory mediators increased while CCL11 decreased in the serum during AE-COPD. Serum was collected during stable visits and during AE-COPD, prior to medication (n = 18 paired samples). Samples were stored at −80 °C prior to analysis by Luminex® assay, except as noted. (a) sICAM-1, (b) sVCAM-1, (c) IL-10, (d) IL-15, (e) GDF-15 (by ELISA), (f) CCL11 are shown. Open circles represent individual subjects; mean ± SEM of the grouped data are shown by the single red circles. Note differences in units of measurement. The dotted line represents the minimal detectable concentration. The Wilcoxon matched-pairs signed rank test was used to determine significant differences between visits
AE-COPD leads to decreased TIMP-1 and increased TIMP-2 production in the sputum
We also analyzed sputum supernatants for 36 different analytes using a combination of Luminex® and ELISA assays (Table 2). Significant increases during AE-COPD were found for CRP and tissue inhibitor of metalloproteinase (TIMP)-2, while TIMP-1 levels in sputum decreased during AE-COPD (Fig. 6). IL-6 and IL-8 levels in sputum also increased slightly during AE-COPD, but did not reach statistical significance (IL-6: stable, 117.1 ± 108.2 pg/mL vs. during AE-COPD, 225.9 ± 193.5 pg/mL; IL-8: stable, 3.7×103 ± 2.6×103 ng/mL vs. AE-COPD, 5.5×103 ± 4.3×103 ng/mL; mean ± SD, p = 0.16 for both analytes).

CRP and TIMP-2 increased in sputum during AE-COPD, whereas TIMP-1 decreased. Sputum was collected during stable visits and during AE-COPD, prior to medication. After processing, sputum supernatants were stored at −80 °C prior to analysis by Luminex. (a) CRP, (b) TIMP-1, (c) TIMP-2 are shown (n = 10 paired samples for CRP and n = 7 paired samples for TIMP-1 and TIMP-2). Open circles represent individual subjects; mean ± SEM of the grouped data are shown by the single red circles. The dotted line represents the minimal detectable concentration. The Wilcoxon matched-pairs signed rank test was used to determine significant differences between visits
Discussion
This observational study of paired samples from COPD subjects in the stable and exacerbated states demonstrates that CD4+ T cells and CD8+ T cells decreased in peripheral blood during AE-COPD. Decreases occurred both in absolute numbers and as a percentage of all leukocytes, and were noted repeatedly in those with multiple AE-COPD. We found no changes during AE-COPD in numbers or percentages of B cells, NK cells or DCs in peripheral blood, although numbers of neutrophils increased, as did expression of CD40 and CD86 by BDCA-1+ DC, compatible with activation. We also demonstrate significant increases during AE-COPD in serum levels of GDF-15, a sensitive marker of cardiopulmonary stress that in other settings predicts reduced long-term survival, and of IL-10, IL-15, sICAM and sVCAM. These results extend the concept that AE-COPD are systemic inflammatory events to which adaptive immune mechanisms contribute.
Determining whether lymphocyte recruitment should be targeted to improve AE-COPD clinical outcomes requires defining whether lung inflammation depends on that recruitment, or conversely can be mediated by the newly recognized and sizeable populations of lung resident lymphocytes [23, 24]. As assessed by bronchoalveolar lavage, CD4+ T cells are reduced in the lungs of active smokers without airflow obstruction [25] and in active smokers regardless of airflow obstruction [26], whereas bronchial biopsies showed increased intraepithelial CD4+ T cells in COPD patients, relative to smokers and never-smokers [27]. In terms of function, we recently showed that lung resident CD4+ T cells in many subjects with stable advanced COPD have very reduced ability to produce inflammatory cytokines, either spontaneously or on polyclonal stimulation, relative to subjects without COPD [28]. Thus, the CD4-dependent component of an AE-COPD should depend heavily on acute recruitment, and on subsequent activation by lung resident DC. Such activation is likely to be intact, because during clinical stability expression of co-stimulatory surface receptors by all three lung DC populations increases in correlation with spirometrically-defined COPD progression [29, 30]. Moreover, lung DC from COPD can polarize peripheral blood CD4+ T cells in vitro [31], suggesting a similar role in the lungs.
By contrast, relative to lung CD8+ T cells from smokers with preserved spirometry, lung resident CD8+ T cells from COPD subjects have greater capacity to secrete proinflammatory cytokines such as IFN-γ and TNF-α [11, 12]. Because such secretion can be triggered by IL-18 without the need for interaction with lung DC [11], lung resident CD8+ T may not only respond appropriately to cognate antigens (e.g., virally-infected lung epithelium), but might also indiscriminately sustain inflammation through bystander activation. Hence, the lung resident CD8+ T cell population, which increases in extent with worsening FEV1 [13, 32, 33], appears quite capable of stimulating lung damage by activating macrophages and by recruiting other inflammatory cell types. However, the peripheral blood of COPD patients also contains an increased proportion of IFN-γ + and TNF-α + CD8+ T cells, relative to healthy never-smokers [34]. Therefore, establishing whether the contribution of CD8+ T cells to inflammation in AE-COPD also depends in part on cells acutely recruited from the blood will be an important future goal.
The large body of experimental data on lymphocyte trafficking [35] implies that the decreases we found in blood CD4+ and CD8+ T cells during AE-COPD reflect recruitment both of naïve and memory lymphocytes into regional lymph nodes and organized lymphoid tissues (which develop within lung parenchyma in advanced COPD [33, 36]) and of effector-memory cells to the lungs. The finding that blood T cell expression of activation markers showed no net change might reflect these competing patterns of migration. It contrasts with evidence of T cell activation on presentation during severe asthma attacks [37]; the same study found that blood CD4+ T cells of five subjects hospitalized for AE-COPD showed increased expression of HLA-DR, which we did not measure, but not of CD25, in agreement with our data [37]. The disparity with the acute asthmatics might relate to the lesser degree of acute stress in our subjects, despite the severity of their underlying lung function in the stable state, or may be due to differences between the two diseases.
Our results in peripheral blood complement two studies showing increased percentages of total lymphocytes (defined by cytospin differential counts) in sputum in this setting [14, 15]. Collectively, these two studies and ours imply that T cell recruitment to the lungs is a component of AE-COPD, without proving causality in pathogenesis. By contrast, no change in percentages of lymphocytes in sputum or in density of CD4+ or CD8+ T cells in endobronchial biopsies was seen in an earlier study, which did, however, find a significant increase during exacerbation in airway wall numbers of CD3+ T cells [16]. Another study of sputum during AE-COPD found decreased CD4:CD8 ratios (assessed by immunofluorescent staining of cytospins), relative to paired samples in the stable state, but no change in percentages of lymphocytes (assessed morphologically) among total sputum leukocytes [17]. Thus, controversy remains about T cell recruitment to the lungs during AE-COPD.
We cannot address this controversy with our own sputum data. At the time of this study, we were not yet using flow cytometry to analyze sputum leukocyte subsets, as we are now doing successfully in stable COPD [20]. Our preliminary results suggest that to quantify lymphocytes in respiratory secretions accurately, flow cytometry will be superior to analysis of cytospins. This question is being examined prospectively in the Subpopulations and Intermediate Outcome Measures in COPD study (SPIROMICS) [38]. Supporting this possibility, paired comparisons of differential counts in bronchoalveolar lavage samples from humans and mice showed that cytospins result in preferential loss of lymphocytes, relative to more gentle sample preparation [39, 40]. Because flow cytometry also extends the identification of lymphocyte subsets and their activation states in sputum or other samples well beyond the capability of Giemsa-stained cytospins, we believe that it has considerable promise to investigate multiple airways diseases.
Our demonstration that serum GDF-15 levels rose in uncomplicated AE-COPD treated successfully in outpatients is significant, as this molecule has emerged as a key biomarker of cardiopulmonary stress, especially involving pulmonary vasculature. GDF-15 is a divergent TGF-beta superfamily member, first cloned from activated macrophages [41] and named Macrophage inhibitory cytokine-1 (MIC-1). However, the same gene was cloned by other strategies, leading to many alternative names [42–45]. GDF-15 plays key regulatory roles in processes as diverse as cell cycle progression, differentiation, maintenance of pregnancy, apoptosis and tumor progression [46, 47]. Serum levels of GDF-15 levels rise modestly in acute coronary syndrome and congestive heart failure, in both conditions accurately predicting one-year mortality [48–50], but are very elevated in acute pulmonary embolus [51] and pulmonary hypertension [52–54]. Elevated GDF-15 levels also predicted long-term mortality in a population-based Swedish male cohort (adjusted odd-ratio of death of 3.38, 95 % CI 1.38–8.26), independently of baseline IL-6 or CRP levels [55]. Hence, this molecule merits additional study as a COPD biomarker.
The elevation we demonstrated in serum IL-15 is significant due to its multiple actions that could potentiate lung inflammation during AE-COPD (reviewed in [56]). These include antigen-independent activation of CD8+ T cells and enhanced effector function of CD8+ T cells, NK cell and NK T cells. IL-15 also attracts and activates neutrophils, while promoting their retention and delaying apoptosis. This last action is shared with multiple types of leukocytes and respiratory epithelial cells, and might explain the previous observations that the fraction of circulating apoptotic neutrophils is deceased during AE-COPD [57, 58], although that finding might also reflect altered clearance. The lack of a parallel change in IL-15 expression in sputum might indicate that its source is not within the lungs, that its elaboration during AE-COPD is directed away from the airway lumen, or potentially that some factors during inflammation inhibited its immunodetection. Dissociation of biomarker concentrations between the peripheral blood and sputum compartments, reflected in our measurements of other biomarkers, is not unprecedented. A study analyzing the expression during the stable state of 100 biomarkers (including IL-15) in bronchoalveolar lavage, bronchial biopsies, serum and induced sputum from healthy smokers (n = 23) and COPD subjects (n = 24) observed no correlations between lung and serum biomarkers [59]. Additional investigation in larger cohorts is needed to determine the most suitable biomarkers to define the pathologic inflammatory processes occurring within the airways during AE-COPD, which may vary with the cause of specific episodes.
Multiple other circulating biomarkers of inflammation are upregulated during AE-COPD (reviewed in [60]). Although our finding of increased sVCAM is novel to our knowledge, the elevations detected in CRP, IL-10 and sICAM agree with some [22, 61, 62] but not all studies [63]. We also observed an increase in mean serum levels of both IL-6 and IL-8 during AE-COPD, as has been seen by some [22, 62, 64], but not all studies [9]; however, neither attained statistical difference from stable visits, likely due to our sample size. We did not see increased MPO, TNF-α, or TNF receptors I and II during AE-COPD, which others have reported.
Induced or spontaneously produced sputum has also been analyzed extensively during AE-COPD, with elevations previously shown for IL-1β, IL-6, TNF-α, TNF receptors I and II, CCL5, CCL4 [9], LTB4 [65] and MMP-9 [14]. We found marked but non-statistically significant elevations in sputum IL-1β concentrations and a non-significant trend towards higher CXCL10 in serum, which a previous analysis determined to be the two biomarkers that individually best discriminated bacterial AE-COPD from viral AE-COPD, respectively [9]. The broad range of these measures in our study could be taken to imply heterogeneous etiologies; because we did not perform microbiological analysis in this study, we cannot address that possibility. The current finding of reduced sputum TIMP-1 during AE-COPD, the only sputum analyte to decrease, agrees with a previous study [14]. By contrast, we noted increased TIMP-2, another inhibitor of MMPs. It is uncertain why the two TIMPs differed, especially as they are produced by multiple parenchymal lung cell types, including bronchial epithelial cells, type II alveolar pneumocytes and smooth muscle cells, plus by neutrophils and macrophages [66]. The functions of TIMP-1 and TIMP-2 during AE-COPD are unknown. Although neither has been directly implicated in lymphocyte recruitment to the lungs, TIMP-1 does appear to regulate CD4+ T cell migration into brain parenchyma in a murine model of viral encephalitis, independently of MMPs [67]. Furthermore, new data is constantly emerging on the ability of TIMPs to affect many other cellular functions, such as growth, migration, and apoptosis [68]. We did not see increases in any sputum biomarkers known to drive lymphocyte recruitment, which may be due to a lack of power, but might also argue that lymphocytes are not recruited to the lungs during AE-COPD. However, two inter-related alternatives should be considered: first, that chemokines responsible for lymphocyte recruitment into lung parenchyma might not be elevated in sputum due to preferential abluminal secretion and binding to matrix; second, that lymphocytes are recruited into lung parenchyma and especially intra-epithelial sites during AE-COPD, but do not transmigrate into airways and so cannot be detected in sputum. The latter possibility merits testing.
Strengths of the current study include the rigorous definition of AE-COPD as an event requiring a clinical decision to treat with steroids, antibiotics or both by a physician blinded to the biomarker data. The chief limitation is the small number of subjects having paired samples from stable and exacerbation visits. Based on the highly productive experience of the East London cohort, we had originally planned a 100 subject cohort. However, shortly after initiation of this study, both our sites joined the NHLBI-sponsored trial of macrolide therapy to prevent AE-COPD [69], which had similar entry criteria. Many potential subjects preferred to enroll in that interventional trial, and we had limited success in crossing over subjects on completion of their year of study drug. Additionally, 13 of our subjects had at least one self-reported AE-COPD for which they sought medical attention locally rather than coming for a study visit.
Conclusions
During event-defined AE-COPD, relative to paired samples collected during clinical stability, neutrophils increased while CD4+ and CD8+ T cells (but not B cells, NK cells or DC) decreased in peripheral blood, both relatively and absolutely and in significant correlation with serum CRP levels. There were also significant increases in serum concentrations of GDF-15, a biomarker of pulmonary vascular stress with known strong independent prognostic value for long-term survival, and of sICAM-1, sVCAM-1, IL-10 and IL-15. These findings support the growing evidence that dysregulated inflammation contributes to the morbidity of AE-COPD and their sustained adverse effect on survival.
During review of this article, GDF-15 was reported to be elevated in 29 subjects hospitalized for AE-COPD, relative to a separate group of subjects with stable COPD and to healthy control subjects [70].
Abbreviations
- AE-COPD:
-
Acute exacerbation of chronic obstructive pulmonary disease
- BCSS:
-
Breathlessness, cough, and sputum scale
- COPD:
-
Chronic obstructive pulmonary disease
- CRP:
-
C-reactive protein
- DC:
-
Dendritic cells
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GDF-15:
-
Growth and differentiation factor 15
- ICS:
-
Inhaled corticosteroids
- NK:
-
Natural killer
- sICAM-1:
-
soluble intracellular adhesion molecule-1
- sVCAM-1:
-
soluble vascular adhesion molecule-1
- TIMP:
-
Tissue inhibitor of metalloproteinase
- UMHS:
-
University of Michigan Health System
- VAAAHS:
-
Veterans Affairs Ann Arbor Healthcare System
References
Donaldson GC, Seemungal TA, Bhowmik A, Wedzicha JA. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57:847–52.
Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157:1418–22.
Singanayagam A, Schembri S, Chalmers JD. Predictors of mortality in hospitalized adults with acute exacerbation of chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2013;10:81–9.
Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R. Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary disease. Thorax. 2005;60:925–31.
Toy EL, Gallagher KF, Stanley EL, Swensen AR, Duh MS. The economic impact of exacerbations of chronic obstructive pulmonary disease and exacerbation definition: a review. COPD. 2010;7:214–28.
Seemungal T, Harper-Owen R, Bhowmik A, Moric I, Sanderson G, Message S, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1618–23.
Patel IS, Seemungal TA, Wilks M, Lloyd-Owen SJ, Donaldson GC, Wedzicha JA. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax. 2002;57:759–64.
MacNee W, Donaldson K. Exacerbations of COPD: environmental mechanisms. Chest. 2000;117:390S–7S.
Bafadhel M, McKenna S, Terry S, Mistry V, Reid C, Haldar P, et al. Acute exacerbations of chronic obstructive pulmonary disease: identification of biologic clusters and their biomarkers. Am J Respir Crit Care Med. 2011;184:662–71.
Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc. 2007;4:512–21.
Freeman CM, Han MK, Martinez FJ, Murray S, Liu LX, Chensue SW, et al. Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol. 2010;184:6504–13.
Freeman CM, Martinez FJ, Han MK, Washko Jr GR, McCubbrey AL, Chensue SW, et al. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir Res. 2013;14:13.
Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, et al. CD8 + ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:711–7.
Mercer PF, Shute JK, Bhowmik A, Donaldson GC, Wedzicha JA, Warner JA. MMP-9, TIMP-1 and inflammatory cells in sputum from COPD patients during exacerbation. Respir Res. 2005;6:151.
Fujimoto K, Yasuo M, Urushibata K, Hanaoka M, Koizumi T, Kubo K. Airway inflammation during stable and acutely exacerbated chronic obstructive pulmonary disease. Eur Respir J. 2005;25:640–6.
Saetta M, Di Stefano A, Maestrelli P, Turato G, Ruggieri MP, Roggeri A, et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med. 1994;150:1646–52.
Tsoumakidou M, Tzanakis N, Chrysofakis G, Kyriakou D, Siafakas NM. Changes in sputum T-lymphocyte subpopulations at the onset of severe exacerbations of chronic obstructive pulmonary disease. Respir Med. 2005;99:572–9.
Rabe KF, Hurd S, Anzueto A, Barnes PJ, Buist SA, Calverley P, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176:532–55.
McCubbrey AL, Sonstein J, Ames TM, Freeman CM, Curtis JL. Glucocorticoids relieve collectin-driven suppression of apoptotic cell uptake in murine alveolar macrophages through downregulation of SIRPalpha. J Immunol. 2012;189:112–9.
Freeman CM, Crudgington S, Stolberg VR, Brown JP, Sonstein J, Alexis NE, et al. Design of a multi-center immunophenotyping analysis of peripheral blood, sputum and bronchoalveolar lavage fluid in the Subpopulations and Intermediate Outcome Measures in COPD Study (SPIROMICS). J Transl Med. 2015;13:19.
Leidy NK, Rennard SI, Schmier J, Jones MK, Goldman M. The breathlessness, cough, and sputum scale: the development of empirically based guidelines for interpretation. Chest. 2003;124:2182–91.
Hurst JR, Donaldson GC, Perera WR, Wilkinson TM, Bilello JA, Hagan GW, et al. Use of plasma biomarkers at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;174:867–74.
Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol. 2013;31:137–61.
Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One. 2011;6, e16245.
Hoser G, Kawiak J, Domagala-Kulawik J, Kopinski P, Droszcz W. Flow cytometric evaluation of lymphocyte subpopulations in BALF of healthy smokers and nonsmokers. Folia Histochem Cytobiol. 1999;37:25–30.
Forsslund H, Mikko M, Karimi R, Grunewald J, Wheelock AM, Wahlstrom J, et al. Distribution of T-Cell subsets in BAL fluid of patients with mild to moderate COPD depends on current smoking status and not airway obstruction. Chest. 2014;145:711–22.
Lofdahl MJ, Roos-Engstrand E, Pourazar J, Bucht A, Dahlen B, Elmberger G, et al. Increased intraepithelial T-cells in stable COPD. Respir Med. 2008;102:1812–8.
Freeman CM, McCubbrey AL, Crudgington S, Nelson J, Martinez FJ, Han MK, et al. Basal gene expression by lung CD4+ T cells in chronic obstructive pulmonary disease identifies independent molecular correlates of airflow obstruction and emphysema extent. PLoS One. 2014;9, e96421.
Freeman CM, Martinez FJ, Han MK, Ames TM, Chensue SW, Todt JC, et al. Lung dendritic cell expression of maturation molecules increases with worsening chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180:1179–88.
Kheradmand F, Shan M, Corry DB. Smoking gun: mature dendritic cells in human lung provide clues to chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180:1166–7.
Shan M, Cheng HF, Song LZ, Roberts L, Green L, Hacken-Bitar J, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1:4ra10.
Turato G, Zuin R, Saetta M. Pathogenesis and pathology of COPD. Respiration. 2001;68:117–28.
Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–53.
Paats MS, Bergen IM, Hoogsteden HC, van der Eerden MM, Hendriks RW. Systemic CD4+ and CD8+ T-cell cytokine profiles correlate with GOLD stage in stable COPD. Eur Respir J. 2012;40:330–7.
Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–20.
van der Strate BW, Postma DS, Brandsma CA, Melgert BN, Luinge MA, Geerlings M, et al. Cigarette smoke-induced emphysema: A role for the B cell? Am J Respir Crit Care Med. 2006;173:751–8.
Corrigan CJ, Hartnell A, Kay AB. T lymphocyte activation in acute severe asthma. Lancet. 1988;1:1129–32.
Couper D, Lavange LM, Han M, Barr RG, Bleecker E, Hoffman EA, et al. Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS). Thorax. 2013;69:491–4.
Saltini C, Hance AJ, Ferrans VJ, Basset F, Bitterman PB, Crystal RG. Accurate quantification of cells recovered by bronchoalveolar lavage. Am Rev Respir Dis. 1984;130:650–8.
Curtis JL, Kaltreider HB. Characterization of bronchoalveolar lymphocytes during a specific antibody-forming cell response in the lungs of mice. Am Rev Respir Dis. 1989;139:393–400.
Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci U S A. 1997;94:11514–9.
Lawton LN, Bonaldo MF, Jelenc PC, Qiu L, Baumes SA, Marcelino RA, et al. Identification of a novel member of the TGF-beta superfamily highly expressed in human placenta. Gene. 1997;203:17–26.
Hromas R, Hufford M, Sutton J, Xu D, Li Y, Lu L. PLAB, a novel placental bone morphogenetic protein. Biochim Biophys Acta. 1997;1354:40–4.
Yokoyama-Kobayashi M, Saeki M, Sekine S, Kato S. Human cDNA encoding a novel TGF-beta superfamily protein highly expressed in placenta. J Biochem. 1997;122:622–6.
Böttner M, Laaff M, Schechinger B, Rappold G, Unsicker K, Suter-Crazzolara C. Characterization of the rat, mouse, and human genes of growth/differentiation factor-15/macrophage inhibiting cytokine-1 (GDF-15/MIC-1). Gene. 1999;237:105–11.
Tong S, Marjono B, Brown DA, Mulvey S, Breit SN, Manuelpillai U, et al. Serum concentrations of macrophage inhibitory cytokine 1 (MIC 1) as a predictor of miscarriage. Lancet. 2004;363:129–30.
Buckhaults P, Rago C, St Croix B, Romans KE, Saha S, Zhang L, et al. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 2001;61:6996–7001.
Bonaca MP, Morrow DA, Braunwald E, Cannon CP, Jiang S, Breher S, et al. Growth differentiation factor-15 and risk of recurrent events in patients stabilized after acute coronary syndrome: observations from PROVE IT-TIMI 22. Arterioscler Thromb Vasc Biol. 2011;31:203–10.
Khan SQ, Ng K, Dhillon O, Kelly D, Quinn P, Squire IB, et al. Growth differentiation factor-15 as a prognostic marker in patients with acute myocardial infarction. Eur Heart J. 2009;30:1057–65.
Anand IS, Kempf T, Rector TS, Tapken H, Allhoff T, Jantzen F, et al. Serial measurement of growth-differentiation factor-15 in heart failure: relation to disease severity and prognosis in the Valsartan Heart Failure Trial. Circulation. 2010;122:1387–95.
Lankeit M, Kempf T, Dellas C, Cuny M, Tapken H, Peter T, et al. Growth differentiation factor-15 for prognostic assessment of patients with acute pulmonary embolism. Am J Respir Crit Care Med. 2008;177:1018–25.
Nickel N, Kempf T, Tapken H, Tongers J, Laenger F, Lehmann U, et al. Growth differentiation factor-15 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:534–41.
Meadows CA, Risbano MG, Zhang L, Geraci MW, Tuder RM, Collier DH, et al. Increased expression of growth differentiation factor-15 in systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2011;139:994–1002.
Nickel N, Jonigk D, Kempf T, Bockmeyer CL, Maegel L, Rische J, et al. GDF-15 is abundantly expressed in plexiform lesions in patients with pulmonary arterial hypertension and affects proliferation and apoptosis of pulmonary endothelial cells. Respir Res. 2011;12:62.
Wiklund FE, Bennet AM, Magnusson PK, Eriksson UK, Lindmark F, Wu L, et al. Macrophage inhibitory cytokine-1 (MIC-1/GDF15): a new marker of all-cause mortality. Aging Cell. 2010;9:1057–64.
Zdrenghea MT, Mallia P, Johnston SL. Immunological pathways in virus-induced COPD exacerbations: a role for IL-15. Eur J Clin Invest. 2012;42:1010–5.
Pletz MW, Ioanas M, de Roux A, Burkhardt O, Lode H. Reduced spontaneous apoptosis in peripheral blood neutrophils during exacerbation of COPD. Eur Respir J. 2004;23:532–7.
Schmidt-Ioanas M, Pletz MW, de Roux A, Lode H. Apoptosis of peripheral blood neutrophils in COPD exacerbation does not correlate with serum cytokines. Respir Med. 2006;100:639–47.
Ropcke S, Holz O, Lauer G, Muller M, Rittinghausen S, Ernst P, et al. Repeatability of and relationship between potential COPD biomarkers in bronchoalveolar lavage, bronchial biopsies, serum, and induced sputum. PLoS One. 2012;7, e46207.
Wouters EF, Groenewegen KH, Dentener MA, Vernooy JH. Systemic inflammation in chronic obstructive pulmonary disease: the role of exacerbations. Proc Am Thorac Soc. 2007;4:626–34.
Dev D, Wallace E, Sankaran R, Cunniffe J, Govan JR, Wathen CG, et al. Value of C-reactive protein measurements in exacerbations of chronic obstructive pulmonary disease. Respir Med. 1998;92:664–7.
Tang Y, Guan Y, Liu Y, Sun J, Xu L, Jiang Y. The role of the serum IL-33/sST2 axis and inflammatory cytokines in chronic obstructive pulmonary disease. J Interferon Cytokine Res. 2014;34:162–8.
Bathoorn E, Liesker JJ, Postma DS, Koeter GH, van der Toorn M, van der Heide S, et al. Change in inflammation in out-patient COPD patients from stable phase to a subsequent exacerbation. Int J Chron Obstruct Pulmon Dis. 2009;4:101–9.
Wedzicha JA, Seemungal TA, MacCallum PK, Paul EA, Donaldson GC, Bhowmik A, et al. Acute exacerbations of chronic obstructive pulmonary disease are accompanied by elevations of plasma fibrinogen and serum IL-6 levels. Thromb Haemost. 2000;84:210–5.
Tufvesson E, Ekberg M, Bjermer L. Inflammatory biomarkers in sputum predict COPD exacerbations. Lung. 2013;191:413–6.
Mocchegiani E, Giacconi R, Costarelli L. Metalloproteases/anti-metalloproteases imbalance in chronic obstructive pulmonary disease: genetic factors and treatment implications. Curr Opin Pulm Med. 2011;17 Suppl 1:S11–19.
Savarin C, Bergmann CC, Hinton DR, Stohlman SA. MMP-independent role of TIMP-1 at the blood brain barrier during viral encephalomyelitis. ASN Neuro. 2013;5, e00127.
Ashutosh, Chao C, Borgmann K, Brew K, Ghorpade A. Tissue inhibitor of metalloproteinases-1 protects human neurons from staurosporine and HIV-1-induced apoptosis: mechanisms and relevance to HIV-1-associated dementia. Cell Death Dis. 2012;3:e332.
Albert RK, Connett J, Bailey WC, Casaburi R, Cooper Jr JA, Criner GJ, et al. Azithromycin for prevention of exacerbations of COPD. N Engl J Med. 2011;365:689–98.
Mutlu LC, Altintas N, Aydin M, Tulubas F, Oran M, Kucukyalin V, Kaplan G, Gurel A. Growth Differentiation Factor-15 Is a novel biomarker predicting acute exacerbation of chronic obstructive pulmonary disease. Inflammation. 2015;445:155-60.
Acknowledgments
The authors thank Dr. Catherine Spino and Glen Feak for database support; Christi Getty and Candace Flaherty for support in patient recruitment; and Mary Freer, Joyce O’Brien and Cat Meyer for administrative support.
Sources of support
CMF: Career Development Award and Merit Review I01 BX001389, Biomedical Laboratory Research & Development Service, Department of Veterans Affairs.
CHM: T32 HL07749, National Heart, Lung and Blood Institute, NIH.
FJM: R01 HL082480, KL2 RR024987, National Heart, Lung and Blood Institute, NIH.
MKH: K24 HL04212, National Heart, Lung and Blood Institute, NIH.
JLC: R01 HL082480, U01 HL098961, National Heart, Lung and Blood Institute, NIH; and Merit Review award I01 CX000911, Clinical Research & Development Service, Department of Veterans Affairs.
Additional support came from a Research Enhancement Award Program from the Biomedical Laboratory Research & Development Service, Department of Veterans Affairs; the Tissue Procurement Core of the University of Michigan Comprehensive Cancer Center, grant P30 CA46952; and the Lung Tissue Research Consortium (Clinical Centers), grant N01 HR046162.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CMF and JLC have grant support from MedImmune, Ltd.
MKH participated in advisory boards for Boehringer Ingelheim, Pfizer, GlaxoSmithKline, Genentech, Novartis, Forest and Medimmune; participated on speaker’s bureaus for Boehringer Ingelheim, Pfizer, GlaxoSmithKline, Grifols Therapeutics, Forest and the National Association for Continuing Education, and WebMD; has consulted for Novartis and United Biosource Corporation; and has received royalties from UpToDate and ePocrates. She has served as an investigator for research sponsored by GlaxoSmithKline.
FJM has been a member of advisory boards and/or consultant for Able Associates, Actelion, Almirall, Bayer, GSK, Ikaria, Janssen, MedImmune, Merck, Pearl, Pfizer and Vertex, American Institute for Research, AstraZeneca, Carden Jennings, Cardiomems, Grey Healthcare, HealthCare Research and Consulting, Merion, Nycomed/Takeda, and Sudler and Hennessey. He has been a member of steering committees for studies sponsored by Actelion, Centocor, Forest, GlaxoSmithKline, Gilead, Mpex, Nycomed/Takeda. He has participated in Food and Drug Administration mock panels for Boehringer Ingelheim, Forest and GSK. He has served on speaker’s bureaus or in continuing medical education activities sponsored by American College of Chest Physicians, American Lung Association, Astra Zeneca, Bayer, William Beaumont Hospital, Boehringer Ingelheim, Center for Health Care Education, CME Incite, Forest, France Foundation, GlaxoSmithKline, Lovelace, MedEd, MedScape/WebMD, National Association for Continuing Education, Network for Continuing Medical Education, Nycomed/Takeda, Projects in Knowledge, St Luke’s Hospital, the University of Illinois Chicago, University of Texas Southwestern, University of Virginia, UpToDate. He has served on DSMBs for Biogen and Novartis. He has received royalties from Castle Connolly and Informa.
The other authors report no potential conflicts of interest.
Authors’ contributions
JLC, FJM and MKH designed and implemented the study and evaluated subjects during visits. LM and DLT recruited, consented, and monitored subjects and collected specimens. JT processed and analyzed specimens. CHM performed data analysis. CMF processed and analyzed specimens and performed data analysis. CMF and JLC wrote the manuscript. All authors read and approved the final manuscript.
Additional file
Additional file 1:
Supplemental materials & methods.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Freeman, C.M., Martinez, C.H., Todt, J.C. et al. Acute exacerbations of chronic obstructive pulmonary disease are associated with decreased CD4+ & CD8+ T cells and increased growth & differentiation factor-15 (GDF-15) in peripheral blood. Respir Res 16, 94 (2015). https://doi.org/10.1186/s12931-015-0251-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-015-0251-1