
Abstract
Aclarubicin (aclacinomycin A) is one of the anthracycline antineoplastic antibiotics with a multifaceted mechanism of antitumor activity. As a second-generation drug, it offers several advantages compared to standard anthracycline drugs such as doxorubicin or daunorubicin, which could position it as a potential blockbuster drug in antitumor therapy. Key mechanisms of action for aclarubicin include the inhibition of both types of topoisomerases, suppression of tumor invasion processes, generation of reactive oxygen species, inhibition of chymotrypsin-like activity, influence on cisplatin degradation, and inhibition of angiogenesis. Therefore, aclarubicin appears to be an ideal candidate for antitumor therapy. However, despite initial interest in its clinical applications, only a limited number of high-quality trials have been conducted thus far. Aclarubicin has primarily been evaluated as an induction therapy in acute myeloid and lymphoblastic leukemia. Studies have indicated that aclarubicin may hold significant promise for combination therapies with other anticancer drugs, although further research is needed to confirm its potential. This paper provides an in-depth exploration of aclarubicin’s diverse mechanisms of action, its pharmacokinetics, potential toxicity, and the clinical trials in which it has been investigated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aclarubicin (ACR), also known as aclacinomycin A, was first isolated from Streptomyces galilaeus culture by T. Oki et al. in 1975 [1]. The microorganism was found in a soil sample collected at Kamiosaki, Japan. As it turned out, the microorganisms produced an antibiotic complex called aclacinomycin, which was then extracted from the cell culture and separated on silica gel. The obtained compounds were examined by NMR and mass spectroscopy. One of them was ACR. Scientists observed ACR ability to inhibit leukemia in mice, which was the impulse to do further research.
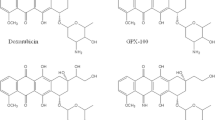
ACR is a second generation anthracycline antineoplastic antibiotic with multidirectional mechanism of antitumor and antiproliferative action. The most important of these is inhibition of topoisomerase II binding to DNA. Moreover, ACR interacts with topoisomerase I in a dose depending manner. ACR also induces the formation of free radicals, cell apoptosis and inhibits angiogenesis, has antimetastatic activity and inhibits the activity of chymotrypsin and the 20S proteasome. Furthermore, it can affect the regulation of erythroid gene expression. One of its main advantages over other anthracyclines, such as doxorubicin (DOX) or daunorubicin (DNR), is its low cardiotoxicity [2]. In terms of chemical structure, ACR, like other anthracycline antibiotics, is a glycoside composed of tetracyclic aglycone with a quinone ring. The aglycone moiety, aklavinone, is linked at the C-1 position of ring A to a trisaccharide which contains three deoxyhexoses: L-rhodosamine, 2-deoxy-L-fucose and L-cinerulose A. This is a feature that distinguishes ACR from doxorubicin or daunorubicin, and other anthracyclines, since the latters generally contain only one aminosaccharide group (Fig. 1).

Structure of aclarubicin
The aim of this work is to review current literature on ACR, with particular emphasis on the multidirectional mechanism of antineoplastic action, pharmacokinetics, toxicity and recently conducted clinical trials.
Molecular mechanism of action of aclarubicin
Interfering with DNA transcription and replication
Anthracyclines are well known for their capacity to bind to DNA. This noncovalent binding process of placing the aglycone of the antibiotic between adjacent base pairs of DNA double helix, called intercalation, results in DNA structure change [1]. Intercalation inhibits cell division and growth by blocking the process of DNA replication. This effect is prominent for quickly proliferating cells, such as cancer [3]. Moreover, ACR influences the activity of both topoisomerases. While doxorubicin and daunorubicin are only able to act on topoisomerase II [4], ACR has a dual mechanism of action, acting as a topoisomerase II catalytic inhibitor as well as a topoisomerase I poison [5]. During cell replication process there is a build-up of tension in the DNA. Topoisomerases are enzymes responsible for altering the topology of the DNA through binding and cleaving the strands of the double helix and later rearranging and re-ligating them in a new position [6]. Anthracycline antibiotics work on topoisomerase II in at least two different mechanisms. They may act by stimulating the formation of DNA-topoisomerase II cleavable complex or inhibing DNA cleavage by preventing association of the enzyme and DNA. Studies have shown that ACR belongs to the latter group of drugs [4]. While action of ACR against topoisomerase I is similar to camptothecin. The drug stabilizes topoisomerase I—DNA cleavage complex which causes damage of the DNA structure. In this mechanism, topoisomerase acts as a ‘poison”—it isn't the lack of enzyme activity that causes the destruction of the genetic material but inhibition of enzyme is a specific point of catalytic cycle, after cleave of DNA strand [6].
What is more, anthracyclines present the ability to induce histone eviction from the DNA, which enhances the generation of DNA breaks. Although this mechanism was not observed when the chromatin was fully condensed. While daunorubicin mostly affected active gene bodies, for ACR, the targeted genomic regions were marked by histone modification H3K27me3, hence recognized as facultative heterochromatin, containing repressed as well as silent genes. The additional sugar moieties of ACR may be responsible for its ability to dissociate nucleosomes from more condensed chromatin structures. Targeting relatively silent or less active regions, such as poised promoters, might be useful in treating tumors with that particular epigenetic state [7].
Antimetastatic potential
Studies have shown that ACR possesses the ability to inhibit cancer cell migration in non-cytotoxic concentrations. Along with decreased cell motility, changes in the cell shape were observed. Cells treated with ACR exhibited very discrete leading lamellae, weaker actin folds distributed along the membrane, vinculin presence only at the cell periphery, loss of polarity and a presence of discontinuous membrane ruffles. This together might be the reason for the impairment of cells migration. The antimetastatic effect has also been achieved by slight increase of cell adhesion. Additionally, the treatment with ACR led to a decrease of cellular distribution and affinity state of β1 integrins—a major class of cell surface receptors which are important for the cell-extracellular matrix interaction and cell migration. The components of integrin signaling, FAK and Src kinase, were also strongly affected by the ACR treatment, with higher phosphorylation level of FAK leading to phosphorylation-dephosphorylation imbalance and suppressed cell motility. These mechanisms together suggest that ACR may affect cell membrane functions and, in addition, suppress the tumor invasion process [8].
Generation of reactive oxygen species
The generation of reactive oxygen species (ROS) is considered to be one of the mechanisms of ACR cytotoxicity. Mitochondrial Respiratory Complex I is responsible for the reduction of anthracyclines to their semichinone derivatives [9] (Fig. 2).

One-electron reduction of anthracycline antibiotics (based on [10] modified)
Those forms in presence of transition metals and molecular oxygen cause oxidative stress and cellular damage by inducing the generation of ROS such as H2O2 or O2•−. Until now, iron was considered as an element essential for oxidative DNA damage [11], but recent studies proved that copper has apoptotic activity as well [12]. The production of ROS can lead to the DNA-damage alongside topoisomerase inhibition and DNA intercalation [13]. Furthermore, the metabolism of ACR and ROS production cause the loss of mitochondrial membrane potential, what might potentiate the cytotoxic effect. Compared with doxorubicin, ACR has the ability to generate ROS in lower concentration (IC50 = 0.274–0.621 for ACR vs. IC50 = 2.842–5.321 for DOX) [14]. ACR can also induce NOS expression and NO release, which may contribute to cardiovascular dysfunction, although not as severe as with other anthracycline antibiotics. Moreover, the activity of NOS also causes a one-electron reduction of anthracyclines, contributing to oxidative stress [15].
Induction of cell death
ACR, similarly to other anthracycline antibiotics, has been observed to induce biochemical changes leading to programmed cell death, promoting apoptosis with the cell membrane remaining intact [16]. These conclusions have been reached by detecting morphological changes in the tumor cells affected by ACR (concentrations up to 500 nM, incubation time 3–24 h), such as cell shrinkage, chromatin condensation, externalization of phosphatidylserine, DNA fragmentation and formation of apoptotic bodies which are postulated as central criteria of programmed cell death [16, 17]. However, it has been observed that the anti-tumor activity of ACR can lead to either apoptosis or necrosis of the cancerous cells, depending on the kind of cell line, the capability of cells to accumulate the drug, the incubation time and the dose of the drug. According to a study on A549, HepG2, and MCF-7 cell lines, while apoptosis is the major mode of cell death during the first 48 h of the treatment, it can be followed by secondary necrosis that has been detected after longer incubation (72–96 h), with ACR doses corresponding to the IC50 values for each cell line (0.27 µM, 0.32 µM and 0.62 µM respectively). The higher ability to accumulate the drug correlated with a higher level of apoptosis [17].
Another study, conducted on human acute lymphoblastic leukemia Jurkat cells, showed that ACR sensitizes cells to TRAIL-induced apoptosis. The reason for this action is the ACR-induced up-regulation of mRNA and protein expression of TRAIL receptor, as well as induction of caspase-8, Fas, and receptor-interacting protein. The action on TRAIL receptor promoter was independent of p53, as opposed to the similar effect achieved during treatment with doxorubicin [18].
Inhibition of chymotrypsin-like activity of the 20S proteasome
The potential anti-tumor activity of ACR is also related to its influence on the process of ubiquitin-dependent protein degradation. This process is carried out by the 20S proteasomes and the 26S proteasomes (whose catalytic center consists of the 20S proteasome). Proteasomes are macromolecular protein complexes directly involved in the breakdown of most polyubiquitinated proteins and polypeptides in eukaryotic cells. They are responsible for the degradation of nuclear oncoproteins and cyclins, which play a key role in regulating the cell cycle. Among many active sites of the proteasome, chymotrypsin-like, trypsin-like, and caspase-like sites can be distinguished [19, 20].
The mechanism of action of ACR differs from that of another anticancer drug, which affects the process of ubiquitin-dependent protein degradation—cisplatin. Cisplatin acts directly on ubiquitin, influences the process of attaching ubiquitin to proteins and polypeptides. In vitro studies on rabbit reticulocyte lysates have shown that ACR acts by inhibiting the activity of the catalytic centers of the 20S proteasomes, without affecting the ubiquitination process (IC50 = 52 µM) [21]. Moreover, ACR at a concentration of 50 µM was shown to significantly inhibit only the chymotrypsin-like activity of the 20S proteasome, without affecting the trypsin-like and caspase-like activity [20, 22]. The effectiveness of this mode of activity in producing an overall anticancer effect is uncertain, primarily because it necessitates relatively high concentrations of ACR for activation.
Inhibition of angiogenesis
Hypoxia-inducible factor- 1 (HIF-1) is a transcription factor responding in cells due to the low oxygen concentration. Under hypoxic conditions, HIF-1 has the ability to translocate to the nucleus, where it initiates the expression of specific genes involved in the tumor progression process such as VEGF [23]. In turn, VEGF is one of the most significant factors that stimulate angiogenesis and neovascularisation, and without its crucial role, this process cannot occur. ACR can inhibit the activity of HIF-1 and consequently inhibits the hypoxic induction of VEGF. Yamazaki et al. conducted an experiment that proved the above statement. They transfected a mammalian cell line with a luciferase reporter gene construct which contained a few copies of HIF-1 binding site. The activity of luciferase under hypoxic conditions was inhibited by ACR (IC50 = 0.021 mg/ml i.e. 25,89 µM) but neither doxorubicin nor daunorubicin showed that effect. In addition, ACR reduced VEGF protein expression in a dose-dependent manner while doxorubicin and daunorubicin showed no such effect. Unlike the above drugs, ACR affects topoisomerase I, which, according to the authors of the study, may have consequences in inhibiting angiogenesis by ACR [24].
Regulation of erythroid gene expression
The concept that leukemic cells are immature due to differentiation blocks and cannot thus control their growth created a therapeutic alternative for the patients [25, 26]. Instead of the destruction of cells, the agents could resume their maturation process. Furthermore, differentiation agents could show reduced toxicity. ACR, as well as some other anthracyclines, in sub-toxic concentration, are considered differentiation inducing drugs. In vitro studies have proven ACR can affect the regulation of erythroid gene expression in leukemic and solid tumor cells (e.g., neuroblastoma, melanoma) [27, 28]. After administration of low doses of ACR to patients with Acute Myelomonocytic or Myeloblastic Leukemia, the rise in the appearance of mature cells was noted. The concentrations causing cell differentiation ranged widely from 30–50 nM [28] to 250–350 nM [27], depending on the cell line used in the experiment. On the other hand, these reports are based on only a single human cases [29, 30]. Patients with terminal acute myeloblastic leukemia were administered 7 to 20 mg/daily of ACR. After the course, an increase in the number of mature neutrophils was noted along with a decrease of leukemia cells. The exact mechanism is still poorly understood; however, a few conclusions have been drawn. ACR acts mainly by regulating the transcription. It involves inducing the expression of erythropoietin genes, such as gamma-globin, PGBD, erythropoietin receptor (EpoR), and overexpression of erythroid transcription factors, such as GATA-1 [31] (Fig. 3).

Molecular pathways initiated by aclarubicin
Chemosensitization and radiosensitization
One of the investigated therapeutic possibilities of ACR is its support in subclinical doses during doxorubicin therapy. ACR is postulated to sensitize cells to the action of DOX. In addition, administering two drugs simultaneously reduces the risk of resistance. Cross-resistance of the anthracyclines: DOX and DNR were observed on the leukemic cell line, but this effect did not apply to ACR. ACR may act as a multi-drug resistance modulator. It has been suggested that ACR may be an inhibitor of proteins involved in drug resistance. The phenomenon of cross-resistance occurs in many cancers, hence great hopes are placed in combination therapy with drug resistance modulators [32]. Yueh-Lun Lee et al. in their research focused on the possibility of sensitizing K562 myeloid leukemia cells to imatinib (at the concentration of 200 nM), by 10 nM ACR co-treatment. Imatinib is a specific Bcr-Abl tyrosine kinase inhibitor and is used as first-line treatment for short-term chronic myelogenous leukemia (CML). The difficulty in treating CML is the resistance of the tumor progenitor cells. ACR induced erythroid differentiation via the mitogen-activated kinase38 (MAPK) pathway. The inhibition of erythroid differentiation by p38MAPK inhibitor, p38MAPK mutation, or p38MAPK knockdown, reduced the effectiveness of ACR/imitanib treatments influence on growth inhibition and apoptosis. These results suggest that ACR-induced differentiated K562 cells are more sensitive to imatinib. Moreover, such a scheme led to inhibition of their growth and induction of apoptosis, down-regulation of Bcr-Abl, Mcl-1, and Bcl-xL, as well as activation of caspase-3 [33].
ACR also shows synergism in combination with arsenic trioxide. Arsenic trioxide is a substance used to treat acute promyelocytic leukemia. This is related to the regulation of apoptosis-related proteins, including Bcl-2 down-regulation and caspases activation. It is also currently being tested for the treatment of other cancers. As2O3 therapy is limited by its side effects, including gastrointestinal and cardiac toxicity. A study by Yongbin Ye et al. showed that administration of a low dose of As2O3 and ACR in a ratio of 40: 1 (e.g. 0.4 mM to 10 nM) gave positive results. This treatment regimen reduced Bcl-2, c-IAP and XIAP, and significantly activated caspase-3 and SMAC. These results indicate activation of apoptotic pathways and arrest of the cell cycle, which gives a chance to improve the effectiveness of the therapy based on the synergy of As2O3 and ACR [34].
Interesting results were obtained in the context of combining ACR with radiotherapy. The results show that ACR reduces the expression of glycoproteins in the plasma membrane, including the epidermal growth factor receptor (EGFR) and Met. Moreover, ACR lowers the level of RTKs (receptor tyrosine kinases) associated with tumor growth, angiogenesis, and increased risk of metastasis. Following this path, studies have been carried out confirming that ACR sensitizes cancer cells to radiation, the survival of which depends on RTK signaling. The results confirm that 2-h pretreatment with 500 nM of ACR lowers the level of RTK protein and increases the sensitivity to radiation [35].
Resistance
The results of in vitro studies carried out on various cell lines (including hepatoma, SCCL and P388 leukemia cells) indicate that the resistance of cancer cells to ACR is lower than that to traditional anthracyclines, such as doxorubicin, daunorubicin or epirubicin. It has been suggested that the two main causes of this phenomenon are related to the increased expression of P-glycoprotein in neoplastic cells and the differences in the mechanism of action between ACR and, DNR and EPI.
To ensure an adequate biological effect, both in in vitro studies and after administration of the above-mentioned anthracyclines to patients, an important aspect is to maintain the optimal concentration of the drug in the tumor cells for a specified time. It has been proven that the resistance of neoplastic cells to these anthracyclines is largely associated with a decrease in the concentration of the drug in the cells, of which the main reason is the increase in the expression of P-glycoprotein in these cells. However, because ACR is a weak substrate of P-gp, high levels of ACR were observed in cells with increased expression of this glycoprotein, while concentrations of other anthracyclines were significantly reduced [36]. The different mechanisms of drug accumulation in cells may also explain the absence or weak cross-resistance to ACR in cells resistant to DOX or DNR [37, 38]. The decreased resistance to ACR is also related to the fact that, unlike the other discussed anthracyclines, ACR activity is not dependent on the intracellular level of topoisomerase II, the amount of which increases in the S phase of the cell cycle. For this reason, the ACR effect is noticeable even with a small number of S-phase cells [39].
Pharmacokinetics
ACR is an intravenous, rapidly metabolized drug with half-life of 13.3 h. The metabolism of ACR takes place in the liver and leads to the formation of inactive aglycons and cytotoxic glycosides, with a half-life of up to 25 h. ACR is mainly excreted in the bile, but about 6% is also excreted in the urine. The volume of distribution determined in preclinical studies in mice is 39.1 l/kg; the drug accumulates in the tissues and is then released from the tissues into the blood [40, 41]. During preliminary studies in humans, it was possible to establish that ACR has a two-compartment drug distribution model, like other anthracyclines. ACR was characterized by volume of distribution of 2073 L/m2 and a the total body clearance of 150 L/m2/h [41].
Clinical trials
Despite the initial interest regarding clinical applications of ACR, few high-quality trials were conducted to evaluate the drug’s efficacy in recent years. The main rationales behind ACR use in oncology are: (i) lack of cross-resistance with other anthracyclines and (ii) lower cardiotoxicity which were postulated after in vitro studies. In theory, these make ACR the perfect choice for therapy of neoplasms that were resistant to previous regimens consisting of anthracyclines or relapsed cases.
Treatment regimens based on ACR alone or in combination with other drugs were mainly evaluated as induction therapy in acute myeloid and lymphoblastic leukemia (AML and ALL). The main outcomes of those studies, regarding the clinical efficacy of tested regimens, are summarised in Table 1, while details on the regimens of the discussed therapies are presented in Table 2.
In several published case series papers ACR was found to be mildly effective in remission induction in patients with refractory or relapsed AML. Despite low CR rates (between 18 and 37%), ACR seems to be a promising candidate for combined therapy regimens, especially considering the characteristic of the evaluated group. Although a significant proportion of enrolled patients were primarily resistant to or relapsed after previous therapy with other anthracyclines, they responded to ACR, which may confirm the lack of cross-resistance between the drugs [42,43,44,45,46]. However, those results should be interpreted with caution due to the small sample size and lack of control groups. In addition, Mathé et al. provided preliminary data regarding the use of ACR in the treatment of refractory ALL and lymphosarcoma [42].
Furthermore, two trials compared ACR in combination with cytosine arabinoside (ara-C) and daunorubicin puls ara-C in patients with newly diagnosed AML. No significant difference was found in CR rate or long-term survival between groups [47].
ACR in combination with etoposide was found to induce remission in some patients with refractory or relapsed AML and was proposed as an alternative treatment for patients with an allergic reaction to ara-C [45, 46]. Moreover, AVA protocol, consisting of ACR, ara-C, and etoposide was proposed as a treatment option for remission induction and consolidation in elderly patients with AML [48]. Fengler et al., on the other hand, evaluated the efficacy of AVA with prednisone in heavily pre-treated children with ALL bone marrow relapse [58].
Ӧberg et al. compared the response to induction regimens consisting of thioguanine, ara-C, and either ACR or DNR in elderly patients with newly diagnosed AML and found no statistically significant difference in CR or long-term cause specific mortality [49].
Furthermore, a randomized trial by Nagura et al. found no statistically significant difference in CR and 7-year DFS between regimens consisting of behenoyl cytosine arabinoside, 6-mercaptopurine, prednisolone and either ACR or DNR used for induction and consolidation in patients under 65 years old with newly diagnosed AML. Although no difference between the two arms was noted in 7-year survival as well, the DNR group seemed to have better survivability in the early years after diagnosis [50].
The successful use of induction and maintenance regimen CAG, comprising ara-C, ACR, and G-CSF (granulocyte colony-stimulating factor), was reported in patients with newly diagnosed or refractory/relapsed AML. Furthermore modified CAG with the addition of decitabine was proposed as induction treatment in elderly AML patients [44, 51, 52].
The efficacy of combination therapy with homoharringtonine (currently known as omacetaxine mepesuccinate—a clinically approved protein synthesis inhibitor targeting ribosomal subunit A), Ara-C, and ACR (HAA regimen) as an option for remission induction and consolidation in patients with de novo or refractory/relapsed AML was suggested by three case series studies [43, 53, 54]. Moreover, the results of a randomised trial conducted by Jin et al., which enrolled patients with de novo AML, proved the superiority of HAA induction regimen over DNR alone [55].
Zhang et al. compared HCAG therapy (CAG + Homoharringtonine) with FLAG (fudarabine, ara-C and G-CSF) as induction regimens in patients with refractory AML and found no statistically significant difference in CR and OOR [56].
Although it is important to note the scarcity of large high-quality studies in the area, the aforementioned trials encourage the use of ACR, especially in AML. The efficacy of ACR alone, however limited, in relapsed cases that were previously treated with other anthracyclines is especially promising. Furthermore, it appears that ACR may be successfully incorporated into combination treatment regimens for better clinical efficacy. Although most of the discussed studies were case series trials, a few randomized trials found no or little difference between widely used AML induction therapy protocols and those containing ACR.
Although cautious optimism may be warranted regarding the potential application of ACR in the treatment of AML or ALL, further high-quality studies are required to precisely establish its clinical efficacy.
Individual papers reported the use of ACR in other hematological diseases—multiple myeloma and myelodysplastic syndrome (MDS) (summarised in Table 1). In the latter case, Harada et al. found no significant differences in therapeutic effects and survival after treatment with either ACR or ara-C [57]. Furthermore, Xu et al. reported HAA regimen to be effective in remission induction for patients with MDS [53].
Similarly, a few studies evaluated the clinical efficacy of ACR in the treatment of solid tumors (Table 3). Generally administered ACR alone was found to have little or no effect in non-small cell lung, small cell lung, breast, pancreatic and gastric cancer, although one study suggested it may have radiosensitizing properties. Conversely, local administration of ACR preparations alone or in combination with other drugs was reported to induce at least some response in this case. This discrepancy may be attributed to pharmacokinetics or cellular uptake of the drug.
Toxicity
Preclinical studies on animal models have proven relatively low toxicity of aclarubicn with maintained antitumor activity (Table 4). In a study conducted on golden hamsters, ACR appeared to cause less apparent changes in the myocardium compared to doxorubicin and daunorubicin. Also, these changes were reversible, degenerative skin changes were not observed. Oki et al. study on beagle dogs indicated weight loss, nausea, and vomiting as adverse effects with 3 months treatment schedule. Myelosuppression also developed and was dose-dependent [79].
Concerning cardiotoxicity, a study on golden hamsters showed that ECG changes after a single intravenous dose were slight to moderate and in multiple administration—slight as well, and reversible after 2 months [80]. Oki et al. study on rabbits have proven these findings. ACR appeared to be more than 10 × less cardiotoxic than doxorubicin as a reference, regarding both acute and chronic cardiotoxicity [79].
A general review on the toxicity of aclarubicn in various clinical studies was included in Table 1.
Myelosuppression, especially thrombocytopenia, with often subsequent infections, seems to be the most common effect. Also, nausea, vomiting, and diarrhea are observed [45]. These findings are supported by Yu et al. study in which all patients developed severe granulocytopenia and thrombocytopenia, as well as often gastrointestinal toxicity (nausea, vomiting) [43]. Jianyong et al. have similar observations with rare non-hematological toxicities [51].
The randomized study in acute myeloid leukemia patients noted patients treated with aclarubicn (HCAG regimen) experienced lower toxicity than patients with flutarabine (FLAG regimen) regardless of the grade of hematological toxicity or the presence of non-hematological toxicity (oral cavity toxicity, hepatic dysfunction, renal impairment, pulmonary infection and gastrointestinal disorder) [56]. No alopecia was recorded, or it was uncommon [42, 58].
Aabo et al. suggested that cardiotoxicity of ACR may be higher than it was presented in preclinical studies with patients developing cardiac dysfunction shown by clinical signs and changes in ECG [83]. Other studies observed no significant cardiotoxicity effect with only slight cardiac events (mild heart arrhythmia in mild heart function defect) and no change in left ventricular ejection fraction in post-treatment examination [45, 48, 58]. However, individual patients can develop acute heart arrhythmia or other cardiac complications causing discontinuation of treatment [54, 84]. A possible explanation for the weak cardiotoxic effect of ACR may be due to the relatively low distribution of the drug in the heart [79]. This aspect seems to be a great advantage of this anthracycline, nevertheless, more studies should be conducted, concerning especially the effect on myocardium.
Regarding the maximum dose, the acceptable toxicity limit appears to be 120–150 mg/m2 per course. Myelosuppression is considered the main dose-limiting toxicity [42]. Higher doses increase the incidence and intensity of myelosuppression, as well gastrointestinal side effects, such as diarrhea or vomiting. Cardiac events also can be more frequent [59, 60, 83].
Conclusions
Due to the complexity of its activity, ACR is a relatively poorly understood anthracycline. Apart from inhibition of topoisomerase I and II it was found to affect several other biochemical pathways of cancer cells, leading to anticancer effects. However, based on the concentration required to active some of these modes of action, only part of these mechanism, seems to be significant in vivo. In pharmacokinetics studies ACR achieved plasma concentration of ca. 0.34 μM, however its high distribution to tissues, high cellular accumulation pattern of anthracyclines and formation of active metabolites suggest possible high concentration of the drug in cancer cells [41]. In vitro submicromolar activities were found in case of apoptosis induction, ROS-generation, cells differentiation, chemosentitization and radiosentitization, therefore these effects are expected to be important part of ACR activity in vivo.ACR seems to be a particularly promising candidate for combination therapies with other anticancer drugs. This is related to the ACR sensitization of cells to other drugs and the lack of cross-resistance between drugs, as well as, prominent cardiac safety profile. However, promising data form clinical trials require further confirmation. ACR require more, recent, high quality, multicentre clinical trials, involving more participants and control groups for better characterization of its safety and efficacy in specific types of cancer. A problem might be a fact that ACR was discovered and patented over 40 years ago, and pharmaceutical companies may be no interest in development of a drug, whose patent protection has expired. While it routinely used in some countries in Asia (e.g. Japan) its entry to other markets is doubtful [85].
Data availability statement
Data available on request.
References:
Skovsgaard T (1987) Pharmacodynamic aspects of aclarubicin with special reference to daunorubicin and doxorubicin. Eur J Haematol 38:7–20. https://doi.org/10.1111/j.1600-0609.1987.tb00018.x
Cresteil T (2017) Aclarubicin. Ref Modul Biomed Sci. https://doi.org/10.1016/B978-0-12-801238-3.97571-8
Mukherjee A, Sasikala WD (2013) Drug-DNA intercalation: From discovery to the molecular mechanism. Dyn Protein Nucleic Acids 92:1–62. https://doi.org/10.1016/B978-0-12-411636-8.00001-8
Jensen PB, Sørensen BS, Sehested M, Demant EJF, Kjeldsen E, Friche E, Hansen HH (1993) Different modes of anthracycline interaction with topoisomerase II. Separate structures critical for DNA-cleavage, and for overcoming topoisomerase II-related drug resistance. Biochem Pharmacol 45:2025–2035. https://doi.org/10.1016/0006-2952(93)90013-M
Hajji N, Mateos S, Pastor N, Domínguez I, Cortés F (2005) Induction of genotoxic and cytotoxic damage by aclarubicin, a dual topoisomerase inhibitor. Mutat Res Genet Toxicol Environ Mutagen 583:26–35. https://doi.org/10.1016/j.mrgentox.2005.01.012
Bush NG, Evans-Roberts K, Maxwell A (2015) DNA Topoisomerases. EcoSal Plus. https://doi.org/10.1128/ecosalplus.ESP-0010-2014
Pang B, De Jong J, Qiao X, Wessels LFA, Neefjes J (2015) Chemical profiling of the genome with anti-cancer drugs defines target specificities. Nat Chem Biol 11:472–480. https://doi.org/10.1038/nchembio.1811
Addadi-Rebbah S, Poitevin S, Fourre N, Polette M, Garnotel R, Jeannesson P (2004) Assessment of the antiinvasive potential of the anthracycline aclacinomycin (Aclarubicin) in a human fibrosarcoma cell line. Int J Oncol 24:1607–1615
Davies KJA, Doroshow JH (1986) Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 261:3060–3067. https://doi.org/10.1016/s0021-9258(17)35746-0
Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L (2004) Anthracyclines: molecular advances and pharmacologie developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56:185–229. https://doi.org/10.1124/pr.56.2.6
Gammella E, Maccarinelli F, Buratti P, Recalcati S, Cairo G (2014) The role of iron in anthracycline cardiotoxicity. Front Pharmacol 5:25. https://doi.org/10.3389/fphar.2014.00025
Mizutani H, Hayashi Y, Hashimoto M, Imai M, Ichimaru Y, Kitamura Y, Ikemura K, Miyazawa D, Ohta K, Ikeda Y, Maeda T, Yoshikawa M, Hiraku Y, Kawanishi S (2019) Oxidative DNA damage and apoptosis induced by aclarubicin, an anthracycline: role of hydrogen peroxide and copper. Anticancer Res 39:3443–51. https://doi.org/10.21873/anticanres.13490
Kania K, Zych A, Jóźwiak Z (2007) Involvement of reactive oxygen species in aclarubicin-induced death of human trisomic and diabetic fibroblasts. Toxicol Vitr 21:1010–1019. https://doi.org/10.1016/j.tiv.2007.02.013
Rogalska A, Koceva-Chyła A, Jóźwiak Z (2008) Aclarubicin-induced ROS generation and collapse of mitochondrial membrane potential in human cancer cell lines. Chem Biol Interact 176:58–70. https://doi.org/10.1016/j.cbi.2008.07.002
Fogli S, Nieri P, Cristina Breschi M (2004) The role of nitric oxide in anthracycline toxicity and prospects for pharmacologic prevention of cardiac damage. FASEB J 18:664–675. https://doi.org/10.1096/fj.03-0724rev
Dartsch DC, Schaefer A, Boldt S, Kolch W, Marquardt H (2002) Comparison of anthracycline-induced death of human leukemia cells: Programmed cell death versus necrosis. Apoptosis 7:537–548. https://doi.org/10.1023/A:1020647211557
Rogalska A, Szwed M, Jóźwiak Z (2010) Aclarubicin-induced apoptosis and necrosis in cells derived from human solid tumours. Mutat Res Genet Toxicol Environ Mutagen 700:1–10. https://doi.org/10.1016/j.mrgentox.2010.04.013
Horinaka M, Yoshida T, Nakata S, Shiraishi T, Tomosugi M, Yoshikawa S, Wakada M, Sakai T (2012) Aclarubicin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis through death receptor 5 upregulation. Cancer Sci 103:282–287. https://doi.org/10.1111/j.1349-7006.2011.02150.x
Kisselev AF, Callard A, Goldberg AL (2006) Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate. J Biol Chem 281:8582–8590. https://doi.org/10.1074/jbc.M509043200
Figueiredo-Pereira ME, Er Chen W, Li J, Johdo O (1996) The antitumor drug aclacinomycin A, which inhibits the degradation of ubiquitinated proteins, shows selectivity for the chymotrypsin-like activity of the bovine pituitary 20 S proteasome. J Biol Chem 271:16455–16459. https://doi.org/10.1074/jbc.271.28.16455
Isoe T, Naito M, Shirai A, Hirai R, Tsuruo T (1992) Inhibition of different steps of the ubiquitin system by cisplatin and aclarubicin. BBA Gen Subj 1117:131–135. https://doi.org/10.1016/0304-4165(92)90070-B
Orlowski RZ, Dees EC (2002) The role of the ubiquitination-proteasome pathway in breast cancer: applying drugs that affect the ubiquitin-proteasome pathway to the therapy of breast cancer. Breast Cancer Res 5:1. https://doi.org/10.1186/bcr460
Carbajo-Pescador S, Ordoñ Ez R, Benet M, Jover R, Garcıá-Palomo A, Mauriz JL, Gonzá Lez-Gallego J (2013) Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br J Cancer 109:83–91. https://doi.org/10.1038/bjc.2013.285
Yamazaki Y, Hasebe Y, Egawa K, Nose K, Kunimoto S, Ikeda D (2006) Anthracyclines, small-molecule inhibitors of hypoxia-inducible factor-1 alpha activation. Biol Pharm Bull 29:1999–2003. https://doi.org/10.1248/bpb.29.1999
Friend C, Scher W, Holland JG, Sato T (1971) Hemoglobin synthesis in murine virus-induced leukemic cells in vitro: stimulation of erythroid differentiation by dimethyl sulfoxide. Proc Natl Acad Sci U S A 68:378–382. https://doi.org/10.1073/PNAS.68.2.378
Sachs L (1978) The differentiation of myeloid leukaemia cells: new possibilities for therapy. Br J Haematol 40:509–517. https://doi.org/10.1111/J.1365-2141.1978.TB05826.X
Steinheider G, Westendorf J, Marquardt H (1986) Induction of erythroid differentiation by the anthracycline antitumor antibiotics, aclacinomycin A, musettamycin and marcellomycin. Leuk Res 10:1233–1239. https://doi.org/10.1016/0145-2126(86)90242-0
Morin MJ, Sartorelli AC (1984) Inhibition of glycoprotein biosynthesis by the inducers of HL-60 cell differentiation, aclacinomycin A and marcellomycin. Cancer Res 44:2807–2812
Sakurai M, Sampi K, Hozumi M (1983) Possible differentiation of human acute myeloblastic leukemia cells by daily and intermittent administration of aclacinomycin-A. Leuk Res 7:139–143. https://doi.org/10.1016/0145-2126(83)90004-8
Sato S, Sakashita A, Ishiyama T, Nakamaki T, Hino K, Tomoyasu S, Tsuruoka N, Honma Y, Hozumi M (1992) Possible differentiation treatment with aclacinomycin A in acute myelomonocytic leukemia refractory to conventional chemotherapy. Anticancer Res 12:371–376
Morceau F, Chénais B, Gillet R, Jardillier JC, Jeannesson P, Trentesaux C (1996) Transcriptional and posttranscriptional regulation of erythroid gene expression in anthracycline-induced differentiation of human erythroleukemic cells. Cell Growth Differ 7:1023–1029
Gajek A, Rogalska A, Koceva-Chyła A (2019) Aclarubicin in subtoxic doses reduces doxorubicin cytotoxicity in human non-small cell lung adenocarcinoma (A549) and human hepatocellular carcinoma (HepG2) cells by decreasing DNA damage. Toxicol In Vitro 55:140–150. https://doi.org/10.1016/J.TIV.2018.12.015
Lee YL, Chen CW, Liu FH, Huang YW, Huang HM (2013) Aclacinomycin A sensitizes K562 chronic myeloid leukemia cells to imatinib through p38MAPK-mediated erythroid differentiation. PLoS ONE 8:e61939–e61939. https://doi.org/10.1371/JOURNAL.PONE.0061939
Ye Y, Xu X, Zhang M, Qiu D, Bai X, Wang J, Weng G, Zhou R, Guo Z, He H, Yi W, He X, Guo K (2015) Low-dose arsenic trioxide combined with aclacinomycin A synergistically enhances the cytotoxic effect on human acute myelogenous leukemia cell lines by induction of apoptosis. Leuk Lymphoma 56:3159–3167. https://doi.org/10.3109/10428194.2015.1011155
Bennett DC, Charest J, Sebolt K, Lehrman M, Rehemtulla A, Contessa JN (2013) High-throughput screening identifies aclacinomycin as a radiosensitizer of EGFR-mutant non-small cell lung cancer. Transl Oncol 6:382–391. https://doi.org/10.1593/TLO.13232
Lehne G, De Angelis P, Clausen OPF, Rugstad HE (1996) Human hepatoma cells rich in P-glycoprotein are sensitive to aclarubicin and resistant to three other anthracyclines. Br J Cancer 74:1719–1729. https://doi.org/10.1038/bjc.1996.621
Jensen PB, Vindeløv L, Roed H, Demant EJF, Sehested M, Skovsgaard T, Hansen HH (1989) In vitro evaluation of the potential of aclarubicin in the treatment of small cell carcinoma of the lung (SCCL). Br J Cancer 60:838–844. https://doi.org/10.1038/bjc.1989.376
Dong J, Naito M, Tatsuta T, Seimiya H, Johdo O, Tsuruo T (1995) Difference between the resistance mechanisms of Aclacinomycin- and Adriamycin-resistant P388 cell lines. Oncol Res 7:245–252
Jensen PB, Hansen HH, Jensen PS, Sørensen BS, Westergaard O, Demant EJF, Friche E, Vindeløv L, Sehested M, Wassermann R (1991) Antagonistic effect of aclarubicin on daunorubicin-induced cytotoxicity in human small cell lung cancer cells: relationship to DNA integrity and topoisomerase II. Cancer Res 51:5093–5099
Wang J, Maitani Y, Takayama K (2002) Antitumor effects and pharmacokinetics of aclacinomycin A carried by injectable emulsions composed of vitamin E, cholesterol, and PEG-lipid. J Pharm Sci 91:1128–1134. https://doi.org/10.1002/JPS.10104
Karanes C, Young JD, Samson MK, Smith LB, Franco LA, Baker LH (1983) Phase I trial of aclacinomycin-A. a clinical and pharmacokinetic study. Invest New Drugs 1:173–179. https://doi.org/10.1007/BF00172077
Mathé G, Bayssas M, Gouveia J, Dantchev D, Ribaud P, Machover D, Misset JL, Schwarzenberg L, Jasmin C, Hayat M (1978) Preliminary results of a phase II trial of aclacinomycin in acute leukaemia and lymphosarcoma. an oncostatic anthracyclin that is rarely cardiotoxic and induces no alopecia. Cancer Chemother Pharmacol 1:259–262. https://doi.org/10.1007/BF00257160
Yu W, Mao L, Qian J, Qian W, Meng H, Mai W, Tong H, Tong Y, Jin J (2013) Homoharringtonine in combination with cytarabine and aclarubicin in the treatment of refractory/relapsed acute myeloid leukemia: a single-center experience. Ann Hematol 92:1091–1100. https://doi.org/10.1007/S00277-013-1758-5
Wang Y, Li W, Chen S, Qiu H, Sun A, Wu D (2011) Salvage chemotherapy with low-dose cytarabine and aclarubicin in combination with granulocyte colony-stimulating factor priming in patients with refractory or relapsed acute myeloid leukemia with translocation (8;21). Leuk Res 35:604–607. https://doi.org/10.1016/J.LEUKRES.2010.11.003
Kern W, Braess J, Grote-Metke A, Kuse H, Fuchs R, Hossfeld DK, Reichle A, Wörmann B, Büchner T, Hiddemann W (1998) Combination of aclarubicin and etoposide for the treatment of advanced acute myeloid leukemia: results of a prospective multicenter phase II trial. German AML Cooperative Group Leukemia 12:1522–1526. https://doi.org/10.1038/SJ.LEU.2401155
Hilbe W, Thaler J, Eisterer W, Ludescher C, Niederwieser D (1994) Treatment of relapsed and refractory acute myelogenous leukaemia with aclacinomycin A (ACA) and etoposide (VP-16). Leuk Res 18:655–658. https://doi.org/10.1016/0145-2126(94)90063-9
De Nully BP, Hoffmann T, Hansen OP, Boesen AM, Grønbæk K, Hippe E, Jensen MK, Thorling K, Storm HH, Pedersen-Bjergaard J (1997) Long-term survival and development of secondary malignancies in patients with acute myeloid leukemia treated with aclarubicin or daunorubicin plus cytosine arabinoside followed by intensive consolidation chemotherapy in a Danish national phase III trial. danish society of haematology study group on AML. Leukemia 11:37–41. https://doi.org/10.1038/SJ.LEU.2400514
Staib P, Lathan B, Knöppel-Schwark S, Tesch H, Voliotis D, Steinmetz HT, Schwonzen M, Wickramanayake PD, Diehl V (1998) Cytosine arabinoside, etoposide and aclarubicin (AVA) for the treatment of acute myeloid leukemia (AML) in elderly patients. Ann Oncol 9:221–223. https://doi.org/10.1023/A:1008235801218
Öberg G, Killander A, Björeman M, Gahrton G, Grimfors G, Gruber A, Hast R, Lerner R, Liliemark J, Mattson S, Paul C, Simonsson B, Stalfelt AM, Stenke L, Tidefelt U, Udén AM, Björkholm M (2002) Long-term follow-up of patients >or=60 yr old with acute myeloid leukaemia treated with intensive chemotherapy. Eur J Haematol 68:376–381. https://doi.org/10.1034/J.1600-0609.2002.00423.X
Nagura E, Kimura K, Yamada K, Ohta K, Maekawa T, Takaku F, Uchino H, Masaoka T, Amaki I, Kawashima K, Kariyone S, Toyama K, Ichimaru M, Nomura T, Sakai Y, Takatsuki K, Hamajima N (1994) Nationwide randomized comparative study of daunorubicin and aclarubicin in combination with behenoyl cytosine arabinoside, 6-mercaptopurine, and prednisolone for previously untreated acute myeloid leukemia. Cancer Chemother Pharmacol 34:23–29. https://doi.org/10.1007/BF00686107
Li J, Chen Y, Zhu Y, Zhou J, Xu Y, Li Y et al (2015) Efficacy and safety of decitabine in combination with G-CSF, low-dose cytarabine and aclarubicin in newly diagnosed elderly patients with acute myeloid leukemia. Oncotarget 6:6448. https://doi.org/10.18632/ONCOTARGET.3361
Suzushima H, Wada N, Yamasaki H, Eto K, Shimomura T, Kugimiya MH, Horikawa K, Nishimura S, Tsuda H, Mitsuya H, Asou N (2010) Low-dose cytarabine and aclarubicin in combination with granulocyte colony-stimulating factor for elderly patients with previously untreated acute myeloid leukemia. Leuk Res 34:610–614. https://doi.org/10.1016/J.LEUKRES.2009.08.010
Xu W, Jin J, Qian W (2010) Homoharringtonine in combination with cytarabine and aclarubicin as induction therapy improves remission and survival of patients with higher risk myelodysplastic syndromes. Chin Med J (Engl) 123:108–110. https://doi.org/10.3760/cma.j.issn.0366-6999.2010.01.020
Jin J, Jiang DZ, Mai WY, Meng HT, Qian WB, Tong HY, Huang J, Mao LP, Tong Y, Wang L, Chen ZM, Xu WL (2006) Homoharringtonine in combination with cytarabine and aclarubicin resulted in high complete remission rate after the first induction therapy in patients with de novo acute myeloid leukemia. Leukemia 20:1361–1367. https://doi.org/10.1038/SJ.LEU.2404287
Jin J, Wang JX, Chen FF, Wu DP, Hu J, Zhou JF et al (2013) Homoharringtonine-based induction regimens for patients with de-novo acute myeloid leukaemia: a multicentre, open-label, randomised, controlled phase 3 trial. Lancet Oncol 14:599–608. https://doi.org/10.1016/S1470-2045(13)70152-9
Zhang YJ, Yu K, Li LJ (2019) Comparison of efficacy of HCAG and FLAG re-induction chemotherapy in acute myeloid leukemia patients of low- and intermediate-risk groups. Clin Transl Oncol 21:1543–1550. https://doi.org/10.1007/S12094-019-02085-Z
Harada M, Shibuya T, Teshima T, Murakawa M, Okamura T, Niho Y et al (1993) A randomized phase II trial of low-dose aclarubicin vs very low-dose cytosine arabinoside for treatment of myelodysplastic syndromes. Leuk Res 17:629–632. https://doi.org/10.1016/0145-2126(93)90066-T
Case DCJ, Ervin TJ, Boyd MA, Bove LG, Sonneborn HL, Paul SD (1987) Phase II study of aclarubicin in acute myeloblastic leukemia. Am J Clin Oncol. https://doi.org/10.1097/00000421-198712000-00014
Machover D, Gastiaburu J, Delgado M, Goldschmidt E, Hulhoven R, Misset JL, de Vassal F, Tapiero H, Dorval T, Ribaud P (1984) Phase I-II study of aclacinomycin for a treatment of acute myeloid leukemia. Biomed Pharmacother 38:328–331
Pedersen-Bjergaard J, Nissen NI, Brincker H, Ellegaard J, Drivsholm A, Freund L, Jensen KB, Jensen MK (1987) Aclarubicin in the treatment of acute nonlymphocytic leukemia refractory to treatment with daunorubicin and cytarabine: a phase II trial. Eur J Haematol 38:55–55. https://doi.org/10.1111/j.1600-0609.1987.tb00023.x
Hansen OP, Pedersen-Bjergaard J, Ellegaard J, Brincker H, Boesen AM, Christensen BE, Drivsholm A, Hippe E, Jans H, Jensen KB (1991) Aclarubicin plus cytosine arabinoside versus daunorubicin plus cytosine arabinoside in previously untreated patients with acute myeloid leukemia: a Danish national phase III trial. The danish society of hematology study group on AML. Denmark Leukemia 5:510–516
Shibuya T, Teshima T, Harada M, Taniguchi S, Okamura T, Okamura SI, Niho Y (1990) Treatment of myelodysplastic syndrome and atypical leukemia with low-dose aclarubicin. Leuk Res 14:161–167. https://doi.org/10.1016/0145-2126(90)90045-B
Fengler R, Buchmann S, Riehm H, Berthold F, Dopfer R, Graf N, Holldack J, Jobke A, Jürgens H, Klingebiel T (1987) Aggressive combination chemotherapy of bone marrow relapse in childhood acute lymphoblastic leukemia containing aclacinomycin-A: a multicentric trial. Haematol Blood Transfus 30:493–496. https://doi.org/10.1007/978-3-642-71213-5_86
Adachi T, Sezaki T, Ishii H, Hasegawa M, Asano K, Takahashi I, Kimura I (1983) Clinical responses and side effects of aclarubicin (ACR) in the treatment of multiple myeloma. Jpn J Cancer Chemother 10:1653–1658
Tapazoglou E, Samson MK, Pazdur R (1985) Phase II evaluation of aclacinomycin-A in patients with adenocarcinoma and large cell carcinoma of the lung. Am J Clin Oncol 8:298–301. https://doi.org/10.1097/00000421-198508000-00004
Jensen PB, Larsen SK, Stilbo I (1992) Phase II study of high-dose aclarubicin in previously treated patients with small-cell lung cancer. Cancer Chemother Pharmacol 30:219–220. https://doi.org/10.1007/BF00686316
Wakabayashi T, Yoshida J, Ishiyama J, Kageyama N (1987) Clinical trials of combination chemotherapy using cis-platinum with aclarubicin in intracranial rhabdomyosarcoma. Jpn J Cancer Chemother 14:2374–2377
Miyamoto T, Morita S, Asakawa H, Koyama T, Dokiya T, Kondo M, Yamashita T, Ohkawa T, Kanehira T (1989) A clinical study of the radiosensitization of aclarubicin. Japan J Cancer Clin 35:472–476
Natale RB, Cody RL, Simon MS, Wheeler RH (1993) An in vivo and in vitro trial of aclarubicin in metastatic breast cancer: a novel approach to the study of analogs. Cancer Chemother Pharmacol 31:485–488. https://doi.org/10.1007/BF00685040
Asbury RF, Cnaan A, Johnson L, Harris J, Zaentz SD, Haller DG (1994) An eastern cooperative oncology group phase II study of single agent DHAD, VP-16, aclacinomycin, or spirogermanium in metastatic pancreatic cancer. Am J Clin Oncol 17:166–169. https://doi.org/10.1097/00000421-199404000-00016
Ito H (1995) Experimental study and clinical application of a new combination chemotherapy with cis-platinum, adriamycin and carboquone in patients with advanced prostate cancer. Nihon Ika Daigaku Zasshi 62:456–468. https://doi.org/10.1272/JNMS1923.62.456
Novik Y, Ryan LM, Haller DG, Asbury R, Dutcher JP, Schutt A (1999) Phase II protocol for the evaluation of new treatments in patients with advanced gastric carcinoma: results of ECOG 5282. Med Oncol 16:261–266. https://doi.org/10.1007/BF02785872
Tokuhashi Y, Kikkawa F, Tamakoshi K, Suganuma N, Kuzuya K, Arii Y, Kawai M, Hattori SE, Kobayashi I, Furuhashi Y, Nakashima N, Tomoda Y (1997) A randomized trial of cisplatin, vinblastine, and bleomycin versus cyclophosphamide, aclacinomycin, and cisplatin in epithelial ovarian cancer. Oncology 54:281–286. https://doi.org/10.1159/000227704
Saito T, Takehara M, Lee R, Fujimoto T, Nishimura M, Tanaka R, Ito E, Adachi K, Kudo R (2004) Neoadjuvant chemotherapy with cisplatin, aclacinomycin A, and mitomycin C for cervical adenocarcinoma—a preliminary study. Int J Gynecol Cancer 14:483–490. https://doi.org/10.1111/J.1048-891X.2004.014309.X
Kobayashi O, Onodera S, Rino Y, Okugawa T, Okada K, Sairenji M, Motohashi H (1994) Evaluation of medicinal lymph node dissection in advanced gastric cancer. Nihon Geka Gakkai Zasshi 95:860–865
Sawada T, Yoshimura Y, Kawakami S (1994) Balloon occluded arterial infusion (BOAI) treatment of patients with locally advanced cervical carcinoma. Anticancer Res 14:2791–2794
Ambiru S, Miyazaki M, Ito H, Kaiho T, Ando K, Hayashi S, Nakajima N (1995) Intraportal infusion of 5-FU and lipiodol-aclarubicin after hepatic resection for colorectal liver metastasis. Nihon Geka Gakkai Zasshi 96:145–152
Kawashima O, Kurihara T, Kamiyoshihara M, Sakata S, Ishikawa S, Morishita Y (1999) Management of malignant pericardial effusion resulting from recurrent cancer with local instillation of aclarubicin hydrochloride. Am J Clin Oncol 22:396–398. https://doi.org/10.1097/00000421-199908000-00015
Oki T, Takeuchi T, Oka S, Umezawa H (1981) New anthracycline antibiotic aclacinomycin a: experimental studies and correlations with clinical trials. Recent Results Cancer Res 76:21–40. https://doi.org/10.1007/978-3-642-81565-2_4
Hori S, Shirai M, Hirano S, Oki T, Inui T, Tsukagoshi S, Ishizuka M, Takeuchi T, Umezawa H (1977) Antitumor activity of new anthracycline antibiotics, aclacinomycin-A and its analogs, and their toxicity. Gann, Japanese J Cancer Res 68:685–90. https://doi.org/10.20772/cancersci1959.68.5_685
Dantchev D, Slioussartchouk V, Paintrand M, Hayat M, Bourut C, Mathé G (1979) Electron microscopic studies of the heart and light microscopic studies of the skin after treatment of golden hamsters with Adriamycin, detorubicin, AD-32, and aclacinomycin. Cancer Treat Rep 63:875–888
Tone H, Nishida H, Takeuchi T, Umezawa H (1985) Experimental studies on aclacinomycin. Drugs Exp Clin Res 11:9–15
Aabo K, Mortensen SA, Skovsgaard T, Gymoese E (1983) Intermittent high-dose aclarubicin in patients with advanced cancer: a phase I study with special reference to cardiac toxicity. Cancer Treat Rep 67:281–282
Rowe JM, Chang AYC, Bennett JM (1988) Aclacinomycin A and etoposide (VP-16-213): an effective regimen in previously treated patients with refractory acute myelogenous leukemia. Blood 71:992–996. https://doi.org/10.1182/blood.v71.4.992.992
Yoshida C, Kondo T, Ito T, Kizaki M, Yamamoto K, Miyamoto T, Morita Y, Eto T, Katsuoka Y, Takezako N, Uoshima N, Imada K, Ando J, Komeno T, Mori A, Ishikawa Y, Satake A, Watanabe J, Kawakami Y, Morita T, Taneike I, Nakayama M, Duan Y, Guijarro BG, Delgado A, Llamas C, Kiyoi H (2022) Real-world treatment patterns and clinical outcomes in patients with AML in Japan who were ineligible for first-line intensive chemotherapy. Int J Hematol 116:89–101. https://doi.org/10.1007/s12185-022-03334-
Funding
Narodowe Centrum Nauki,2017/25/N/NZ7/01382,Kamil Piska,Uniwersytet Jagielloński Collegium Medicum,N42/DBS/000322,Kamil Piska
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Murzyn, A., Orzeł, J., Obajtek, N. et al. Aclarubicin: contemporary insights into its mechanism of action, toxicity, pharmacokinetics, and clinical standing. Cancer Chemother Pharmacol (2024). https://doi.org/10.1007/s00280-024-04693-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00280-024-04693-1