
Abstract
Naringenin is a plant polyphenol, widely explored due to its interesting biological activities, namely anticancer, antioxidant, and anti-inflammatory. Due to its potential applications and attempt to overcome the industrial demand, there has been an increased interest in its heterologous production. The microbial biosynthetic pathway to produce naringenin is composed of tyrosine ammonia-lyase (TAL), 4-coumarate-CoA ligase (4CL), chalcone synthase (CHS), and chalcone isomerase (CHI). Herein, we targeted the efficient de novo production of naringenin in Escherichia coli by performing a step-by-step validation and optimization of the pathway. For that purpose, we first started by expressing two TAL genes from different sources in three different E. coli strains. The highest p-coumaric acid production (2.54 g/L) was obtained in the tyrosine-overproducing M-PAR-121 strain carrying TAL from Flavobacterium johnsoniae (FjTAL). Afterwards, this platform strain was used to express different combinations of 4CL and CHS genes from different sources. The highest naringenin chalcone production (560.2 mg/L) was achieved by expressing FjTAL combined with 4CL from Arabidopsis thaliana (At4CL) and CHS from Cucurbita maxima (CmCHS). Finally, different CHIs were tested and validated, and 765.9 mg/L of naringenin was produced by expressing CHI from Medicago sativa (MsCHI) combined with the other previously chosen genes. To our knowledge, this titer corresponds to the highest de novo production of naringenin reported so far in E. coli.
Key points
• Best enzyme and strain combination were selected for de novo naringenin production.
• After genetic and operational optimizations, 765.9 mg/L of naringenin was produced.
• This de novo production is the highest reported so far in E. coli.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
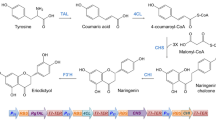
Plants produce secondary metabolites, including polyphenols, as a defense mechanism to respond to biotic and abiotic stresses. Naringenin is a plant-derived polyphenolic compound that has several recognized biological activities, including antioxidant, anti-inflammatory, and anticancer (Ghofrani et al. 2015; Jin et al. 2017; Kataoka et al. 2020; Wadhwa et al. 2020; Shi et al. 2021). As other plant secondary metabolites, naringenin is produced and accumulated in very low amounts in plants, and its production is affected by climatic, seasonal, and geographical variations. Moreover, its extraction from native plants is considered unsustainable and difficult due to complex downstream processing that leads to low production yields. Additionally, the chemical synthesis is not considered an environmentally friendly and economic process to produce this compound as it uses toxic compounds and expensive substrates (Liu et al. 2017; Rainha et al. 2020; Gomes et al. 2022a, 2022b). Therefore, in the last years, the interest in the heterologous production of naringenin has increased to satisfy its industrial demand. The heterologous microbial production of naringenin requires the expression of several enzymes. Naringenin can be produced from the aromatic amino acids l-phenylalanine and l-tyrosine (Fig. 1) that are derived from the shikimate pathway. Through the l-phenylalanine route, the action of both phenylalanine ammonia-lyase (PAL) and cinnamic acid 4-hydroxylase (C4H) is essential to produce p-coumaric acid. Alternatively, when l-tyrosine is used as a substrate, only tyrosine ammonia-lyase (TAL) must be expressed to catalyze this conversion step. Then, p-coumaric acid is converted by 4-coumarate-CoA ligase (4CL) into p-coumaroyl-CoA being then converted into naringenin chalcone by chalcone synthase (CHS) using three molecules of malonyl-CoA as extender unit. Afterwards, chalcone isomerase (CHI) catalyzes the final conversion of naringenin chalcone into naringenin (Pandey et al. 2016; Yonekura-Sakakibara et al. 2019).

Naringenin biosynthetic pathway. Both l-phenylalanine and l-tyrosine can be used as starter substrates. The extender substrate of this pathway is malonyl-CoA. This pathway is composed of the following enzymes: 4-coumarate-CoA ligase (4CL), chalcone isomerase (CHI), chalcone synthase (CHS), cinnamate 4-hydroxylase (C4H), phenylalanine ammonia-lyase (PAL), and tyrosine ammonia-lyase (TAL)
The biosynthetic pathway responsible for naringenin production has already been engineered in microbial chassis like Escherichia coli, Yarrowia lipolytica, Saccharomyces cerevisiae, and Streptomyces spp. (Koopman et al. 2012; Lv et al. 2019; Lyu et al. 2019; Dunstan et al. 2020; Palmer et al. 2020; Zhou et al. 2020b; Liu et al. 2021; Zhang et al. 2021; Ye et al. 2023). In E. coli, the highest production of naringenin was reported by Zhou et al. (2020b) in a strain modified both to improve malonyl-CoA and l-tyrosine availability. In shake-flask and fed-batch reactor experiments, 485 mg/L and 585 mg/L of naringenin were produced, respectively. However, these production levels are not enough to reach an industrial production of naringenin using E. coli as a microbial host. Using Y. lipolytica as a host, Palmer et al. (2020) reported the production of naringenin in a malonyl-CoA-boosted strain. In test tube fermentations, 124.1 mg/L of naringenin was produced. This production was further improved to 357 mg/L and 898 mg/L in batch and fed-batch reactors, respectively (Palmer et al. 2020). As far as we know, the highest naringenin titer reported so far was achieved using a modified S. cerevisiae strain with improved malonyl-CoA availability (Zhang et al. 2021). In that study, 1129.44 mg/L of naringenin was obtained in a fed-batch bioreactor fermentation (Zhang et al. 2021). More recently, the heterologous production of naringenin in Streptomyces albidoflavus was reported. However, only 22.4 mg/L of naringenin was produced (Ye et al. 2023).
Despite the promising production levels achieved so far, several optimizations are still required to improve the production levels and achieve industrial scale. Beyond the titers, it is also important to have one process with high yields and productivity/rates. The optimization of these three metrics (titer, yield, and productivity/rates) is mandatory to achieve a scalable and cost-effective process at an industrial scale. The target values for these metrics vary depending on the specific compound produced (Wehrs et al. 2019; Olsson et al. 2022). In this study, we aimed to design, assemble, and validate an efficient pathway for de novo production of naringenin in E. coli. A step-by-step optimization was performed by testing different genes from different organisms and validated by performing production experiments and evaluating the production levels of the different intermediaries. The TAL step was also evaluated in three different E. coli strains to choose the best strain to reach higher production levels and to be further used for the validation of the next pathway steps. After choosing the best combination of the pathway enzymes and further performing optimizations in the production experiment parameters (i.e., time and carbon source concentration), 765.9 mg/L of naringenin was achieved corresponding to the highest de novo production level reported so far in E. coli.
Materials and methods
Strains, plasmids, and chemicals
E. coli NZY5α (NZYTech—MB00401) was used as a cloning and plasmid propagation strain. The expression and validation of the p-coumaric acid production pathway were performed in E. coli BL21 (DE3) (NZYTech—MB006), E. coli K-12 MG1655 (DE3) (Nielsen et al. 2010), and E. coli M-PAR-121 (Koma et al. 2020). The heterologous biosynthetic pathways to produce naringenin chalcone and naringenin were expressed in E. coli M-PAR-121. Table 1 shows the features of all the strains and plasmids that were used in this study. The plasmids pCBJ280, pBADMod1-CHS, pYS454, and pBbE2c-Gm4CL1 were kindly provided by Dr. Christian Bille Jendresen (Jendresen et al. 2015), Dr. Claudia Schmidt-Dannert (Watts et al. 2004), Dr. Toru Nakayama (Waki et al. 2020), and Dr. Mark Dunstan (Dunstan et al. 2020), respectively.
Lysogeny broth (LB) Miller medium and super optimal broth with catabolite repression (SOC) medium were purchased from NZYTech. Isopropyl β-d-1-thiogalactopyranoside (IPTG) used to induce protein expression was also purchased from NZYTech. p-Coumaric acid and l-tyrosine were obtained from Sigma-Aldrich, naringenin chalcone from Chengdu Biopurify Phytochemicals, and naringenin from Alfa Aesar. Glucose (Acros), NH4Cl (Panreac), NaCl (NZYTech), Na2HPO4 (Chem-Lab), KH2PO4 (Riel-deHaën), CaCO3 (Panreac), MgSO4 (Labkem), CaCl2 (Panreac), thiamine (Thermo Fisher Scientific), methionine (Panreac), nicotinic acid (Acros Organics), pyridoxine (Fisher BioReagents), biotin (Merck), folic acid (Panreac), riboflavin (Panreac), and pantothenic acid (Sigma-Aldrich) were used in the preparation of M9 minimal medium. For strain selection, the antibiotics kanamycin (NZYTech), ampicillin (VWR), chloramphenicol (NZYTech), and spectinomycin (Alfa Aesar) were used. For protein analysis, a sample buffer was prepared using Tris–HCl buffer pH 6.8 (Tris-base (Fisher Scientific) and HCl (VWR)), glycerol (Fisher Scientific), sodium dodecyl sulfate (SDS) (Fisher Scientific), bromophenol blue (Sigma-Aldrich), and β-mercaptoethanol (AppliChem). Coomassie Blue R-250 (Fisher Scientific) was used for gel staining. Ethyl acetate used for compound extraction from cell broth was purchased from Fisher Scientific. Acetonitrile, trifluoroacetic acid, and sulfuric acid, used in high-performance liquid chromatography (HPLC), were purchased from Fisher Scientific.
Construction of plasmids
TAL from Flavobacterium johnsoniae (FjTAL) was amplified by polymerase chain reaction (PCR) from pCBJ280 and cloned into pRSFDuet-1. 4CL-1 from Glycine max (Gm4CL) was amplified from pBbE2c-Gm4CL1. 4CL-2 from Petroselinum crispum (Pc4CL) was amplified from pETM6-Pc4CL-CmCHS-MsCHI. 4CL from Vitis vinifera (Vv4CL) was removed from pETM6-Vv4CL-CmCHS-MsCHI by restriction digestion with NdeI and XhoI enzymes. All the 4CL genes were subsequently cloned into the pACYCDuet-1 vector. Regarding the CHS genes, CHS from Arabidopsis thaliana (AtCHS) was amplified by PCR from pBADMod1-CHS. CHS from Petunia hybrida (PhCHS) was removed from the plasmid pETM6-At4CL-PhCHS-CmCHI by restriction digestion with NdeI and KpnI enzymes. CHS from Cucurbita maxima (CmCHS) was also removed from pETM6-Vv4CL-CmCHS-MsCHI by restriction digestion with the enzymes NdeI and XhoI. Both PhCHS and CmCHS were cloned into pCDFDuet-1. CmCHS was also cloned into the pRSFDuet-1 vector. CHI from A. thaliana (AtCHI), CHI from Medicago sativa (MsCHI), and CHI from C. maxima (CmCHI) were amplified from pKYS454 and pETM6-Pc4CL-CmCHS-MsCHI and pETM6-At4CL-PhCHS-CmCHI, respectively, and cloned into pRSFDuet-1. To construct pRSFDuet_FjTAL_CmCHS, CmCHS was firstly cloned into the multiple cloning site (MCS) 2 of pRSFDuet-1, and FjTAL was further cloned into the MCS1 of the constructed plasmid (pRSFDuet_CmCHS). To construct pRSFDuet_At4CL_CmCHS, 4CL from A. thaliana (At4CL) was amplified by PCR from pACYCDuet_At4CL and cloned into pRSFDuet_CmCHS. To construct pACYCDuet_FjTAL_At4CL, At4CL was cloned into the MCS2 of pACYCDuet-1. FjTAL was further cloned into the MCS1 of the constructed plasmid. AtCHI, MsCHI, and CmCHI were cloned into pACYCDuet_At4CL to construct pACYCDuet_At4CL_AtCHI, pACYCDuet_At4CL_MsCHI, and pACYCDuet_At4CL_CmCHI, respectively. Tables S1 and S2 hold the sequences of all the genes and primers (Metabion/Eurofins) used, respectively.
NucleoSpin® Plasmid Miniprep Kit (Macherey–Nagel) was used for the isolation of plasmid DNA. All the genes were amplified by PCR using Phusion High-Fidelity DNA Polymerase (Thermo Scientific). The DNA fragments obtained were purified using the NucleoSpin® Gel and PCR Clean-up Kit from Macherey–Nagel. A NanoDrop One instrument (Thermo Scientific) was used to quantify the gene fragments and plasmid DNA. Afterwards, the fragments were digested using the appropriate restriction enzymes (Table S1) (Thermo Scientific) for 3 h at 37 °C. After digestion, NucleoSpin® Gel and PCR Clean-up Kit was used for their purification. Ligation reactions were performed at 22 °C for 1 h 30 min using T4 DNA ligase (Thermo Scientific). Ligation products were further transformed into E. coli NZY5α competent cells (NZYTech) by heat shock method. After transformation, the colonies were tested by colony PCR. Then, the plasmid was extracted and digested as a second confirmation step. Then, all the constructs were verified by DNA sequencing (Eurofins). After confirmation, the plasmids were transformed into chemically competent cells from the specific E. coli expression strain.
Protein analysis
To confirm the expression of FjTAL and all the 4CL, CHS, and CHI genes, E. coli M-PAR-121 carrying the desired plasmids were grown in LB Miller media at 37 °C. When an optical density at 600 nm (OD600nm) of 0.6 was reached (set as time 0 h), 10 mL of the sample was taken and centrifuged. Then, IPTG (final concentration of 0.1 mM) was added to the culture to induce the protein expression. The remaining culture was incubated for 6 h at 26 °C. At this time point (time 6 h), 10 mL of sample was also taken and centrifuged. The pellets were resuspended in 1 mL of Tris–HCl buffer (10 mM, pH 7.8). The cells were disrupted using a microtip probe linked to a Vibra-Cell processor (Sonics) by sonication on ice using the following conditions: 35% amplitude and 3 s ON plus 5 s OFF for a total of 7 min ON. Then, samples were centrifuged. The supernatant was recovered corresponding to the soluble phase. The pellet was resuspended in 1 mL of Tris–HCl buffer (10 mM, pH 7.8) corresponding to the insoluble phase. To quantify the amount of protein of both fractions, the Pierce Coomassie (Bradford) Protein Assay Kit was used following the manufacturer’s instructions. Protein fractions were mixed with 2 × sample buffer (65.8 mM Tris–HCl buffer pH 6.8, 26.3% glycerol, 2.1% SDS, 5% β-mercaptoethanol, and 0.01% bromophenol blue) and then denaturated at 100 °C for 5 min. Then, these fractions were subjected to SDS polyacrylamide gel electrophoresis (SDS-PAGE) to evaluate the expression levels. The SDS-PAGE gel was composed of 10% running gel and 4% stacking gel. The gel staining was performed using 0.2% (w/v) Coomassie Blue R-250 (Fisher Scientific) for 15 min. The gel was further destained with distilled water overnight. The protein ladders used were Color Prestained Protein Standard, Broad Range (10–250 kDa) (NEB) and NZYColour Protein Marker II (NZYTech).
Metabolite production experiments
E. coli expression strain carrying different plasmids was cultivated overnight in LB Miller medium supplemented with the respective antibiotics, at 37 °C and 200 rpm. Then, 250-mL flasks containing 50 mL of LB Miller were inoculated using this preculture to a normalized initial OD600nm of 0.1. The culture was incubated at 37 °C and 200 rpm until reaching an OD600nm of 0.9. When this OD was achieved, the expression of the heterologous protein was induced by adding IPTG (final concentration of 0.1 mM). The culture was incubated at 26 °C during 5 h. Then, the cells were centrifuged at 8000 rpm for 5 min, and the pellet was resuspended in the M9 minimal medium. M9 minimal medium is composed by 3 g/L KH2PO4, 6 g/L Na2HPO4, 0.5 g/L NaCl, 1 g/L NH4Cl, 110 mg/L MgSO4, 15 mg/L CaCl2, 340 mg/L thiamine, 5 g/L CaCO3, and vitamins (12.2 mg/L nicotinic acid, 10.8 mg/L pantothenic acid, 2.8 mg/L pyridoxine, 0.84 mg/L riboflavin, 0.12 mg/L biotin, and 0.084 mg/L folic acid). M9 media was also supplemented with 40 g/L glucose, unless otherwise specified. l-Tyrosine (3 mM) was supplemented to the media when required. The M9 media was also supplemented with the required antibiotics (kanamycin (50 µg/mL), spectinomycin (100 µg/mL), and/or chloramphenicol (25 µg/mL) depending on the specific experiment). Unless otherwise specified, the production experiments were conducted in triplicate for 63 h. Supernatant samples (1 mL) were collected during the experiment. These samples were filtered and used to analyze p-coumaric acid and glucose. Whole broth samples (culture broth with cells—1 mL) were also collected to be further extracted and perform naringenin chalcone and naringenin analysis.
Metabolite extraction
Since naringenin chalcone and naringenin are not completely diffused to the production medium and are mostly present inside of the E. coli cells, it was necessary to extract them from the cells to enable the analysis of these compounds. Whole broth (1 mL) was mixed with ethyl acetate (1:1 ratio). To achieve an efficient extraction of those compounds from the cells, the samples were vortexed for 2 min. Subsequently, the samples were centrifuged at 10,000 rpm for 2 min to separate the phases, and the organic phase (upper phase) was then transferred to a new tube. These steps of extraction were repeated until the pellet lost the yellow color (Rodrigues et al. 2020). The solvent was then evaporated in the fume hood to concentrate the extracts. The extracts were then resuspended in acetonitrile. The amount of acetonitrile added to the extracts varied between different samples and time points, ranging from 300 µL to 1 mL. For samples with very high production levels, additional dilution with acetonitrile was performed to fit within the calibration curve. Afterwards, the samples were analyzed by ultra-HPLC (UHPLC) (Rodrigues et al. 2020).
Analytical methods
For glucose quantification, samples were analyzed by HPLC using a JASCO system associated with an RI detector (RI-2031). The column used to analyze the samples was the Aminex HPX-87H from Bio-Rad that was kept at 60 °C. The mobile phase used was 5 mM H2SO4 with a flow rate of 0.5 mL/min. Glucose was detected at a retention time of 10.9 min.
For p-coumaric acid, naringenin chalcone, and naringenin analysis, UHPLC was used. The chromatographic system was composed of a Kinetex® 2.6 µm Polar C18 100 Å LC column (150 × 4.6 mm) (Phenomenex) and the Shimadzu Nexera X2 system (Shimadzu Corporation, Kyoto, Japan) (LC-30AD pump unit, CBM-20A system controller, DGU20A 5R degasser unit, SIL-30AC autosampler unit, SPD-M20A detector unit, CTO-20AC column oven). Two mobile phases were used, namely 0.1% (v/v) of trifluoroacetic acid in water (mobile phase A) and acetonitrile (mobile phase B). Using a constant follow rate of 1 mL/min, the following gradient was used: 10–40% mobile phase B for 11 min and 40–70% for 2 min, 70–10% for 8 min and 10% mobile phase B for an additional 4 min. p-Coumaric acid was quantified based on the peak areas at 310 nm, and the retention time of the compound was 7.1 min. Naringenin chalcone and naringenin were detected at 290 nm, and the retention times were 12.0 min and 12.6 min, respectively.
Statistical analysis
The results are presented as the mean value of three independent experiments ± standard deviation. Data statistical analysis was performed using GraphPad Prism Software, Inc., version 8.0.1. Ordinary one-way ANOVA tests were used to evaluate the statistical significance when required. The differences were considered significant when the p-value was < 0.05.
Results
Design and validation of an optimized heterologous E. coli strain to produce p-coumaric acid
In the phenylpropanoid pathway, both l-phenylalanine and l-tyrosine routes can be used to produce p-coumaric acid. l-Phenylalanine route requires the action of PAL and C4H enzymes. C4H is a plant membrane-bound cytochrome P450-dependent hydroxylase. These enzymes are difficult to express in bacteria since this microorganism lacks an endomembrane system, thus resulting in protein instability and insolubility (Watts et al. 2004; Gomes et al. 2022b). For this reason, the l-tyrosine route was chosen in this work to produce p-coumaric acid. Moreover, this route only requires the action of one enzyme to catalyze this step which is advantageous to reduce a possible metabolic burden imposed to the cells.
TAL from Rhodotorula glutinis (RgTAL) and FjTAL were selected to perform this first step of the biosynthetic pathway since they have been widely reported for the production of several polyphenolic compounds (Wu et al. 2014; Jendresen et al. 2015; Rodrigues et al. 2015a, 2020, 2022; Dunstan et al. 2020). Moreover, three different E. coli strains were tested to express both TAL genes: E. coli BL21 (DE3), E. coli K-12 MG1655 (DE3), and E. coli M-PAR-121 (Fig. 2). E. coli BL21 (DE3) and K-12 MG1655 (DE3) are common E. coli expression strains widely used in the biotechnology industry as platform strains to express the most diverse types of heterologous proteins (Castiñeiras et al. 2018). E. coli M-PAR-121 is a tyrosine-overproducing strain that has been derived from MG1655 (DE3) (Koma et al. 2020).

p-Coumaric acid production by Escherichia coli BL21 (DE3), K-12 MG1655 (DE3), and M-PAR-121 strains expressing tyrosine ammonia-lyase (TAL) gene from Flavobacterium johnsoniae (FjTAL) or TAL gene from Rhodotorula glutinis (RgTAL). The production experiments in E. coli M-PAR-121 were carried out using glucose as the sole substrate. The production experiments in E. coli BL21 (DE3) and K-12 MG1655 (DE3) were carried out in a first approach using glucose as the sole substrate and then with the supplementation of 3 mM l-tyrosine. Results correspond to the average of three independent experiments ± standard deviation
As presented in Fig. 2, the highest production levels of p-coumaric acid using glucose as substrate were obtained when the E. coli M-PAR-121 strain expressed either FjTAL or RgTAL. Moreover, as a proof of concept, a production experiment was carried out with supplementation of 3 mM of l-tyrosine to verify if the strains BL21 (DE3) and K-12 MG1655 (DE3) were able to achieve the same production levels obtained in M-PAR-121 without supplementation of l-tyrosine (Fig. 2). As can be observed in Fig. 2, the production of p-coumaric acid in both strains with supplementation of l-tyrosine did not reach the same production levels as in E. coli M-PAR-121 strain. However, higher production of p-coumaric acid was observed compared with the experiments performed with only glucose. Moreover, the production levels were slightly higher for the E. coli K-12 MG1655 (DE3) comparing with the ones achieved in E. coli BL21 (DE3). Regarding the expression of FjTAL or RgTAL, it was not possible to observe statistically significant differences in the production levels of p-coumaric acid in this production experiments with l-tyrosine supplementation.
The highest production of p-coumaric acid obtained in these experiments (2.54 g/L) was achieved for the E. coli M-PAR-121 strain expressing FjTAL (Fig. 2). The expression of FjTAL enzyme in E. coli M-PAR-121 strain was also validated through SDS-PAGE gel (Fig. S1). The production titer obtained in our study is equivalent to the one previously reported by Jones et al. (2017) (2.51 g/L). However, the productivity of our process was significantly higher compared with the one reported in their study (0.04 g/L/h vs 0.02 g/L/h). Considering these results, the M-PAR-121 strain expressing FjTAL was chosen as the platform strain to construct and validate the following steps of the heterologous pathway (4CL, CHS, and CHI).
Design and validation of an optimized heterologous E. coli strain to produce naringenin chalcone
After selecting the best p-coumaric acid producer, 4CL and CHS steps were constructed and validated to choose the best combination of enzymes to produce naringenin chalcone from glucose. At4CL, Gm4CL, Vv4CL, and Pc4CL were selected to test the conversion of p-coumaric acid into p-coumaroyl-CoA. AtCHS, PhCHS, and CmCHS were selected to convert p-coumaroyl-CoA into naringenin chalcone. These genes were selected based on previous reports of heterologous production of naringenin and other polyphenols available in the literature (Xu et al. 2011; Wu et al. 2015; Jones et al. 2016; Dunstan et al. 2020; Rodrigues et al. 2020, 2022; Zhou et al. 2020b). The efficient individual expression of all these genes in E. coli M-PAR-121 was evaluated using SDS-PAGE (Figs. S2-S3). Then, considering the four different 4CL genes and the three different CHS genes, it was possible to construct 12 different pathway combinations (Fig. 3a).

Naringenin chalcone production by Escherichia coli M-PAR-121 expressing 12 different biosynthetic pathways. a Schematic representation of the 12 different biosynthetic pathways. Tyrosine ammonia-lyase (TAL) gene from Flavobacterium johnsoniae (FjTAL) that was previously selected was expressed in the pRSFDuet-1 vector. The different 4-coumarate-CoA ligase (4CL) genes and the different chalcone synthase (CHS) genes were expressed in the pACYCDuet-1 vector and in the pCDFDuet-1 vector, respectively. b Naringenin chalcone production from 40 g/L of glucose by the 12 different constructed E. coli M-PAR-121 strains. c Profile of glucose consumption and p-coumaric acid and naringenin chalcone production for the E. coli M-PAR-121 strain expressing pRSFDuet_FjTAL, pACYCDuet_At4CL, and pCDFDuet_CmCHS (strain 9). The production experiments were maintained for 63 h. Results correspond to the average of three independent experiments ± standard deviation
These combinations were tested in the E. coli M-PAR-121 strain holding pRSFDuet_FjTAL, and the production of naringenin chalcone using glucose as substrate was evaluated (Fig. 3b). After 63 h of fermentation, it was observed that the highest production of naringenin chalcone (320.4 mg/L) was obtained when FjTAL, At4CL, and CmCHS genes were expressed (strain 9 from Fig. 3a). Moreover, as can be observed in Fig. 3b, when the CmCHS gene was expressed, a higher production of naringenin chalcone was obtained comparing directly with the other correspondent pathways (with the same 4CL gene and different CHS gene). The production profiles of p-coumaric acid and naringenin chalcone, as well as the profile of glucose consumption for strain 9, are presented in Fig. 3c. Regarding p-coumaric acid and naringenin chalcone production profiles, an increase in the titers of both products along the fermentation time was observed. Additionally, high amounts of p-coumaric acid were still accumulated at the end of fermentation (1046.9 mg/L). The same behavior was observed for the other strains (data not shown).
Regarding glucose, its concentration has decreased during the production experiment time, as expected. However, only 15 g/L of glucose was consumed, remaining almost 25 g/L of glucose at the end of the process.
With the aim of improving the productivity of the constructed strain, a new strategy was designed to optimize the pathway balancing and to reduce the metabolic burden of the cells that can impair the production levels. This metabolic burden is imposed by the replication and maintenance of plasmids. The replication of plasmids and its maintenance implies the use of cell supplies, namely carbon building blocks and energy molecules (Silva et al. 2012; Rodrigues et al. 2020). Hence, the cell metabolic burden was reduced by cloning the three genes in only two plasmids instead of the three plasmids previously used. Considering that Duet plasmids (Novagen) have two multiple cloning sites containing two T7 promoters, two genes were cloned in one plasmid, and the other gene was cloned alone in another plasmid. The genes present in strain 9 were tested in different combinations in pRSFDuet-1 and pACYCDuet-1, and naringenin chalcone production was further evaluated (Fig. 4a).

Evaluation of the effect on the production of naringenin chalcone of the reduction of cell’s metabolic burden. a Naringenin chalcone production from 40 g/L of glucose by the control strain (holding three plasmids) and by the three different Escherichia coli M-PAR-121 strains constructed to reduce the metabolic burden of the cells (holding two plasmids) after 63 h of fermentation. b Profile of naringenin chalcone and p-coumaric acid production, and glucose consumption for the E. coli M-PAR-121 carrying pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL. Results correspond to the average of three independent experiments ± standard deviation
Comparing with the control (strain expressing pRSFDuet_FjTAL, pACYCDuet_At4CL, and pCDFDuet_CmCHS), it is possible to conclude that only one strategy to reduce the metabolic burden of the cells (E. coli M-PAR-121 expressing pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL) led to a significant improvement in the production of naringenin chalcone (≈1.7-fold improvement compared to the control). The other two strategies resulted in lower production levels of naringenin chalcone. Moreover, a lower accumulation of p-coumaric acid comparing to the control strain was observed during the production experiment (Fig. S4). The optimal constructed strain only consumed 16 g/L of glucose during the production experiment. Moreover, 560.2 mg/L of naringenin chalcone was produced. However, a significant amount of p-coumaric acid (758.8 mg/L) is still accumulated at the end of the experiment (Fig. 4b). The UHPLC chromatogram of this sample can be observed in Fig. S5.
Design and validation of an optimized heterologous E. coli strain to produce naringenin
CHI catalyzes the last step of the biosynthetic pathway responsible to produce naringenin. Several CHI genes derived from different species were tested in combination with FjTAL, At4CL, and CmCHS to optimize this step of the pathway and achieve the final production of naringenin from glucose. AtCHI, MsCHI, and CmCHI were selected since they have been reported on the literature for the successful heterologous production of naringenin and also other flavonoids (Wu et al. 2015; Jones et al. 2016; Li et al. 2019; Wang et al. 2019; Dunstan et al. 2020; Zhou et al. 2020a). The expression of these genes in E. coli M-PAR-121 was individually tested and evaluated by SDS-PAGE gel (Fig. S6). Then, the CHI genes were cloned into the pACYCDuet_At4CL vector. The constructed vectors were expressed in the E. coli M-PAR-121 holding pRSFDuet_FjTAL_CmCHS. Production of naringenin from glucose was evaluated using the three constructed strains (Fig. 5).

Naringenin production by Escherichia coli M-PAR-121 expressing three different biosynthetic pathways after 63 h and 120 h of fermentation. Results correspond to the average of three independent experiments ± standard deviation
At 63 h of fermentation, the three constructed strains produced 319.4 to 361.9 mg/L of naringenin, and no statistically significant differences between the tested pathways could be found. Furthermore, the naringenin production levels did not reach a plateau at 63 h, and high concentrations of p-coumaric acid were still available to be converted into the final product (Fig. S7). Considering that, the production experiment was prolonged until 120 h to also evaluate if there would be differences over time on the production levels for the different biosynthetic pathways tested (Fig. 5).
By expressing pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL_MsCHI plasmids, 689.5 mg/L of naringenin was produced at 120 h (Fig. 5). This production represents ≈2.2-fold production improvement comparing to the one obtained at 63 h, and a higher productivity was also obtained in this production experiment (5.8 mg/L/h vs 5.1 mg/L/h). The consumption of glucose and the profile of the production of p-coumaric acid and naringenin are presented in Fig. S8. Along the experiment, a high p-coumaric acid accumulation was observed similarly to what occurred on the other production experiments. Additionally, only 17 g/L of glucose was consumed during this production experiment by the E. coli M-PAR-121 strain expressing pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL_MsCHI.
After increasing the fermentation time, optimizations in the media components need to be performed to try to improve the production process. Since it was found that glucose accumulates at the end of all our production experiments, reducing glucose concentration was considered. Different concentrations of glucose were tested (40 g/L (control), 30 g/L, 20 g/L, and 10 g/L), and its effect on naringenin production was assessed (Fig. 6).

Evaluation of the effect of the carbon source on the levels of production of naringenin. a Naringenin production by Escherichia coli M-PAR-121 expressing the plasmids pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL_MsCHI using different concentrations of glucose as carbon source (40 g/L, 30 g/L, 20 g/L, and 10 g/L). b Profile of naringenin and p-coumaric acid production and glucose consumption for the E. coli M-PAR-121 carrying pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL_MsCHI when the production experiment was performed with 30 g/L of glucose. Results correspond to the average of three independent experiments ± standard deviation
As shown in Fig. 6a, the highest production of naringenin was achieved when 30 g/L of glucose was used in the production experiment. Using this concentration, 765.9 mg/L of naringenin was produced, with a productivity of 6.4 mg/L/h and a yield of 0.03 g/g. This result represents a ≈1.1-fold improvement on the production levels compared to the control. The UHPLC chromatogram of this sample can be observed in Fig. S5. Beyond improving production levels, the use of a lower glucose concentration also has the advantage of reducing the production costs associated to the process. Additionally, it was possible to verify that significant p-coumaric acid amounts are still accumulated at the end of the production experiments (433.4 mg/L) and remain available to be subsequently converted into naringenin (Fig. 6b). Although the higher production titer and productivity were achieved using 30 g/L of glucose, higher production yields were reached using 20 g/L of glucose and 10 g/L of glucose as carbon source (0.03 g/g and 0.04 g/g, respectively). This naringenin production (765.9 mg/L) represents the highest de novo production of naringenin achieved so far in E. coli.
Discussion
Naringenin is a plant polyphenolic compound with several known bioactivities and potential applications. Consequently, the industrial interest on this compound has been increasing, and the design of microbial cell factories able to produce this compound has been considered an alternative solution to reach its industrial production. By performing a step-by-step validation and optimization of the biosynthetic pathway, we have constructed an optimized E. coli strain able to produce de novo naringenin with the highest production levels reported so far in the literature using E. coli as a microbial chassis. This step-by-step optimization started by the validation of the first step of the biosynthetic pathway, catalyzed by TAL, to produce p-coumaric acid. Herein, two different TAL genes (RgTAL or FjTAL) were expressed in three different E. coli strains (Fig. 2). From the three tested strains, E. coli M-PAR-121 was the one that was able to produce the highest p-coumaric acid amounts. This strain was previously engineered to boost the l-tyrosine production pool. Therefore, it can produce high amounts of l-tyrosine from glucose being available to be converted into p-coumaric acid. This strain was constructed by Koma et al. (2020) by integrating two genes of the central metabolic pathway and eight genes of the shikimate pathway. Moreover, it is a plasmid-free strain which facilitates its use for the construction of metabolic pathways, and it was able to produce twofold l-tyrosine comparing with other previously constructed l-tyrosine plasmid-free overproducing strains (Koma et al. 2020). For all the strains, the highest production levels of p-coumaric acid were achieved when FjTAL was expressed. This was expected since FjTAL has a lower Km than RgTAL for l-tyrosine (6.7 µM vs 380 µM), as reported by Jendresen et al. (2015) and Zhou et al. (2016). FjTAL has also a very low ability to accept l-phenylalanine comparing with its ability to accept l-tyrosine with a Kcat/Km ratio of 2400 (Jendresen et al. 2015). In contrast, RgTAL has only Kcat/Km ratio of 10 (Zhou et al. 2016). These catalytic properties demonstrate that the FjTAL enzyme is highly specific to accept tyrosine and has a high catalytic efficiency (Jendresen et al. 2015; Zhou et al. 2016; Haslinger and Prather 2020). Moreover, it was also recently shown by Virklund et al. (2023) that FjTAL has a low affinity for p-coumaric acid being less prone to suffer product inhibition. To verify if E. coli K-12 MG1655 (DE3) and E. coli BL21 (DE3) can reach the same production levels than E. coli M-PAR-121, l-tyrosine was supplemented to the production media. Although the production of p-coumaric acid has increased comparing with the production levels achieved in the experiments performed with only glucose, the production on both strains did not reach the same production levels that were achieved by the E. coli M-PAR-121 without any supplementation of l-tyrosine (Fig. 2). This result reinforces that M-PAR-121 has a high pool of l-tyrosine being produced due to the modifications performed in its central metabolism that is consequently available for p-coumaric acid production. In fact, the E. coli M-PAR-121 strain was reported to be able to produce 2.2 g/L of tyrosine that corresponds to 12.1 mM (Koma et al. 2020). Considering this endogenous production of tyrosine, the supplementation of 3 mM of l-tyrosine in the BL21 (DE3) and K-12 MG1655 (DE3) strains did not meet the levels of endogenous production of the M-PAR-121 strain. This discrepancy may explain why these strains could not reach the production levels achieved in the M-PAR-121 strain. This l-tyrosine concentration (3 mM) was chosen based on previous reports of heterologous production of hydroxycinnamic acids and other polyphenols in E. coli (Katsuyama et al. 2008; Rodrigues et al. 2015a, 2020; Haslinger and Prather 2020). The supplementation of 12 mM of l-tyrosine to match the amount produced by the M-PAR-121 strain would not be reasonable, as this substrate is expensive and would increase the costs associated with the production process. Nevertheless, since the main goal of our work was to achieve the final production of naringenin without supplementing expensive precursors, the E. coli M-PAR-121 strain expressing FjTAL was selected as platform strain to test the next steps of the pathway. Moreover, comparing FjTAL and RgTAL efficiency in these production experiments with l-tyrosine supplementation, it was not possible to observe differences statistically significant in the production levels. This result demonstrates that both TAL enzymes have a good performance when l-tyrosine concentrations are not limiting, which has been previously reported (Haslinger & Prather (2020)).
After choosing this strain, 4CL and CHS steps were validated by testing genes from different organisms (Fig. 3). Regarding 4CL, our study showed higher production levels when At4CL was expressed. Similarly, Jones et al. (2016) reported higher productions of naringenin and eriodyctiol in all the pathways holding the At4CL gene independently of the CHS and CHI expressed. Moreover, regarding the 4CL genes from A. thaliana identified so far (Ehlting et al. 1999), the isoform used in our study (4CL1) holds a low Km for p-coumaric acid, showing its specificity for this substrate. Notwithstanding, the 4CL step is always considered a critical step in the phenylpropanoid pathway. This enzyme catalyzes the conversion of p-coumaric acid into p-coumaroyl-CoA in two steps. The first step corresponds to the adenylate formation and the second one to the thioester formation. These two steps require the presence of adenosine triphosphate (ATP), coenzyme A (CoA), and Mg2+. Since these molecules are also used by key metabolic pathways, it is expected that they are less available to be used on the phenylpropanoid pathway, possibly being a limiting factor to achieve higher production levels (Ehlting et al. 2001; Lavhale et al. 2018). Moreover, 4CL enzymes are not completely efficient on the conversion of the hydroxycinnamic acid to its derived CoA ester (Ehlting et al. 1999, 2001; Lavhale et al. 2018). Considering that, improving their activity through the construction of mutant versions of the 4CL enzyme can be an interesting approach to be employed. For example, Xiong et al. (2017) have designed a random mutagenesis library of the At4CL1 and have found one mutant with a 1.7-fold higher catalytic efficiency for p-coumaric acid comparing to the wild-type enzyme, leading also to a significant improvement in the heterologous production of resveratrol and naringenin. This strategy can also be considered in the future to improve the production levels (Xiong et al. 2017). Regarding the CHS step, the highest productions were achieved when CmCHS was expressed (Fig. 3b). This result demonstrates the higher efficiency of this gene to convert p-coumaroyl-CoA into naringenin chalcone. Moreover, it also suggests that CmCHS is more efficient than the other tested CHS genes to compete with the primary metabolism pathways that use malonyl-CoA, namely the fatty acid biosynthesis pathway.
After the identification of the best pathway enzymes to produce naringenin chalcone, strategies to reduce the metabolic burden of the cells and to balance the pathway were tested. Only one of these strategies led to an increase in the production levels (Fig. 4). In this strategy (E. coli M-PAR-121 expressing pRSFDuet_FjTAL_CmCHS and pACYCduet_At4CL), the FjTAL gene was maintained on a high-copy plasmid, resulting in higher p-coumaric acid production during the experiment. The At4CL gene remained in its original plasmid backbone (pACYCDuet-1). Additionally, the CmCHS gene was transferred from a medium-copy plasmid (pCDFDuet-1) to a high-copy plasmid (pRSFDuet-1), likely enhancing the conversion of p-coumaroyl-CoA to naringenin chalcone. In the other two strategies, the FjTAL gene was moved to a low-copy plasmid (pACYCDuet-1). The rationale behind these strategies was to reduce p-coumaric acid accumulation and observe its impact on the production of the final compound. As anticipated, there was a lower accumulation of p-coumaric acid during the experiment, which seems to have limited naringenin chalcone production. Moreover, in both strategies, the At4CL gene was moved to the pRSFDuet-1 vector (pRSFDuet_At4CL_CmCHS) and to the MCS2 of the pACYCDuet-1 vector (pACYCDuet_FjTAL_At4CL). Previous research performed by our group also found lower curcumin production when the At4CL gene was moved from the pACYCDuet-1 vector to the pRSFDuet-1 vector (Rodrigues et al. 2020). This finding supports the idea that maintaining At4CL in MCS1 of pACYCDuet-1 is critical for the efficient conversion of p-coumaric acid to p-coumaroyl-CoA (Rodrigues et al. 2020).
Naringenin chalcone has been previously demonstrated to spontaneously cyclize to form naringenin (Mol et al. 1985). However, it was found that this self-cyclization mostly occurs under basic conditions, with a higher cyclization rate at a pH of 7.5. Moreover, self-cyclization is dramatically reduced at pH values ≤6.5, with only a 10% rate of self-cyclization observed at these pH values (Mol et al. 1985). In our experiment, the initial pH of the M9 media is 6.5, maintained constant throughout the production experiment due to the presence of CaCO3 in suspension, which is added for pH maintenance. CaCO3 is commonly used in acid-forming microbial processes to maintain the pH at around 6.5 (Salek et al. 2015; Ronoh et al. 2022). Therefore, significant cyclization is not expected to be observed in our experiment. This could be confirmed by UHPLC. The representative chromatogram of the naringenin chalcone production experiment (Fig. S5) shows a clear peak corresponding to naringenin chalcone at 12.0 min and only a very small peak at 12.6 min that corresponds to the naringenin retention time. Since the peak corresponding to naringenin in the samples is very small, the self-cyclization of naringenin chalcone into naringenin was considered negligible. This lack of significant cyclization demonstrates that the expression of CHI is essential for converting naringenin chalcone to naringenin and completing the pathway. Consequently, the CHI step was evaluated through the expression of three different CHI from different origins (Fig. 5). After increasing fermentation time, MsCHI was found to be more efficient leading to a higher production of naringenin. Similarly, Leonard et al. (2007) reported higher amounts of naringenin, pinocembrin, and eriodyctiol by expressing MsCHI instead of other CHI not compared here (e.g., CHI from P. hybrida) (Leonard et al. 2007). After testing different glucose concentrations, 765.9 mg/L of naringenin was produced (Fig. 6). During the naringenin production experiments, only the naringenin peak was detected by UHPLC demonstrating that MsCHI was highly efficient to convert naringenin chalcone into naringenin (Fig. S5). The fact that only one peak was found in the samples of the naringenin production experiments supports the previous finding that CHI activity to catalyze the naringenin chalcone cyclization is 107-fold more efficient than self-cyclization (Cheng et al. 2018).
Compared to previous reports in the literature, the naringenin production herein obtained (765.9 mg/L) is a great achievement in E. coli being the highest reported so far using this microorganism. The highest production previously reported was 485 mg/L in shake flask experiments and 585 mg/L in a fed-batch bioreactor using an E. coli strain modified both to improve tyrosine and malonyl-CoA flux (Zhou et al. 2020a). Comparing with the production achieved in Y. lipolytica (898 mg/L), the productivity of our production process was significantly higher (6.4 mg/L/h vs 3.0 mg/L/h) (Palmer et al. 2020). Beyond Y. lipolytica, the highest naringenin titer reported so far was achieved in S. cerevisiae strain (Zhang et al. 2021). Zhang et al. (2021) modified a previously constructed platform S. cerevisiae strain holding the naringenin pathway to improve its malonyl-CoA flux and have compared the production levels between the modified and non-modified strain. The non-modified strain was able to produce 703.5 mg/L of naringenin. In contrast, the modified strain was able to produce 1129.4 mg/L. This difference in the production levels between both strains suggests that improving malonyl-CoA levels is necessary to increase the final production of naringenin. However, these production levels were achieved at a fed-batch bioreactor scale being not possible to compare with the production levels obtained in our study. In our work, we have performed one extensive step-by-step optimization of the biosynthetic pathway. Several genes from different sources were tested to achieve the highest production levels in each step. We believe that this step-by-step optimization allowed us to find a combination of genes able to produce high amounts of naringenin. As far as we know, this is the first report of naringenin production using this specific combination of genes of the pathway (FjTAL, At4CL, CmCHS, and MsCHI). Beyond the extensive exploitation of different genes for each step, we have also tested different E. coli strains. The production levels of one of the key intermediates of the pathway (p-coumaric acid) were significantly higher in the E. coli M-PAR-121. The utilization of this strain able to produce high levels of l-tyrosine can also be an advantage comparing with previous reports of heterologous production of naringenin in E. coli. We believe that the combination of the new pathway genes and the use of a highly efficient tyrosine-overproducing strain is the main reason for our high production levels. This highlights the importance of testing different chassis and pathway genes to find the optimal combination. Although we achieved these production levels without improving the malonyl-CoA availability, we believe that this factor can also be a rate-limiting step in our work hampering higher production levels of naringenin since three molecules of this compound are required in this pathway to perform the extension reaction (Fig. 1). Malonyl-CoA is naturally synthesized by E. coli and participates in key metabolic processes, namely in the production of phospholipids and fatty acids. Moreover, its synthesis is tightly regulated, and it is present at low levels inside of the E. coli cells, which limits its utilization on the biosynthetic pathways responsible for the production of heterologous compounds (Milke and Marienhagen 2020). Several different strategies have already been used to increase malonyl-CoA availability in engineered strains and successfully improve the production levels of naringenin (Xu et al. 2011; Wu et al. 2014, 2015; Zhou et al. 2020b, 2020a). In the future, it will be interesting to improve the malonyl-CoA synthesis by overexpressing key genes involved in its synthesis, for example, the malonate assimilation pathway genes matB and matC from Rhizobium trifolii and the native acetyl-CoA carboxylase to improve malonate and acetyl-CoA conversion into malonyl-CoA, respectively (Xu et al. 2011; Wu et al. 2014). Another interesting alternative that can be considered is the overexpression of the native pyruvate dehydrogenase and phosphoglycerate kinase genes to improve the synthesis of the acetyl-CoA intermediate (Xu et al. 2011; Wu et al. 2014). The deletion of genes of the tricarboxylic acid cycle and glycolysis should also be considered, namely the knockout of genes corresponding to fatty acids synthases (fabB and fabF), fumarase, succinyl-CoA synthetase, and acetaldehyde dehydrogenase (Wu et al. 2015). The combination of these overexpression and deletion strategies should be tested in the future in our strain to evaluate the production levels.
In conclusion, by performing a step-by-step optimization, E. coli M-PAR-121 expressing pRSFDuet_FjTAL_CmCHS and pACYCDuet_At4CL_MsCHI was selected as the best producer of naringenin (769.5 mg/L–6.4 mg/L/h) (Fig. 6). To our knowledge, this is the first time that this optimized combination of genes was used to produce naringenin. Moreover, this naringenin production level corresponds to the highest production reported so far using E. coli. However, there is still a long way to go to achieve the production of this compound at an industrial scale, since a higher titer, yield, and productivity should be required to attain a cost-effective industrial production process. To optimize the metabolic flux of the pathway in the future, considerations should include improving the efficiency of the 4CL step and optimizing CoA and malonyl-CoA availability at the genetic level. Optimizations at the operational conditions level, namely the use of a single production media instead of using the combination of LB with M9, should also be considered to further implement a production process at a larger scale.
Data availability
The data supporting the findings of this study is available within the article and its supplementary material.
References
Castiñeiras TS, Williams SG, Hitchcock AG, Smith DC (2018) E. coli strain engineering for the production of advanced biopharmaceutical products. FEMS Microbiol Lett 365:1–10. https://doi.org/10.1093/femsle/fny162
Cheng AX, Zhang X, Han XJ, Zhang YY, Gao S, Liu CJ, Lou HX (2018) Identification of chalcone isomerase in the basal land plants reveals an ancient evolution of enzymatic cyclization activity for synthesis of flavonoids. New Phytol 217:909–924. https://doi.org/10.1111/nph.14852
Dunstan MS, Robinson CJ, Jervis AJ, Yan C, Carbonell P, Hollywood KA, Currin A, Swainston N, Le FR, Micklefield J, Faulon J-L, Breitling R, Turner N, Takano E, Scrutton NS (2020) Engineering Escherichia coli towards de novo production of gatekeeper (2S)-flavanones: naringenin, pinocembrin, eriodictyol and homoeriodictyol. Synth Biol 5:1–11. https://doi.org/10.1093/synbio/ysaa012
Ehlting J, Büttner D, Wang Q, Douglas CJ, Somssich IE, Kombrink E (1999) Three 4-coumarate:coenzyme A ligases in Arabidopsis thaliana represent two evolutionarily divergent classes in angiosperms. Plant J 19:9–20. https://doi.org/10.1046/j.1365-313X.1999.00491.x
Ehlting J, Shin JJK, Douglas CJ (2001) Identification of 4-coumarate:coenzyme A ligase (4CL) substrate recognition domains. Plant J 27:455–465. https://doi.org/10.1046/j.1365-313X.2001.01122.x
Ghofrani S, Joghataei MT, Mohseni S, Baluchnejadmojarad T, Bagheri M, Khamse S, Roghani M (2015) Naringenin improves learning and memory in an Alzheimer’s disease rat model: insights into the underlying mechanisms. Eur J Pharmacol 764:195–201. https://doi.org/10.1016/j.ejphar.2015.07.001
Gomes D, Rainha J, Rodrigues LR, Rodrigues JL (2022a) Yeast synthetic biology approaches for the production of valuable polyphenolic compounds. In: Harzevili FD (ed) Synthetic biology of yeasts. Springer Nature, Cham, pp. 119–156.
Gomes D, Rodrigues LR, Rodrigues JL (2022b) Perspectives on the design of microbial cell factories to produce prenylflavonoids. Int J Food Microbiol 367:109588. https://doi.org/10.1016/j.ijfoodmicro.2022.109588
Haslinger K, Prather KLJ (2020) Heterologous caffeic acid biosynthesis in Escherichia coli is affected by choice of tyrosine ammonia lyase and redox partners for bacterial cytochrome P450. Microb Cell Fact 19:1–12. https://doi.org/10.1186/s12934-020-01300-9
Jendresen CB, Stahlhut SG, Li M, Gaspar P, Siedler S, Förster J, Maury J, Borodina I, Nielsen AT (2015) Highly active and specific tyrosine ammonia-lyases from diverse origins enable enhanced production of aromatic compounds in bacteria and Saccharomyces cerevisiae. Appl Environ Microbiol 81:4458–4476. https://doi.org/10.1128/AEM.00405-15
Jin L, Zeng W, Zhang F, Zhang C, Liang W (2017) Naringenin ameliorates acute inflammation by regulating intracellular cytokine degradation. J Immunol 199:3466–3477. https://doi.org/10.4049/jimmunol.1602016
Jones JA, Vernacchio VR, Sinkoe AL, Collins SM, Ibrahim MHA, Lachance DM, Hahn J, Koffas MAG (2016) Experimental and computational optimization of an Escherichia coli co-culture for the efficient production of flavonoids. Metab Eng 35:55–63. https://doi.org/10.1016/j.ymben.2016.01.006
Jones JA, Vernacchio VR, Collins SM, Shirke AN, Xiu Y, Englaender JA ... Koffas MA (2017) Complete biosynthesis of anthocyanins using E. coli polycultures. MBio 8(3):10–1128. 7. https://doi.org/10.1128/mBio.00621-17
Kataoka H, Saeki A, Hasebe A, Shibata K, ichiro, Into T, (2020) Naringenin suppresses Toll-like receptor 2-mediated inflammatory responses through inhibition of receptor clustering on lipid rafts. Food Sci Nutr 9:963–972. https://doi.org/10.1002/fsn3.2063
Katsuyama Y, Matsuzawa M, Funa N, Horinouchi S (2008) Production of curcuminoids by Escherichia coli carrying an artificial biosynthesis pathway. Microbiology 154:2620–2628. https://doi.org/10.1099/mic.0.2008/018721-0
Koma D, Kishida T, Yoshida E, Ohashi H, Hayato Yamanaka KM, Nagamori E, Ohmoto T (2020) Chromosome engineering to generate plasmid-free phenylalanine- and tyrosine-overproducing Escherichia coli strains that can be applied in the generation of aromatic- compound-producing bacteria. Appl Environ Microbiol 86:1–24
Koopman F, Beekwilder J, Crimi B, van Houwelingen A, Hall RD, Bosch D, van Maris AJA, Pronk JT, Daran JM (2012) De novo production of the flavonoid naringenin in engineered Saccharomyces cerevisiae. Microb Cell Fact 11:1–15. https://doi.org/10.1186/1475-2859-11-155
Lavhale SG, Kalunke RM, Giri AP (2018) Structural, functional and evolutionary diversity of 4-coumarate-CoA ligase in plants. Planta 248:1063–1078. https://doi.org/10.1007/s00425-018-2965-z
Leonard E, Lim KH, Saw PN, Koffas MAG (2007) Engineering central metabolic pathways for high-level flavonoid production in Escherichia coli. Appl Environ Microbiol 73:3877–3886. https://doi.org/10.1128/AEM.00200-07
Li H, Gao S, Zhang S, Zeng W, Zhou J (2021) Effects of metabolic pathway gene copy numbers on the biosynthesis of (2S)-naringenin in Saccharomyces cerevisiae. J Biotechnol 325:119–127. https://doi.org/10.1016/j.jbiotec.2020.11.009
Li J, Tian C, Xia Y, Mutanda I, Wang K, Wang Y (2019) Production of plant-specific flavones baicalein and scutellarein in an engineered E. coli from available phenylalanine and tyrosine. Metab Eng 52:124–133. https://doi.org/10.1016/j.ymben.2018.11.008
Liu X, Ding W, Jiang H (2017) Engineering microbial cell factories for the production of plant natural products: from design principles to industrial-scale production. Microb Cell Fact 16:1–9. https://doi.org/10.1186/s12934-017-0732-7
Liu X, Li L, Zhao GR (2021) Systems metabolic engineering of Escherichia coli coculture for de novo production of genistein. ACS Synth Biol 11:1746–1757. https://doi.org/10.1021/acssynbio.1c00590
Lv Y, Marsafari M, Zhou J, Xu P (2019) Optimizing oleaginous yeast cell factories for flavonoids and hydroxylated flavonoids biosynthesis. ACS Synth Biol 8:2514–2523. https://doi.org/10.1021/acssynbio.9b00193
Lyu X, Zhao G, Ng KR, Mark R, Chen WN (2019) Metabolic engineering of Saccharomyces cerevisiae for de novo production of kaempferol. J Agric Food Chem 67:5596–5606. https://doi.org/10.1021/acs.jafc.9b01329
Milke L, Marienhagen J (2020) Engineering intracellular malonyl-CoA availability in microbial hosts and its impact on polyketide and fatty acid synthesis. Appl Microbiol Biotechnol 104:6057–6065. https://doi.org/10.1007/s00253-020-10643-7
Mol JNM, Robbins MP, Dixon RA, Veltkamp E (1985) Spontaneous and enzymic rearrangement of naringenin chalcone to flavanone. Phytochemistry 24:2267–2269. https://doi.org/10.1016/S0031-9422(00)83023-X
Nielsen DR, Yoon SH, Yuan CJ, Prather KLJ (2010) Metabolic engineering of acetoin and meso-2,3-butanediol biosynthesis in E. coli. Biotechnol J 5:274–284. https://doi.org/10.1002/biot.200900279
Olsson L, Rugbjerg P, Torello Pianale L, Trivellin C (2022) Robustness: linking strain design to viable bioprocesses. Trends Biotechnol 40:918–931. https://doi.org/10.1016/j.tibtech.2022.01.004
Palmer CM, Miller KK, Nguyen A, Alper HS (2020) Engineering 4-coumaroyl-CoA derived polyketide production in Yarrowia lipolytica through a β-oxidation mediated strategy. Metab Eng 57:174–181. https://doi.org/10.1016/j.ymben.2019.11.006
Pandey RP, Parajuli P, Koffas MAG, Sohng JK (2016) Microbial production of natural and non-natural flavonoids: pathway engineering, directed evolution and systems/synthetic biology. Biotechnol Adv 34:634–662. https://doi.org/10.1016/j.biotechadv.2016.02.012
Rainha J, Gomes D, Rodrigues LR, Rodrigues JL (2020) Synthetic biology approaches to engineer Saccharomyces cerevisiae towards the industrial production of valuable polyphenolic compounds. Life 10:56. https://doi.org/10.3390/life10050056
Rodrigues JL, Araújo RG, Prather KLJ, Kluskens LD (2015a) Production of curcuminoids from tyrosine by a metabolically engineered Escherichia coli using caffeic acid as an intermediate. Biotechnol J 10:1–27. https://doi.org/10.1002/biot.201400637
Rodrigues JL, Araújo RG, Prather KLJ, Kluskens LD, Rodrigues LR (2015b) Heterologous production of caffeic acid from tyrosine in Escherichia coli. Enzyme Microb Technol 71:36–44. https://doi.org/10.1016/j.enzmictec.2015.01.001
Rodrigues JL, Gomes D, Rodrigues LR (2022) Challenges in the heterologous production of furanocoumarins in Escherichia coli. Molecules 27:1–20. https://doi.org/10.3390/molecules27217230
Rodrigues JL, Gomes D, Rodrigues LR (2020) A combinatorial approach to optimize the production of curcuminoids from tyrosine in Escherichia coli. Front Bioeng Biotechnol 8:1–15. https://doi.org/10.3389/fbioe.2020.00059
Ronoh DK, Swart RM, Nicol W, Brink H (2022) The effect of pH, metal ions, and insoluble solids on the production of fumarate and malate by Rhizopus delemar in the presence of CaCO3. Catalysts 12. https://doi.org/10.3390/catal12030263
Salek SS, van Turnhout AG, Kleerebezem R, van Loosdrecht MCM (2015) pH control in biological systems using calcium carbonate. Biotechnol Bioeng 112:905–913. https://doi.org/10.1002/bit.25506
Shi X, Luo X, Chen T, Guo W, Liang C, Tang S, Mo J (2021) Naringenin inhibits migration, invasion, induces apoptosis in human lung cancer cells and arrests tumour progression in vitro. J Cell Mol Med 25:2563–2571. https://doi.org/10.1111/jcmm.16226
Silva F, Queiroz JA, Domingues FC (2012) Evaluating metabolic stress and plasmid stability in plasmid DNA production by Escherichia coli. Biotechnol Adv 30:691–708. https://doi.org/10.1016/j.biotechadv.2011.12.005
Virklund A, Jendresen CB, Nielsen AT, Woodley JM (2023) Characterization of tyrosine ammonia lyases from Flavobacterium johnsonian and Herpetosiphon aurantiacus. Biotechnol J 2023:1–8. https://doi.org/10.1002/biot.202300111
Wadhwa R, Paudel KR, Chin LH, Hon CM, Madheswaran T, Gupta G, Panneerselvam J, Lakshmi T, Singh SK, Gulati M, Dureja H, Hsu A, Mehta M, Anand K, Devkota HP, Chellian J, Chellappan DK, Hansbro PM, Dua K (2020) Anti-inflammatory and anticancer activities of naringenin-loaded liquid crystalline nanoparticles in vitro. J Food Biochem 45:1–14. https://doi.org/10.1111/jfbc.13572
Waki T, Mameda R, Nakano T, Yamada S, Terashita M, Ito K, Tenma N, Li Y, Fujino N, Uno K, Yamashita S, Aoki Y, Denessiouk K, Kawai Y, Sugawara S, Saito K, Yonekura-Sakakibara K, Morita Y, Hoshino A, Takahashi S, Nakayama T (2020) A conserved strategy of chalcone isomerase-like protein to rectify promiscuous chalcone synthase specificity. Nat Commun 11:1–14. https://doi.org/10.1038/s41467-020-14558-9
Wang D, Wang X, Tong W, Cui Y, Li X, Sun H (2019) Umbelliferone alleviates lipopolysaccharide-induced inflammatory responses in acute lung injury by down-regulating TLR4/MyD88/NF-κB signaling. Inflammation 42:440–448. https://doi.org/10.1007/s10753-018-00953-4
Watts KT, Lee PC, Schmidt-Dannert C (2004) Exploring recombinant flavonoid biosynthesis in metabolically engineered Escherichia coli. ChemBioChem 5:500–507. https://doi.org/10.1002/cbic.200300783
Wehrs M, Tanjore D, Eng T, Lievense J, Pray TR, Mukhopadhyay A (2019) Engineering robust production microbes for large-scale cultivation. Trends Microbiol 27:524–537. https://doi.org/10.1016/j.tim.2019.01.006
Wu J, Du G, Chen J, Zhou J (2015) Enhancing flavonoid production by systematically tuning the central metabolic pathways based on a CRISPR interference system in Escherichia coli. Sci Rep 5:1–14. https://doi.org/10.1038/srep13477
Wu J, Zhou T, Du G, Zhou J, Chen J (2014) Modular optimization of heterologous pathways for de novo synthesis of (2S)-naringenin in Escherichia coli. PLoS ONE 9:1–9. https://doi.org/10.1371/journal.pone.0101492
Xiong D, Shikun L, Wu J, Liang C, Wang W, Wang W, Jin J-M, Tang S-Y (2017) Improving key enzyme activity in phenylpropanoid pathway with a designed biosensor. Metab Eng 40:115–123. https://doi.org/10.1016/j.ymben.2017.01.006
Xu P, Ranganathan S, Fowler ZL, Maranas CD, Koffas MAG (2011) Genome-scale metabolic network modeling results in minimal interventions that cooperatively force carbon flux towards malonyl-CoA. Metab Eng 13:578–587. https://doi.org/10.1016/j.ymben.2011.06.008
Ye S, Magadán-Corpas P, Pérez-Valero Á, Villar CJ, Lombó F (2023) Metabolic engineering strategies for naringenin production enhancement in Streptomyces albidoflavus J1074. Microb Cell Fact 22:167. https://doi.org/10.1186/s12934-023-02172-5
Yonekura-Sakakibara K, Higashi Y, Nakabayashi R (2019) The origin and evolution of plant flavonoid metabolism. Front Plant Sci 10:1–16. https://doi.org/10.3389/fpls.2019.00943
Zhang Q, Yu S, Lyu Y, Zeng W, Zhou J (2021) Systematically engineered fatty acid catabolite pathway for the production of (2 S)-naringenin in Saccharomyces cerevisiae. ACS Synth Biol 10:1166–1175. https://doi.org/10.1021/acssynbio.1c00002
Zhou S, Hao T, Zhou J (2020a) Fermentation and metabolic pathway optimization to de novo synthesize (2S)-naringenin in Escherichia coli. J Microbiol Biotechnol 30:1574–1582. https://doi.org/10.4014/JMB.2008.08005
Zhou S, Liu P, Chen J, Du G, Li H, Zhou J (2016) Characterization of mutants of a tyrosine ammonia-lyase from Rhodotorula glutinis. Appl Microbiol Biotechnol 100:10443–10452. https://doi.org/10.1007/s00253-016-7672-8
Zhou S, Yuan S-F, Nair P, Alper H, Deng Y, Zhou J (2020b) Development of a growth coupled dynamic regulation network balancing malonyl-CoA node to enhance (2-S)-naringenin synthesis in E. coli. BioRxiv 7:1–29. https://doi.org/10.1101/2020.07.07.19263
Acknowledgements
The authors thank Dr. Daisuke Koma for kindly providing the E. coli M-PAR-121 strain (Koma et al. 2020). The authors also thank Dr. Christian Bille Jendresen (Jendresen et al. 2015), Dr. Claudia Schmidt-Dannert (Watts et al. 2004), Dr. Toru Nakayama (Waki et al. 2020), and Dr. Mark Dunstan (Dunstan et al. 2020) for kindly providing the plasmids pCBJ280, pBADMod1-CHS, pYS454, and pBbE2c-Gm4CL1, respectively.
Funding
Open access funding provided by FCT|FCCN (b-on). This study was supported by the Portuguese Foundation for Science and Technology (FCT) under the scope of the strategic funding of UIDB/04469/2020 unit with https://doi.org/10.54499/UIDB/04469/2020. D.G. acknowledges FCT for her grant SFRH/BD/04433/2020.
Author information
Authors and Affiliations
Contributions
DG performed the experiments and data analysis. DG and JR conceived, designed, and drafted the manuscript. JR and LR coordinated the study and provided feedback and suggestions on the manuscript. LR obtained financial support. All authors gave final approval for publication.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gomes, D., Rodrigues, J.L. & Rodrigues, L.R. Step-by-step optimization of a heterologous pathway for de novo naringenin production in Escherichia coli. Appl Microbiol Biotechnol 108, 435 (2024). https://doi.org/10.1007/s00253-024-13271-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-024-13271-7