
Abstract
Oxygen is crucial for converting glucose to gluconic acid catalyzed by glucose oxidase (Gox). However, industrial gluconic acid production faces oxygen supply limitations. To enhance Gox efficiency, Vitreoscilla hemoglobin (VHb) has been considered as an efficient oxygen transfer carrier. This study identified GoxA, a specific isoform of Gox in the industrial gluconic acid-producing strain of Aspergillus niger. Various forms of VHb expression in A. niger were tested to improve GoxA’s catalytic efficiency. Surprisingly, the expression of free VHb, both intracellularly and extracellularly, did not promote gluconic acid production during shake flask fermentation. Then, five fusion proteins were constructed by linking Gox and VHb using various methods. Among these, VHb-GS1-GoxA, where VHb’s C-terminus connected to GoxA’s N-terminus via the flexible linker GS1, demonstrated a significantly higher Kcat/Km value (96% higher) than GoxA. Unfortunately, the expression of VHb-GS1-GoxA in A. niger was limited, resulting in a low gluconic acid production of 3.0 g/L. To overcome the low expression problem, single- and dual-strain systems were designed with tools of SpyCatcher/SpyTag and SnoopCatcher/SnoopTag. In these systems, Gox and VHb were separately expressed and then self-assembled into complex proteins. Impressively, the single-strain system outperformed the GoxA overexpression strain S1971, resulting in 23% and 9% higher gluconic acid production under 0.6 vvm and 1.2 vvm aeration conditions in the bioreactor fermentation, respectively. The successful construction of Gox and VHb fusion or complex proteins, as proposed in this study, presents promising approaches to enhance Gox catalytic efficiency and lower aerodynamic costs in gluconic acid production.
Key points
• Overexpressing free VHb in A. niger did not improve the catalytic efficiency of Gox
• The VHb-GS1-GoxA showed an increased Kcat/Km value by 96% than GoxA
• The single-strain system worked better in the gluconic acid bioreactor fermentation
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucose oxidase (Gox) is an enzymatic catalyst with significant industrial importance, primarily used for the conversion of glucose into gluconic acid (Bankar et al. 2009; Bauer et al. 2022). This enzymatic reaction involves electron transfer, with oxygen serving as the electron acceptor and the generation of hydrogen peroxide as a byproduct. The reaction products, gluconic acid and hydrogen peroxide, possess unique properties, such as antibacterial effects, making Gox highly valuable in diverse industries, including food, feed, pharmaceuticals, and healthcare (Bankar et al. 2009; Bauer et al. 2022). Furthermore, the electron transfer properties of Gox have garnered considerable interest in the field of biosensor development. By harnessing the enzymatic capabilities of Gox, biosensors can be designed to detect glucose levels accurately and efficiently, finding applications in medical diagnostics, glucose monitoring, and various other analytical systems (Choi et al. 2011; Fu et al. 2018; Mano 2019).
Gox is primarily utilized in industrial applications to produce gluconic acid. Gluconic acid is commonly produced through submerged fermentation using Aspergillus niger, a fungus that secretes Gox, or enzymatic catalysis employing commercial Gox (Dubey et al. 2017; Singh and Kumar 2007; Zhang et al. 2016). In recent years, several studies have focused on protein engineering, chemical modification, and immobilization techniques to enhance the stability, catalytic activity, and reusability of Gox (Dubey et al. 2017; Jiang et al. 2021; Kovacevic et al. 2019; Mu et al. 2019; Yu et al. 2021b). These efforts aim to improve the production efficiency and reduce the overall cost of gluconic acid. Moreover, it is worth noting that the Gox-catalyzed reaction is highly dependent on oxygen availability, which has been identified as a limiting factor in industrial production processes (Canete-Rodriguez et al. 2016; Singh and Kumar 2007). Previous research has shown that optimizing oxygen transfer and concentration can significantly enhance the production of gluconic acid (Bankar et al. 2009; Dubey et al. 2017; Khurshid et al. 2013). Such optimization may result in higher aeration costs during actual production, thereby limiting the economic benefits of gluconic acid products. Therefore, there is a need to explore and develop more efficient methods for oxygen transfer to enhance the catalytic activity of Gox. By addressing this challenge, it is possible to improve the overall performance and economic feasibility of Gox-mediated gluconic acid production.
Vitreoscilla hemoglobin (VHb) exhibits a strong capacity for carrying oxygen and possesses terminal oxidase and peroxidase activities (Stark et al. 2012). VHb has emerged as a valuable tool for oxygen transport, and numerous studies had extensively reviewed its properties and applications (Stark et al. 2015; Webster et al. 2021; Yu et al. 2021a). It is widely used in microbial, animal, and plant systems to promote cell growth and enhance the production of various bio-products, including alcohols, antibiotics, organic acids, and polysaccharides (Hofmann et al. 2009; Liu et al. 2017; Stark et al. 2015; Tang et al. 2020; Zhang et al. 2020). Furthermore, VHb has been found to enhance the secretion of proteins such as Lipase, Coenzyme Q10, and L-asparaginase (Kwon et al. 2005; Mora-Lugo et al. 2015; Stark et al. 2015). VHb facilitates the direct delivery of oxygen to oxygenases, suggesting that its oxygen-carrying capacity may potentially improve the catalytic efficiency of Gox.
Gox belongs to the group of oxidases, a subclass of oxidoreductases that require molecular oxygen for their catalytic process. Several studies have focused on the role of VHb in enhancing the catalytic activity of various oxidases (Khang et al. 2003; Liu et al. 2021; Seo et al. 2011; Wang et al. 2022). Previous research demonstrated that fusing VHb with d-amino acid oxidases (DAOs) from Rhodotorula gracilis or Trigonopsis variabilis significantly improved the catalytic efficiency of DAOs in synthesizing 7-aminocephalosporanic acid (Khang et al. 2003; Ma et al. 2009; Seo et al. 2011). In a recent study by Wang et al. (2022), the effect of VHb on the catalytic efficiency of five oxidases, including DAO from T. variabilis (TvDAO), Gox, alcohol oxidase from Pichia pastoris (AOX), p-hydroxymandelate synthase from Amycolatopsis orientalis (AoHMS), and l-glutamate oxidase (LGOX), was systematically investigated. Different forms of VHb were tested, including the addition of free or immobilized VHb into the catalytic systems or fusion of VHb with oxidases. The fusion of VHb with TvDAO and LGOX resulted in higher conversion rates. The chimeric protein VHb-TvDAO exhibited 1.3-fold higher activity compared to immobilized TvDAO on the same resin. The yield of α-ketoglutarate increased from 81 to 97% with the VHb-LGOX fusion protein in bioreactor biotransformation. Moreover, both free and immobilized forms of VHb were found to improve the catalytic efficiency of TvDAO, GOD, and AOX when added to the catalytic systems under specific oxygen supply conditions. For example, the addition of 100 mg of immobilized VHb increased the activities of immobilized Gox and AOX by 1.8-fold and 2.5-fold, respectively. Similarly, the activity of Gox increased by 40% and 60%, and the activity of immobilized AOX increased by 120% and 400% when 0.5 mg and 1.0 mg of free VHb were added (Wang et al. 2022). Although VHb showed a positive effect on Gox, both immobilized and free forms of VHb may be not suitable for commercial applications due to the difficulty of producing VHb proteins in large quantities at affordable costs. Therefore, the fusion protein of Gox and VHb appeared to have more practical application potential. However, Wang et al. (2022) did not further investigate the fusion of Gox and VHb, and currently, few report was also found on the construction of fusion proteins between Gox and VHb.
In this study, the Gox present in an industrial A. niger strain used for industrial production of gluconic acid was initially identified. Subsequently, VHb fusion proteins were designed and constructed to enhance the activity of Gox. However, direct fusion expression of Gox and VHb resulted in a significant decrease in protein secretion. To overcome this limitation, novel approaches utilizing the SpyCatcher/SpyTag and SnoopCatcher/SnoopTag systems were developed to improve the catalytic efficiency of Gox in bioreactor. The research findings provide new insights and potential technical solutions for the commercial application of Gox.
Materials and methods
Strains of A. niger and culture conditions
The parent strain A. niger S1691 was employed for expressing the genes of Gox, VHb, and fusion proteins. Strain S1691 was derived by deleting the genes of four Gox or glucose dehydrogenase (gox1 EHA27180.1, gox2 EHA25730.1, gox3 EHA19699.1, gox4 EHA23687.1) in strain S469. In A. niger ATCC1015, strain S469 was created by overexpressing the Cre enzyme under the control of the Tet-on inducible promoter (Xu et al. 2019). In this study, spore preparation, transformant screening, and gene knock-out phenotype screening of A. niger were performed using potato dextrose agar (PDA), complete medium (CM), and minimal medium (MM), respectively, following previously described methods (Cao et al. 2020). All A. niger strains were cultivated at 28 °C. For plasmid construction and genetic transformation of A. niger, Escherichia coli JM109 and Agrobacterium tumefaciens AGL-1 were utilized, with both being grown in LB medium at 37 °C. When required for strain and plasmid construction, the medium was supplemented with 250 μg/mL hygromycin B or 100 μg/mL kanamycin. The A. niger strains used in this study are listed in Table S1.
Plasmid construction
In this study, the plasmids used are listed in Table S2, and detailed information about the primers can be found in supplement material Table S3. The construction of these plasmids was carried out using the ClonExpress II One Step Cloning Kit (C112-01/02) from Nanjing Vazyme Biotech Co., Ltd. (Nanjing, Jiangsu Province, China). The coding sequences of GoxA-D genes were amplified from cDNA of S1565, whereas the coding sequences of VHb and various fusion proteins were artificially synthesized after codon optimization. The coding sequences of GoxA-D, VHb and various fusion proteins were provided in the supplement material.
The plasmids for expressing GoxA-D, VHb, SPgalA-VHb, GoxA-GS1-VHb, GoxA-GS3-VHb, GoxA-EK1-VHb, GoxA-EK3-VHb, VHb-GS1-GoxA, SPgalA-His-VHb-GS1-GoxA, and SPgalA-His-GoxA were constructed using the same method as previously described in a prior study (Xu et al. 2019). For example, the GoxA coding gene was obtained by amplifying it using the primer pair goxA-F/R and then inserted into the plasmid pLH454, resulting in the creation of pLHGA1. Subsequently, the goxA expression cassette, including the gpdA promoter and trpC terminator, was amplified from pLHGA1 using the primer pair P924-454-L/R and then joined to pLH924, generating the plasmid pLHGA2. Finally, the pLHGA2 vector was utilized for the overexpression of GoxA.
The plasmids used for constructing the single- and dual-strain systems were created using the same method. For instance, to construct the plasmid for expressing SPgalA-His-VHb-SpyCatcher, the coding sequence of SPgalA-His-VHb-SpyCatcher was amplified using the primer pair VSP-F/R and then inserted into the plasmid pLH1080 (Liu et al. 2023), resulting in the formation of pLHVSP. Similarly, to generate the plasmid for expressing SPgalA-Spytag-GoxA, the coding sequence of SPgalA-Spytag-GoxA was amplified using the primer pair GSP-F/R and then inserted into pLH454, leading to the creation of pLHSPG. Subsequently, the SPgalA-His-VHb-Spycacther expression cassette was amplified using the primer pair P1080-L/R and ligated into pLHGSP, resulting in the production of pLHVSPG. The pLHVSPG vector was employed for the co-expression of His-VHb-SpyCatcher and Spytag-GoxA.
Construction of strains
A previously established protocol was employed for the genetic manipulation of A. niger (Cao et al. 2020) Agrobacterium-mediated transformation was utilized to introduce the plasmid into A. niger, while the Cre-loxP-based genetic system was implemented to facilitate recombination at loxP-hph-loxP loci, resulting in the creation of the hph-excision strain.
The strains expressing GoxA-D, VHb, SPgalA-VHb, GoxA-GS1-VHb, GoxA-GS3-VHb, GoxA-EK1-VHb, GoxA-EK3-VHb, VHb-GS1-GoxA, SPgalA-His-VHb-GS1-GoxA, and SPgalA-His-GoxA were generated using the homologous recombination method, as previously described (Cao et al. 2020). To overexpress GoxA, plasmid pLHGA2 was introduced into A. niger S191, and transformants were obtained on CM plates supplemented with cefotaxime sodium (100 μg/mL), hygromycin B (250 μg/mL), ampicillin (100 μg/mL), and streptomycin (100 μg/mL) at 28 °C for 5 days. Putative mutants were identified as hygromycin B-resistant and glufosinate ammonium-sensitive transformants, which were further isolated using PDA plates with hygromycin B (250 μg/mL) and MM with glufosinate ammonium (1000 μg/mL). The confirmation of the mutants was performed using PCR analysis with four primer pairs (Amytest-F1/R1, Amytest-F2/R2, Amytest-F1/gpdA-R, hph-F2/Amytest-R2, and Amytest-F1/Amytest-R2) (as shown in Fig. S1). The verified mutant was named S1971. The same method was used to generate additional mutants, including S2167, S1990, S2102, S2193, S2195, S2294, S2298, S2249, S2250, and S2637. S1971 underwent excision of the hgh using a Cre-loxP system, resulting in the generation of S2143. Plasmids pLHVHB and pLHSVHB were transformed into the S2141 strain, leading to the creation of strains S2241 and S2246, respectively.
For the single-strain system, the plasmid pLHVGSP was introduced into strain S1691. Transformants were subsequently screened on PDA plates with 250 μg/mL hygromycin B. The incorporation of the SPgalA-His-VHb-Spycacther and SPgalA-Spytag-GoxA co-expression cassette was verified through PCR analysis using the primer pairs VSP-F/R and GSP-F/R. The co-expression cassette was randomly inserted into various loci of the genome through non-homologous recombination. A total of 10 verified strains were random selected for gluconic acid production. The strain with the highest output, A. niger S2643, was named. The strains S2648 and S2650 for the dual-strain system were also created using the same method as the single-strain system.
Gluconic acid fermentation in shake flask and bioreactor
The glucose acid shake flask fermentation and bioreactor fermentation methods were slightly modified according to the methods provided by Shandong Fuyang Biotechnology Co., Ltd., China. The composition of the shake flask fermentation medium was as follows: 200 g/L glucose, 0.65 g/L KH2PO4, 1 g/L corn steep liquor, 0.02 g/L MgSO4, 0.2 g/L urea, 20 g/L CaCO3 (added after adjusting the pH to 5.5). For the gluconic acid fermentation in the shake flask, the process involved inoculating 2 × 106 conidia/mL of A. niger mutants into 50 mL of the fermentation medium in 250-mL Erlenmeyer flasks. The flasks were then incubated at 28 °C and 150~250 rpm for a duration of 3 days.
For gluconic acid bioreactor fermentation, 2 × 106 conidia/mL of A. niger mutants was inoculated into the bioreactor fermentation seed medium (200 g/L glucose, 0.3 g/L KH2PO4, 3 g/L corn steep liquor, 0.385 g/L MgSO4, 0.2 g/L urea, pH to 6.5). Seed cultivation was performed in shake flask at 28 °C and 220 rpm. After 24 h of cultivation, the seed culture with 4000 U of Gox activity was collected and inoculated into a 2 L bioreactor containing 1.26 L of bioreactor fermentation medium (300 g/L glucose, 0.158 g/L KH2PO4, 1 g/L corn steep liquor, 0.02 g/L MgSO4, 0.1 g/L urea). The final volume of the fermentation liquid was adjusted to 70% of the bioreactor capacity with sterile water. The bioreactor fermentation was performed at a 0.6 or 1.2 vvm aeration rate, 400 r/min, and 28 °C for 84 h. The pH was maintained and controlled at 5.3 by using 30% NaOH.
Protein fermentation in shake flask
Protein fermentation was carried out following the methodology from the previous report (van den Berg et al. 2012). First, 2 × 106 conidia/mL of A. niger mutants was inoculated into 50 mL pre-culture medium (100 g/L corn steep solids, 1 g/L NaH2PO4·H2O, 0.5 g/L MgSO4·7H2O, 10 g/L glucose, pH was adjusted to 6.0) in 250-mL shake flask. After growth for 24 h at 28 °C and 200 rpm, 5 mL of pre-culture was transferred to 45 mL protein fermentation medium (150 g/L maltose, 60/L g soya peptone, 1 g/L NaH2PO4·H2O, 15 g/L MgSO4·7H2O, 0.08 g/L Tween 80, 20 g/L MES, 1 g/L l-arginine, pH was adjusted to 6.0) in 250-mL shake flask with baffle and foam ball. The protein fermentation process was then carried out at 28 °C and 200 rpm for a total duration of 5 days.
Enzyme purification
The enzyme purification method was adapted from a previous report (Mu et al. 2019) with some modifications. Firstly, the protein fermentation supernatant was collected by passing it through eight layers of gauze and then subjected to high-speed centrifugation at 12,000 ×g for 25 min. Subsequently, imidazole was added to the supernatant to achieve a final concentration of 10 mM. The resulting mixture was then combined with 2 mL of Ni2+-nitrilotriacetic agarose (Qiagen, Duesseldorf, Germany) per liter. To ensure proper equilibration, the mixture was gently stirred at 4 °C for 30 min before being loaded into a column. The resin in the column was washed first with two column volumes of buffer (50 mM acetic acid/sodium acetate, 100 mM NaCl, pH 5.0), followed by washing with 10 column volumes of the same buffer containing 20 mM imidazole. For elution of the target enzymes, His-GoxA and His-VHb-GS1-GoxA, a solution of 250 mM imidazole in buffer was used. The fractions containing the purified enzymes were collected.
Western blot analysis
The purified enzymes or crude enzymes were separated using SDS-PAGE, then transferred onto a PVDF membrane and used for Western blotting following standard procedures (Tang et al. 2012). Specifically, SDS-PAGE was utilized to separate protein samples (150 V, 20 mA for each gel, 1.5 h). The separated proteins were transferred from the gel to the PVDF membrane using a semidry electrophoresis transfer cell (Beijing Liuyi Instrument Factory, China). Subsequently, the membrane was blocked for 1 h at room temperature with skimmed milk (Inner Mongolia Yili Industrial Group Co., Ltd., China) dissolved in Tris-buffer. After removing the skimmed milk solution from the membrane, the membrane was treated with His-Tag Antibody (1:5000, T0009, Affinity Bioscience, China), incubated for 12 h at 4 °C, and then the His-Tag antibody was recovered. The membrane was washed three times with TBST buffer (T1081, Solarbio, China), shaking and washing for 5 min each time. Next, Goat Anti Mouse IgG (H + L) HRP (1:5000, S0002, Affinity Bioscience, China) was used to treat the membrane and incubated at room temperature for 1 h. The Goat Anti Mouse IgG (H+L) HRP was also recovered, and the membrane was washed three times with TBST buffer for 10 min each time. Finally, the chemiluminescence reaction was followed by scanning.
Enzyme assay and enzymatic kinetic assays
The enzyme assay for Gox was conducted following a previously published method with slight modifications (Liu et al. 2020). Specifically, 2.4 mL of sodium phosphate buffer (0.1 M, pH 5.5) containing 0.21 mM o-dianisidine (Sigma, USA) was combined with 0.1 mL of horseradish peroxidase (90 U/mL) and 0.5 mL of a 0.1 M β-D-glucose solution in a 5-mL centrifuge tube. The mixture was then incubated at 37 °C for 10 min. Next, 0.1 mL of the enzyme solution was added and immediately mixed. The absorbance change was measured at 500 nm using a spectrophotometer after 3 min. The absorbance change value was used to calculate the enzyme activity. To establish a standard curve, the commercial Gox (Sigma-Aldrich, Product Number: G2133) was utilized. The commercial Gox was diluted in buffer to create gradient enzyme solutions with concentrations of 0.4, 0.6, 0.8, 1.0, 2.0, and 4.0 U/mL, and these solutions were then used to measure the standard curve. The total protein concentration was assessed using a BCA Protein Assay Kit (TaKaRa, T9300A), and the measurements were conducted in accordance with the guidelines provided by the manufacturer.
The enzymatic kinetic assays for His-GoxA and His-VHb-GS1-GoxA were conducted according to a previously published method (Jiang et al. 2021). The enzyme activities were measured at various concentrations of glucose ranging from 5 to 200 mM under standard conditions. All assays were carried out and analyzed in triplicate. The kinetic parameters, Km (Michaelis constant) and Vmax (maximum reaction rate), were determined by fitting Michaelis-Menten plots using Sigmaplot 1.0 software.
Enzyme catalysis experiment
Crude enzyme catalysis experiment: 20 mL sodium phosphate buffer (0.1 M, pH 5.5) with 2 U/mL crude enzyme and 10 g/L d-glucose at 37 °C and 0~200 rpm for 1 h. The catalytic process of Gox produces a substantial quantity of hydrogen peroxide. To promote the seamless advancement of the reaction and prevent potential harm to Gox, it is necessary to further decompose the hydrogen peroxide. When employing purified enzyme for catalysis experiments, supplementary peroxidase is incorporated to guarantee the unhindered progression of the reaction. Thus, purified enzyme catalysis experiment: 20 mL sodium phosphate buffer (0.1 M, pH 5.5) with 1 U/mL purified enzyme, 10 g/L d-glucose, and 2 U/mL catalase at 37 °C and 0~200 rpm for 1 h.
Solid culture medium staining experiment
After 3 days of growth on solid coloration medium, the prepared coloration mixture was poured into the petri dishes, and the dishes were gently swirled to ensure complete coverage. After cooling, the petri dishes were placed in a 37 °C incubator, and the colonies’ color changes were continuously observed. The composition of the solid coloration medium was as follows: 80 g/L glucose, 5 g/L yeast extract, 0.7 g/L KH2PO4, 1 g/L corn syrup, 0.2 g/L MgSO4·7H2O, 0.2 g/L urea, 1.8% agar, and 10 g/L CaCO3, with the pH adjusted to 7.0. The coloration mixture was prepared by dissolving 1% agar in 10 mL of deionized water, heating it until fully dissolved, then naturally cooling it to around 55 °C. Subsequently, 2 mL of 18% glucose solution, 200 μL of 10 g/L O-dianisidine methanol solution, and 400 μL of 90 U/mL horseradish peroxidase were added to the mixture.
HPLC analysis of gluconic acid
The gluconic acid determination method utilized the previously reported citric acid determination method (Cao et al. 2020). Gluconic acid was qualitatively analyzed using High-Performance Liquid Chromatography (HPLC) with an Agilent 1260 In-finity II Prime system, equipped with an organic acid column (Aminex HPX-87H, 300 mm × 7.8 mm). The column temperature was maintained at 65 °C, and the mobile phase consisted of 5 mM H2SO4, with a flow rate of 0.6 mL/min. Detection was performed using a UV wavelength of 210 nm. Standard gluconic acid samples, ranging from 0.5 to 10 g/L, were prepared using gluconic acid standard solutions for HPLC analysis. Subsequently, a standard curve was established to determine the gluconic acid concentration in the samples.
Measurement of CO-difference spectra for VHb detection
VHb could be detected through carbon monoxide (CO) difference spectra, as demonstrated by Mo et al. 2016 with slight modifications. The samples were derived from the supernatant of shaker flask cultures of various strains on the third day of gluconic acid fermentation. For CO-difference spectral analysis, the sample underwent protein reduction with sodium dithionite (Na2S2O4, 2.5 g/L) and was subsequently divided into two aliquots. One aliquot was subjected to CO gas bubbling to coordinate with VHb, while the other, untreated aliquot served as the calibration control for differential spectrometry analysis. The prepared 100 μL sample was transferred to a 96-well plate. Utilizing a microplate reader, spectra were meticulously recorded within the 400 to 500 nm range.
Statistical analysis
The experiments were conducted in triplicate or quadruplicate (only for the measurement of CO-difference spectra). A t-test was performed to assess significant differences among the groups, with a p-value of < 0.05 (∗) indicating statistical significance. Data were expressed as the mean ± standard deviation.
GenBank accession number for the coding sequences of various proteins
The GenBank accession numbers for the coding sequences of various proteins were listed as follows: GoxA, OR347817; GoxB, OR347818; GoxC, OR347819; GoxD, OR347820; VHb , OR347821; SPglaA-VHb, OR347822; GoxA-GS1-VHb, OR347823; GoxA-GS3-VHb, OR347824; GoxA-EK1-VHb, OR347825; GoxA-EK3-VHb, OR347826; SPglaA-VHb-GS1-GoxA, OR347827; SPglaA-His-GoxA, OR347828; SPglaA-His-VHb-GS1-GoxA, OR347829; SPgalA-His-VHb-SpyCatcher, OR347830; SPgalA-His-VHb-SnoopCatcher, OR347831; SPgalA-Spytag-GoxA, OR347832; SPgalA-Snooptag-GoxA, OR347833.
Results
Identification of a novel Gox from industrial A. niger
An A. niger strain, utilized for industrial production of gluconic acid, was obtained from Shandong Fuyang Biotechnology Co., Ltd. located in China. This specific industrial strain was designated as A. niger S1565. The morphological characteristics of strain S1565 are depicted in Fig. 1c. The genomic sequence of strain S1565 was acquired via genome resequencing, employing the genome of A. niger ATCC1015 (GenBank GCA_000230395.2) as a reference template. Subsequently, the genome database of strain S1565 was screened using a known A. niger Gox sequence (PDB ID: 1CF3) as the query sequence through a local blastp alignment program. Four genes, namely goxA, goxB, goxC, and goxD, were identified as putative Goxs encoding genes within strain S1565. Detailed gene sequences and corresponding amino acid sequences of these four Goxs can be found in the supplement material. Sequence analysis was further conducted by aligning the amino acid sequences encoded by these four genes with the previously reported enzymes. GoxA exhibited a remarkable similarity to the known Gox from A. niger (PDB ID: 1CF3), with only three differing amino acid residues (V189T, S384A, S594A) (Fig. S2). Similarly, GoxB demonstrated a high sequence similarity of 97% to the Gox derived from A. niger CBS513.88 (ACCESSION NO. XP_001395420). Additionally, both GoxC and GoxD displayed significant sequence similarities of 99% and 99%, respectively, to the glucose dehydrogenases (GDH) reported by Sode et al. in A. niger (Sode et al. 2017) and Iwasa et al. in A. phoenicis (Iwasa et al. 2018). The evolutionary relationships of these enzymes were illustrated in Fig. 1a.

Functional identification of Goxs in industrial gluconic acid production strain of A. niger S1565. a Neighbor-joining phylogenetic tree of Goxs from different microorganisms, established using MEGA software version 10.1.7. b Gluconic acid production by GoxA-D overexpression strains and parent strain S1691 after 3 days of fermentation at 28 °C and 200 rpm in shake flasks. c Solid culture medium staining experiments of GoxA-D overexpression strains, parent strain S1691, and industrial strain S1565 after 3 days of cultivation at 28 °C. S1971 overexpressed GoxA, S1990 overexpressed GoxC, S2102 overexpressed GoxD, and S2167 overexpressed GoxB. The values were depicted in b as the mean ± standard deviation (n = 3). Asterisk (*) denoted statistical significance, where p < 0.05
Due to the unavailability of antibiotics suitable for screening S1565, which made it challenging to genetically modify the industrial strain, goxA, goxB, goxC, and goxD were expressed individually in another derivative strain of A. niger, S1691 (derived from A. niger ATCC1015), to ascertain their functionality as Gox. The S1691 strain had been genetically modified to delete the homologs of the four genes gox1-4 mentioned (GenBank EHA27180.1, EHA25730.1, EHA19699.1, and EHA23687.1) to eliminate any potential interference. The expression cassettes of goxA, goxB, goxC, and goxD were integrated into the α-amylase A (GenBank EHA19519.1) locus of the S1691 genome, resulting in the generation of the following strains: S1971 (ΔamyA::PgpdA-godA-TtrpC), S2167 (ΔamyA::PgpdA-godB-TtrpC), S1990 (ΔamyA::PgpdA-godC-TtrpC), and S2102 (ΔamyA::PgpdA-godD-TtrpC). Subsequently, the colonies of the four strains grown on solid agar plates were subjected to a colorimetric assay to determine the production of Gox. As depicted in Fig. 1c, only S1971 exhibited a conspicuous brown color change, akin to the industrial strain S1565, while the other three strains and the parental strain S1691 displayed no discernible alteration in color. This outcome conclusively indicated that S1971, harboring the goxA gene, was capable of producing Gox, whereas the overexpression of goxB, goxC, and goxD failed to yield Gox. To further corroborate these findings, a gluconic acid shake flask fermentation experiment was conducted. After 3 days of fermentation, it was observed that only S1971 produced an impressive 75.2 g/L of gluconic acid, while the fermentation broths of the other strains exhibited no detectable gluconic acid production Fig. 1b. The results of the shake flask fermentation were consistent with the outcomes of the solid agar plate colorimetric assay, thus firmly establishing goxA as the gene encoding Gox in the industrial strain S1565. The strain S1971 expressing goxA was used for further investigation of the role of VHb on gluconic acid production in A. niger.
Effect of free VHb overexpression on gluconic acid production in A. niger
Oxygen transfer plays a pivotal role in gluconic acid production. Wang et al. (2022) demonstrated that the addition of free VHb can enhance the catalytic activity of Gox in in vitro experiments (Wang et al. 2022). Based on this finding, our study aimed to investigate the effect of overexpressing the free form of VHb in the strain S1971 on gluconic acid production and Gox activity during shake flask fermentation. Given the distribution of Gox in both the intracellular and extracellular compartments of A. niger, two VHb overexpression strains were engineered based on S1971. The first strain, S2241 (PgpdA-vgb-TtrpC), specifically expresses VHb intracellularly. In the second overexpression strain, S2246 (PgpdA-spglaA-vgb-TtrpC), the N-terminus of VHb is linked to a signal peptide, SPglaA, derived from A. niger glucoamylase GlaA (GenBank: EHA21384.1).
A 3-day shake flask fermentation of gluconic acid was conducted at different rotational speeds to investigate the acid production differences among the S1971, S2241, and S2246 strains. The results indicated that, under the same rotational speed conditions, there were no significant variations in gluconic acid yield among the three strains. The CO-difference spectra of the supernatants obtained from shake flask gluconic acid fermentation on the third day were analyzed for strains S1971, S2241, and S2246. Notably, only the fermentation supernatants of S2246 exhibited discernible VHb activity, characterized by absorption peaks within the 410~440 nm range (Fig. S3a). However, as the rotational speed decreased from 250 to 150 rpm, a noticeable decline in gluconic acid production was observed for all strains, with reductions of 61%, 64%, and 62% for S1971, S2241, and S2246, respectively (Fig. 2a). In shake flask fermentation systems, rotational speed directly influences the efficiency of oxygen mass transfer. The decrease in rotational speed leads to a decline in oxygen mass transfer efficiency. Consequently, the reduced availability of oxygen negatively impacts the catalytic efficiency of Gox, thereby decreasing the production of gluconic acid. This observation highlights the importance of oxygen mass transfer in the gluconic acid fermentation process. In conclusion, the VHb overexpression strategies employed did not enhance gluconic acid production in the shake flask fermentation.

Effects of free VHb overexpression on gluconic acid fermentation in shake flasks at 28 °C and 200 rpm for 3 days (a) and extracellular Gox enzyme activity during protein fermentation at 28 °C and 200 rpm for 5 days (b). S1971 overexpressed GoxA. S2241 specifically expressed VHb intracellularly. In S2246 (PgpdA-spglaA-vgb-TtrpC), the N-terminus of VHb is linked to a signal peptide, SPglaA, derived from A. niger glucoamylase GlaA (GenBank: EHA21384.1). The values were presented as the mean ± standard deviation (n = 3). Asterisk (*) denoted statistical significance, where p < 0.05
Due to the low nitrogen content in the gluconic acid fermentation medium, it may not provide sufficient support for the synthesis of VHb in large quantities. Therefore, a protein fermentation medium was utilized to conduct a 5-day fermentation of S1971, S2241, and S2246 strains, followed by the measurement of extracellular Gox activity in the supernatant. As shown in Fig. 2b, the results demonstrated that the crude enzyme activity of Gox in the fermentation broth of S2241 and S2246 reached 409.2 and 343.6 U/mL, respectively, which were 2.1-fold and 1.7-fold higher than that of S1971. While these strains showed no significant difference in gluconic acid production during glucose fermentation, a substantial variation in the total Gox enzyme activity secreted during protein fermentation was observed. This observed difference could be attributed to VHb’s oxygen transfer capability enhancing the catalytic efficiency of Gox or variations in Gox secretion. However, three experimental findings contradicted the notion that VHb’s oxygen transfer capability contributes to the catalytic efficiency of Gox. First, whether VHb was expressed intracellularly or secreted extracellularly, there was minimal difference in gluconic acid production between S2241, S2246, and S1971, indicating that VHb did not significantly influence gluconic acid synthesis. Second, during protein fermentation, whether VHb was expressed intracellularly or secreted extracellularly, the extracellular total enzyme activity of S2241 and S2246 significantly increased. This suggested that VHb, whether inside or outside the cell, similarly enhanced the total enzyme activity of Gox, independent of VHb’s oxygen transfer capability. Third, CO-difference spectra measuring VHb functionality revealed no detectable VHb activity outside the cell for S2241. This implied that the increased extracellular Gox total enzyme activity in S2241 is not dependent on VHb’s oxygen transfer for the catalytic efficiency of Gox. Consequently, the expression of VHb enhanced the Gox secretion in the strain during protein fermentation. This phenomenon of VHb overexpression promoting protein synthesis and secretion has been extensively reported (Yu et al. 2021a). Consequently, the hypothesis of enhancing oxygen mass transfer to increase gluconic acid production through the overexpression of free VHb in A. niger had proven to be unsuccessful.
Fusion VHb to enhance catalytic efficiency of Gox in A. niger
To overcome limitations associated with overexpressing free VHb in A. niger, we developed a series of fusion proteins of Gox and VHb to enhance catalytic efficiency. Tandem fusion was considered a viable approach for constructing the fusion protein, with careful consideration given to the order of fusion partners and linker sequence. Recent reports suggest that fusing an additional domain to Gox’s C-terminus using a flexible linker had minimal impact on Gox activity (Choi et al. 2011; Gong et al. 2021; Joensuu et al. 2010; Kovacevic et al. 2019). Choi et al. (2011) constructed a fusion protein by linking Gox with the peptide R5 (SSKKSGSYSGSKGSKRRIL). Directly connecting R5 to Gox’s N-terminus resulted in activity loss, while linking Gox’s C-terminus with R5 using a flexible short peptide (GGGS) restored activity (Choi et al. 2011). Joensuu et al. fused the hydrophobin protein HFBI to Gox’s C-terminus using a linker peptide (SGSVTSTSKTTATASKTSTST) derived from A. niger glucoamylase G1, with no impact on Gox-HFBI fusion protein activity (Joensuu et al. 2010). Kovacevic et al. fused the fluorescent protein yGFP to Gox’s C-terminus using a flexible linker peptide (GSSG), resulting in slightly reduced activity (Kovacevic et al. 2019). Gong et al. successfully fused the carbohydrate-binding module family 2 (CBM2) with a natural short peptide (NL) from β-glucosidase to Gox’s C-terminus, obtaining the fusion protein Gox-NL-CBM2, which maintained activity but exhibited significantly reduced expression levels (Gong et al. 2021). Therefore, linking VHb to the C-terminus of Gox appears to be a preferred choice.
First, we considered constructing four fusion proteins by fusing VHb to the C-terminus of Gox using different lengths of flexible linkers (GS1 and GS3) and rigid linkers (EK1 and EK3). The four fusion proteins were named GoxA-GS1-VHb, GoxA-GS3-VHb, GoxA-EK1-VHb, and GoxA-EK3-VHb. These fusion proteins were overexpressed in the S1691 strain, resulting in the generation of four strains: S2193 (ΔamyA::PgpdA-godA-gs1-vgb-TtrpC), S2195 (ΔamyA::PgpdA-godA-gs3-vgb-TtrpC), S2294 (ΔamyA::PgpdA-godA-ek1-vgb-TtrpC), and S2298 (ΔamyA::PgpdA-godA-ek3-vgb-TtrpC). After 2 days of growth on solid agar plates, no strains showed color changes after a 24-h incubation period (Fig. 3a). Furthermore, after a 3-day gluconic acid fermentation, the gluconic acid concentrations in the cultures of the four strains remained below 1 g/L (Fig. 3b). Additionally, after 5 days of protein fermentation, the crude enzyme activity assay in the culture supernatants of the four strains showed minimal color changes, indicating no detection of Gox activity (Fig. 3b). The CO-difference spectra of supernatants derived from shake flask gluconic acid fermentation on the third day were scrutinized for strains S2193, S2195, S2294, and S2298. Importantly, it was observed that the fermentation supernatants from these four strains displayed no discernible VHb activity (Fig. S3b). These observations raise concerns that the strategy of linking the N-terminus of VHb to the C-terminus of Gox was unsuccessful, possibly leading to the fusion protein losing the activity of Gox, which contradicts previous reports.

Functional characterization of different forms of GoxA and VHb fusion proteins. a Solid culture medium staining experiments of the overexpressed fusion protein strains after 3 days of cultivation at 28 °C. b Gluconic acid production during shake flask fermentation by the overexpressed fusion protein strains at 28 °C and 200 rpm after 3 days. c Enzyme catalysis experiments using crude enzyme solutions obtained from the protein fermentation of strains S1971, S2246, and S2249 under different rotational speeds at 37 °C for 1 h. S1971 overexpressed GoxA. S2246 overexpressed GoxA and secreted VHb. S2193, S2195, S2294, S2298, and S2249 overexpressed the fusion proteins GoxA-GS1-VHb, GoxA-GS3-VHb, GoxA-EK1-VHb, GoxA-EK3-VHb, and VHb-GS1-GoxA, respectively. The values were presented as the mean ± standard deviation (n = 3). Asterisk (*) denoted statistical significance, where p < 0.05
Next, we made an attempt to construct the fusion protein VHb-GS1-GoxA by linking VHb to the N-terminus of Gox using the flexible linker GS1. This resulted in the strain S2249 (ΔamyA::PgpdA-spglaA-vgb-gs1-godA-TtrpC). Following a 3-day gluconic acid shake flask fermentation, strain S2249 exhibited a gluconic acid production of 3.0 g/L (Fig. 3b). Furthermore, after a 5-day protein fermentation, the culture supernatant of S2249 demonstrated an Gox activity of 21.0 U/mg protein (Fig. 3b). It was evident that strain S2249 displayed significantly improved gluconic acid production and enzyme activity compared to the previous four fusion protein expression strains. The results from the CO-difference spectra also revealed that the fermentation supernatants of S2249 exhibited VHb activity (Fig. S3c). However, the gluconic acid yield of S2249 still remained considerably lower than that of strain S1971.
In order to preliminarily evaluate the catalytic performance of VHb-GS1-GoxA compared to GoxA, the culture supernatants of S1971 and S2249 were collected in protein fermentation, and catalytic experiments were conducted under different rotational speeds. The experimental results demonstrated that at 0 rpm, 100 rpm, and 200 rpm, 50 U of VHb-GS1-GoxA crude enzyme solution catalyzed the production of 0.40 g/L, 1.02 g/L, and 1.95 g/L of gluconic acid within 1 h, respectively (Fig. 3c). But GoxA yielded 0.16 g/L, 0.58 g/L, and 1.30 g/L of gluconic acid under the same conditions (Fig. 3c). Notably, VHb-GS1-GoxA exhibited superior catalytic efficiency to GoxA at each rotational speed, resulting in a 152%, 77%, and 50% increase in gluconic acid titers at 0 rpm, 100 rpm, and 200 rpm, respectively. Moreover, the extent of improvement was more pronounced at lower rotational speeds.
To further evaluate the catalytic performance of VHb-GS1-GoxA, we constructed strains expressing His-tagged proteins His-VHb-GS1-GoxA and His-GoxA, designated as S2250 (ΔamyA::PgpdA-spglaA-his-vgb-gs1-godA-TtrpC) and S2637 (ΔamyA::PgpdA-spglaA-his-godA-TtrpC), respectively. After protein purification from the culture supernatants of S2250 and S2637, purified His-VHb-GS1-GoxA and His-GoxA proteins were obtained (Fig. 4a). Enzyme kinetic analysis revealed that His-GoxA exhibited Vmax, Km, and Kcat/Km values of 476.5 U/mg, 44.8 mM, and 23.1 s−1·mM−1, respectively, while His-VHb-GS1-GoxA showed Vmax, Km, and Kcat/Km values of 461.7 U/mg, 27.5 mM, and 45.3 s−1·mM−1, respectively (Fig. 4b). Remarkably, His-VHb-GS1-GoxA demonstrated a 96% increase in Kcat/Km compared to His-GoxA. Here, Vmax referred to the maximum enzymatic reaction rate. Km was a constant that remains stable when environmental conditions are constant. A smaller Km indicated a lower substrate concentration required for the enzyme reaction, signifying a higher affinity between the enzyme and substrate. Kcat, also known as turnover number, is calculated by dividing Vmax by the enzyme concentration. Thus, Kcat measured the quantity of substrate converted by a single enzyme molecule in one second. The ratio of Kcat/Km was the most crucial parameter assessing enzyme catalytic efficiency. A higher Kcat/Km value indicates better catalytic performance of the enzyme. The Kcat/Km value for the fusion protein His-VHb-GS1-GoxA is 1.96 times higher than that of His-GoxA. This demonstrated that the catalytic efficiency of the fusion protein His-VHb-GS1-GoxA was superior to that of His-GoxA.

Characterization of purified His-VHb-GS1-GoxA and His-GoxA. a Western blot analysis of purified His-VHb-GS1-GoxA and His-GoxA. b Kinetic parameters of purified His-VHb-GS1-GoxA and His-GoxA. c Enzyme catalysis experiments using purified His-VHb-GS1-GoxA, His-GoxA, and commercial Gox (comGox) proteins at different rotational speeds. The values were presented as the mean ± standard deviation (n = 3). Asterisk (*) denoted statistical significance, where p < 0.05
Subsequently, catalytic experiments were performed at different rotational speeds to assess the catalytic efficiency. The activities of His-GoxA, His-VHb-GS1-GoxA, and commercial Gox (comGox) were maintained at 1 U/mL in reaction systems, and catalase (2 U/mL) was added to eliminate hydrogen peroxide. As shown in Fig. 4c, at 0 rpm, His-VHb-GS1-GoxA showed gluconic acid production of 2.4 g/L, surpassing the values of comGox (1.1 g/L) and His-GoxA (1.4 g/L) by 54% and 42%, respectively. At 100 rpm, His-VHb-GS1-GoxA produced 5.0 g/L of gluconic acid, which represented a 50% and 34% increase compared to comGox (2.5 g/L) and His-GoxA (3.3 g/L), respectively. Similarly, at 200 rpm, His-VHb-GS1-GoxA achieved gluconic acid production of 5.3 g/L, outperforming comGox (3.2 g/L) and His-GoxA (4.3 g/L) by 40% and 20%, respectively. These findings indicated that His-VHb-GS1-GoxA exhibited superior catalytic efficiency compared to His-GoxA and comGox.
In summary, VHb-GS1-GoxA demonstrated superior catalytic capabilities compared to GoxA. However, the strain S2249 overexpressing VHb-GS1-GoxA exhibited significantly lower gluconic acid production in shake flask fermentation compared to the strain S1971 overexpressing GoxA. This suggests that the expression and secretion of VHb-GS1-GoxA in A. niger are considerably lower than that of GoxA, highlighting a critical challenge that needs to be addressed.
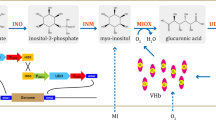
Expression fusion proteins using SpyTag/Catcher and SnoopTag/Catcher in A. niger
Due to the longer peptide chains of fusion proteins, their overexpression may be limited by the need for more intricate protein expression regulation. To address this challenge, two strategies were designed for the assembly of the fusion proteins, utilizing the principles of specific and efficient isopeptide bond formation through SpyCatcher/SpyTag and SnoopCatcher/SnoopTag reactions (Fig. 5) (Hatlem et al. 2019). The first strategy involved a single-strain system, where strain S2643 simultaneously expressed SpyTag-Gox and His-VHb-SpyCatcher proteins, allowing for the intracellular and extracellular self-assembly of the His-VHb-Spy-Gox enzyme complex. The second strategy involved a dual-strain system comprising strains S2648 and S2650. During co-fermentation, strain S2648 secreted SpyTag-Gox and His-VHb-SnoopCatcher proteins, while strain S2650 secreted SnoopTag-Gox and His-VHb-SpyCatcher proteins. It was anticipated that in the extracellular fermentation broth, SpyTag-Gox and His-VHb-SpyCatcher would self-assemble to form the His-VHb-Spy-Gox enzyme complex, while SnoopTag-Gox and His-VHb-SnoopCatcher would self-assemble to form the His-VHb-Snoop-Gox enzyme complex.

Design of single- and dual-strain systems using SpyCatcher/SpyTag and SnoopCatcher/SnoopTag tools
Subsequently, gluconic acid shake flask fermentation experiments were conducted at different rotational speeds for both the single-strain and dual-strain systems. In the dual-strain system, three fermentation setups were performed: individual fermentations of S2648 and S2650, as well as co-fermentation of S2648 and S2650. The gluconic acid yields in the three setups were almost similar at 250 rpm, ranging from 107.5 to 118.2 g/L (Fig. 6A), slightly lower than the yield of S1971 (123.9 g/L) (Fig. 6a). Similarly, no significant differences in gluconic acid yields were observed at rotational speeds of 200 rpm and 150 rpm (Fig. 6a). The co-fermentation of S2648 and S2650 did not exhibit any advantages over the individual fermentations of S2648 and S2650.

Gluconic acid production in single- and dual-strain systems under different rotational speeds during 3-day shake flask fermentation at 28 °C (a) and a comparison of gluconic acid fermentation between single-strain system and S1971 strain in bioreactor at 28 °C for 84 h (b). Single-strain system: S2643, dual-strain system: S2648 and S2650. The values were presented as the mean ± standard deviation (n = 3). Asterisk (*) denoted statistical significance, where p < 0.05
For the single-strain system, gluconic acid production was measured at rotational speeds of 150 rpm, 200 rpm, and 250 rpm. The gluconic acid yields for S2643 under these conditions were 36.1 g/L, 51.3 g/L, and 63.7 g/L, respectively (Fig. 6a). Although the shake flask yield of the single-strain system was lower than that of the dual-strain system, it was less affected by a decrease in rotational speed. At rotational speeds of 200 rpm and 150 rpm, the gluconic acid yields in the single-strain system were 81% and 57% of that at 250 rpm, respectively. The gluconic acid yields in the dual-strain system at rotational speeds of 200 rpm and 150 rpm were 53% and 36 % of that at 250 rpm, respectively. The CO-difference spectra of supernatants collected from shake flask gluconic acid fermentation on the third day were examined for individual strains S2643, S2648, S2650, and the dual-strain system (S2648$S2650). Remarkably, VHb activity was only evident in the fermentation supernatants of S2643, as indicated by discernible absorption peaks falling within the 410~440 nm range. These results suggested that VHb in the dual-strain system did not work on gluconic acid fermentation, while the single-strain system played a more substantial role. To further investigate this, Western blot experiments were conducted on the protein fermentation supernatants of the single-strain and dual-strain systems (Fig. S4). The results confirmed the formation of the His-VHb-Spy-GoxA enzyme complex in the single-strain system, while no complex was detected in the dual-strain system. This finding aligned with the gluconic acid fermentation results.
To evaluate the advantage of the single-strain system in gluconic acid production, we conducted gluconic acid fermentation experiments in 2-L bioreactors using S1971, which expressing GoxA, and the single-strain system S2643. The spores of both strains were inoculated into shake flasks for 1-day seed culture incubation. Afterwards, seed cultures with a total Gox activity of 4000 U were transferred to the bioreactors for an 84-h fermentation. The results, depicted in Fig. 6b, revealed that at an airflow rate of 1.2 vvm, S1971 and S2643 produced gluconic acid at concentrations of 120.6 g/L and 131.1 g/L, respectively. When the airflow rate was reduced to 0.6 vvm, the gluconic acid yields decreased to 87.1 g/L for S1971 and 107.1 g/L for S2643. Under well-aerated conditions (1.2 vvm), S2643 exhibited a slightly higher gluconic acid yield, with a 9% increase compared to S1971. Under low oxygen conditions (0.6 vvm), S2643 demonstrated a 23% higher gluconic acid yield than S1971. These findings suggested that S2643 possessed enhanced oxygen utilization capabilities during the reactor fermentation process for gluconic acid production when compared to S1971.
Discussion
Gluconic acid, a traditionally significant organic acid, finds extensive applications in the food, feed, beverage, textile, pharmaceutical, and construction industries (Canete-Rodriguez et al. 2016). The current industrial production of gluconic acid heavily relies on the submerged fermentation method using A. niger. However, the enzyme-catalyzed production has emerged as a potent alternative, with both methods fundamentally dependent on the proficient performance of Gox. Despite this, the industrial-scale production of gluconic acid faces challenges concerning cost-effectiveness and environmental sustainability. To address these issues, researchers had been dedicated to exploring novel Goxs or implementing protein engineering modifications to develop high-performance and stable Goxs, thereby enhancing the economic viability of gluconic acid industrial production (Bauer et al. 2022; Dubey et al. 2017; Khatami et al. 2022). In this study, we focused on investigating the A. niger strain commonly employed in practical gluconic acid industrial production and successfully identified a new Gox within its genome. Our catalytic experiment in shake flasks revealed that the GoxA, encoded by this novel gene, exhibited remarkably superior catalytic efficiency compared to the commercially available Gox (Sigma-Aldrich, Product Number: G2133). This observation indicated that the amino acid sequence differences (V189T, S384A, S594A) between GoxA and the previously reported Gox from A. niger (PDB ID: 1CF3) likely served as potential sites for enhancing Gox catalytic performance. This finding held new promise for innovative protein engineering strategies aimed at optimizing Gox in the context of gluconic acid production.
It was worth noting that the catalytic efficiency of Gox did not solely depend on the inherent properties of the enzyme itself but also on the influence of other factors in the catalytic system. For instance, oxygen acted as one of the direct substrates involved in Gox catalytic reactions, making the concentration and mass transfer efficiency of oxygen in the solution a critical factor affecting Gox activity. Previous studies typically focused on optimizing the fermentation process to enhance oxygen gas-liquid mass transfer efficiency (Singh and Kumar 2007). However, some research suggested that improving the enzyme’s catalytic efficiency could also be achieved by harnessing the oxygen delivery ability of VHb (Isarankura-Na-Ayudhya et al. 2010; Khang et al. 2003; Ma et al. 2009; Wang et al. 2022). Wang et al. (2022) demonstrated that adding a certain concentration of free VHb could significantly enhance Gox catalytic efficiency in an in vitro reaction system (Wang et al. 2022). Nevertheless, this approach faced challenges in achieving cost-effective industrial applications, as VHb was currently difficult to express at extremely high levels and in large quantities. This study explored the construction of fusion proteins combining VHb and GoxA. Among these fusion proteins, VHb-GS1-GoxA exhibited superior catalytic performance compared to GoxA alone, with a 96% increase in Kcat/Km value. However, unlike GoxA, VHb-GS1-GoxA was not effectively expressed and secreted in A. niger, resulting in a low overall extracellular enzyme activity. To address this problem, single- and dual-strain systems were further designed using the tools of SpyCatcher/SpyTag and SnoopCatcher/SnoopTag. These systems facilitated the individual expression of VHb and GoxA, followed by self-assembly to form the composite protein. The single-strain system was demonstrated to be effective in gluconic acid fermentation within the reactor in this study.
However, a fundamental issue remained as the overexpression and secretion of VHb and VHb-containing fusion proteins in A. niger appeared to be limited. Firstly, the expression of free VHb did not enhance gluconic acid production, which was inconsistent with the results reported by (Wang et al. 2022) in their in vitro catalytic experiments. However, the expression of free VHb did promote an increase in extracellular Gox total enzyme activity during protein fermentation and CO-difference spectra measurement, confirming the physiological function of VHb in A. niger. Thus, we speculated that the expression level of VHb in A. niger was much lower than the concentration of VHb added in vitro catalytic experiments (Wang et al. 2022). Secondly, the low secretion of VHb-GS1-GoxA provided direct evidence of this limitation. Thirdly, although the single-strain system designed using the tool SpyCatcher/SpyTag achieved good results in gluconic acid production experiments in the bioreactor, Western blot analysis failed to detect the presence of His-VHb-SpyCatcher and His-VHb-SnoopCatcher in either the single-strain or dual-strain systems, indicating very low secretion levels of these two VHb proteins. In the dual-strain system, His-VHb-Spy-Gox and His-VHb-Snoop-Gox composite proteins were not detected, suggesting that spontaneous formation of composite proteins does not occur extracellularly in the dual-strain system. This may be constrained by the low secretion of His-VHb-SpyCatcher and His-VHb-SnoopCatcher. Moreover, CO-difference spectra indicated the absence of VHb activity extracellularly in the dual-strain system. Based on these observations, the low secretion of His-VHb-SpyCatcher and His-VHb-SnoopCatcher emerged as a critical constraint in extracellular composite protein formation. Enhancing the expression and secretion of VHb in A. niger became a crucial issue and the next direction for our further research. Potential strategies to address this issue may involve the optimization of signal peptides, enhancing gene copy numbers, regulation of the protein quality control system, and exploring more suitable hosts, such as Pichia pastoris, which was also one of the potential solutions.
In conclusion, the fusion of VHb and Gox proteins had proven to be a promising strategy to enhance Gox enzymatic activity by utilizing VHb’s oxygen delivery capability. Among the engineered fusion proteins, VHb-GS1-GoxA showed superior catalytic performance. However, its expression and secretion in A. niger were significantly limited. To address this challenge, we developed a single-strain system based on the SpyCatcher/SpyTag platform, enabling independent expression of VHb and GoxA, followed by self-assembly to form the composite enzyme. This single-strain system demonstrated favorable performance in gluconic acid fermentation within the bioreactor. These discoveries provide novel insights and lay the foundation for improving the catalytic efficiency of Gox. Moreover, our research offered potential technical solutions to enhance the industrial production efficiency of gluconic acid and reduce aeration costs, thereby contributing to the overall economic benefits of gluconic acid.
Data availability
All data generated during this study are included in this published article and its supplementary information file.
References
Bankar SB, Bule MV, Singhal RS, Ananthanarayan L (2009) Glucose oxidase--an overview. Biotechnol Adv 27(4):489–501. https://doi.org/10.1016/j.biotechadv.2009.04.003
Bauer JA, Zamocka M, Majtan J, Bauerova-Hlinkova V (2022) Glucose oxidase, an enzyme “Ferrari”: its structure, function, production and properties in the light of various industrial and biotechnological applications. Biomolecules 12(3):472. https://doi.org/10.3390/biom12030472
Canete-Rodriguez AM, Santos-Duenas IM, Jimenez-Hornero JE, Ehrenreich A, Liebl W, Garcia-Garcia I (2016) Gluconic acid: properties, production methods and applications-an excellent opportunity for agro-industrial by-products and waste bio-valorization. Process Biochem 51(12):1891–1903. https://doi.org/10.1016/j.procbio.2016.08.028
Cao W, Yan L, Li M, Liu X, Xu Y, Xie Z, Liu H (2020) Identification and engineering a C4-dicarboxylate transporter for improvement of malic acid production in Aspergillus niger. Appl Microbiol Biotechnol 104(22):9773–9783. https://doi.org/10.1007/s00253-020-10932-1
Choi O, Kim BC, An JH, Min K, Kim YH, Um Y, Oh MK, Sang BI (2011) A biosensor based on the self-entrapment of glucose oxidase within biomimetic silica nanoparticles induced by a fusion enzyme. Enzyme Microb Technol 49(5):441–445. https://doi.org/10.1016/j.enzmictec.2011.07.005
Dubey MK, Zehra A, Aamir M, Meena M, Ahirwal L, Singh S, Shukla S, Upadhyay RS, Bueno-Mari R, Bajpai VK (2017) Improvement strategies, cost effective production, and potential applications of fungal glucose oxidase (GOD): current updates. Front Microbiol 8:1032. https://doi.org/10.3389/fmicb.2017.01032
Fu LH, Qi C, Lin J, Huang P (2018) Catalytic chemistry of glucose oxidase in cancer diagnosis and treatment. Chem Soc Rev 47(17):6454–6472. https://doi.org/10.1039/c7cs00891k
Gong W, Han Q, Chen Y, Wang B, Shi J, Wang L, Cai L, Meng Q, Zhang Z, Liu Q, Yang Y, Yang J, Zheng L, Li Y, Ma Y (2021) A glucose biosensor based on glucose oxidase fused to a carbohydrate binding module family 2 tag that specifically binds to the cellulose-modified electrode. Enzyme Microb Technol 150:109869. https://doi.org/10.1016/j.enzmictec.2021.109869
Hatlem D, Trunk T, Linke D, Leo JC (2019) Catching a SPY: using the SpyCatcher-SpyTag and related systems for labeling and localizing bacterial proteins. Int J Mol Sci 20(9):2129. https://doi.org/10.3390/ijms20092129
Hofmann G, Diano A, Nielsen J (2009) Recombinant bacterial hemoglobin alters metabolism of Aspergillus niger. Metab Eng 11(1):8–12. https://doi.org/10.1016/j.ymben.2008.07.002
Isarankura-Na-Ayudhya C, Yainoy S, Tantimongcolwat T, Bulow L, Prachayasittikul V (2010) Engineering of a novel chimera of superoxide dismutase and Vitreoscilla hemoglobin for rapid detoxification of reactive oxygen species. J Biosci Bioeng 110(6):633–637. https://doi.org/10.1016/j.jbiosc.2010.07.001
Iwasa H, Ozawa K, Sasaki N, Kinoshita N, Yokoyama K, Hiratsuka A (2018) Fungal FAD-dependent glucose dehydrogenases concerning high activity, affinity, and thermostability for maltose-insensitive blood glucose sensor. Biochem Eng J 140:115–122. https://doi.org/10.1016/j.bej.2018.09.014
Jiang X, Wang Y, Wang Y, Huang H, Bai Y, Su X, Zhang J, Yao B, Tu T, Luo H (2021) Exploiting the activity-stability trade-off of glucose oxidase from Aspergillus niger using a simple approach to calculate thermostability of mutants. Food Chem 342:128270. https://doi.org/10.1016/j.foodchem.2020.128270
Joensuu JJ, Conley AJ, Lienemann M, Brandle JE, Linder MB, Menassa R (2010) Hydrophobin fusions for high-level transient protein expression and purification in Nicotiana benthamiana. Plant Physiol 152(2):622–633. https://doi.org/10.1104/pp.109.149021
Khang YH, Kim IW, Hah YR, Hwangbo JH, Kang KK (2003) Fusion protein of Vitreoscilla hemoglobin with D-amino acid oxidase enhances activity and stability of biocatalyst in the bioconversion process of cephalosporin C. Biotechnol Bioeng 82(4):480–488. https://doi.org/10.1002/bit.10592
Khatami SH, Vakili O, Ahmadi N, Soltani Fard E, Mousavi P, Khalvati B, Maleksabet A, Savardashtaki A, Taheri-Anganeh M, Movahedpour A (2022) Glucose oxidase: applications, sources, and recombinant production. Biotechnol Appl Biochem 69(3):939–950. https://doi.org/10.1002/bab.2165
Khurshid S, Kafiat T, Hanif U, Ulfat M, Qureshi MZ, Bashir T, Sajid A, Ishtiaq S, Ismayil T (2013) Application of glucose oxidase for the production of metal gluconates by fermentation. Afr J Biotechnol 12:6766–6775. https://doi.org/10.5897/AJB12.2796
Kovacevic G, Ostafe R, Balaz AM, Fischer R, Prodanovic R (2019) Development of GFP-based high-throughput screening system for directed evolution of glucose oxidase. J Biosci Bioeng 127(1):30–37. https://doi.org/10.1016/j.jbiosc.2018.07.002
Kwon SY, Choi YJ, Kang TH, Lee KH, Cha SS, Kim GH, Lee HS, Kim KT, Kim KJ (2005) Highly efficient protein expression and purification using bacterial hemoglobin fusion vector. Plasmid 53(3):274–282. https://doi.org/10.1016/j.plasmid.2004.11.006
Liu D, Ke X, Hu ZC, Zheng YG (2021) Improvement of pyrroloquinoline quinone-dependent d-sorbitol dehydrogenase activity from Gluconobacter oxydans via expression of Vitreoscilla hemoglobin and regulation of dissolved oxygen tension for the biosynthesis of 6-(N-hydroxyethyl)-amino-6-deoxy-alpha-l-sorbofuranose. J Biosci Bioeng 131(5):518–524. https://doi.org/10.1016/j.jbiosc.2020.12.013
Liu D, Wan N, Zhang F, Tang YJ, Wu SG (2017) Enhancing fatty acid production in Escherichia coli by Vitreoscilla hemoglobin overexpression. Biotechnol Bioeng 114(2):463–467. https://doi.org/10.1002/bit.26067
Liu J, Shanshan Z, Wenhao L, Guanyi W, Zhoujie X, Wei C, Weixia G, Hao L (2023) Engineering a phosphoketolase pathway to supplement cytosolic acetyl-CoA in Aspergillus niger enables a significant Increase in citric acid production. J Fungi (Basel) 9(5):504. https://doi.org/10.3390/jof9050504
Liu Z, Yuan M, Zhang X, Liang Q, Yang M, Mou H, Zhu C (2020) A thermostable glucose oxidase from Aspergillus heteromophus CBS 117.55 with broad pH stability and digestive enzyme resistance. Protein Expr Purif 176:105717. https://doi.org/10.1016/j.pep.2020.105717
Ma XF, Yu HM, Cheng W, Hui L LQ, ZY S (2009) Triple fusion of D-amino acid oxidase from Trigonopsis variabilis with polyhistidine and Vitreoscilla hemoglobin. World J Microb Biot 25:1353–1361. https://doi.org/10.1007/s11274-009-0022-6
Mano N (2019) Engineering glucose oxidase for bioelectrochemical applications. Bioelectrochemistry 128:218–240. https://doi.org/10.1016/j.bioelechem.2019.04.015
Mo Q, Zhang H, Liu Q, Tang X, Zhao L, Zan X, Song Y (2016) Enhancing nosiheptide production in Streptomyces actuosus by heterologous expression of haemoprotein from Sinorhizobium meliloti. Lett Appl Microbiol 62(6):480–487. https://doi.org/10.1111/lam.12575
Mora-Lugo R, Madrigal M, Yelemane V, Fernandez-Lahore M (2015) Improved biomass and protein production in solid-state cultures of an Aspergillus sojae strain harboring the Vitreoscilla hemoglobin. Appl Microbiol Biotechnol 99(22):9699–9708. https://doi.org/10.1007/s00253-015-6851-3
Mu Q, Cui Y, Tian Y, Hu M, Tao Y, Wu B (2019) Thermostability improvement of the glucose oxidase from Aspergillus niger for efficient gluconic acid production via computational design. Int J Biol Macromol 136:1060–1068. https://doi.org/10.1016/j.ijbiomac.2019.06.094
Seo YM, Khang YH, Yun H (2011) Kinetic resolution of 3-fluoroalanine using a fusion protein of D-amino acid oxidase with Vitroscilla hemoglobin. Biosci Biotechnol Biochem 75(4):820–822. https://doi.org/10.1271/bbb.110122
Singh OV, Kumar R (2007) Biotechnological production of gluconic acid: future implications. Appl Microbiol Biotechnol 75(4):713–722. https://doi.org/10.1007/s00253-007-0851-x
Sode K, Loew N, Ohnishi Y, Tsuruta H, Mori K, Kojima K, Tsugawa W, LaBelle JT, Klonoff DC (2017) Novel fungal FAD glucose dehydrogenase derived from Aspergillus niger for glucose enzyme sensor strips. Biosens Bioelectron 87:305–311. https://doi.org/10.1016/j.bios.2016.08.053
Stark BC, Dikshit KL, Pagilla KR (2012) The biochemistry of Vitreoscilla hemoglobin. Comput Struct Biotechnol J 3:e201210002. https://doi.org/10.5936/csbj.201210002
Stark BC, Pagilla KR, Dikshit KL (2015) Recent applications of Vitreoscilla hemoglobin technology in bioproduct synthesis and bioremediation. Appl Microbiol Biotechnol 99(4):1627–1636. https://doi.org/10.1007/s00253-014-6350-y
Tang Q, Hu Y, Kang L, Wang CZ (2012) Characterization of glucose-induced glucose oxidase gene and protein expression in Helicoverpa armigera larvae. Arch Insect Biochem Physiol 79(2):104–119. https://doi.org/10.1002/arch.21015
Tang R, Weng C, Peng X, Han Y (2020) Metabolic engineering of Cupriavidus necator H16 for improved chemoautotrophic growth and PHB production under oxygen-limiting conditions. Metab Eng 61:11–23. https://doi.org/10.1016/j.ymben.2020.04.009
van den Berg BA, Reinders MJT, Hulsman M, Wu L, Pel HJ, Roubos JA, D dR (2012) Exploring sequence characteristics related to high-level production of secreted proteins in Aspergillus niger. PLoS One 7(10):e45869. https://doi.org/10.1371/journal.pone.0045869
Wang Q, Zheng H, Tao R, Li Q, Jiang Y, Yang S (2022) Vitreoscilla hemoglobin enhances the catalytic performance of industrial oxidases in vitro. Appl Microbiol Biotechnol 106(9-10):3657–3667. https://doi.org/10.1007/s00253-022-11974-3
Webster DA, Dikshit KL, Pagilla KR, Stark BC (2021) The discovery of Vitreoscilla hemoglobin and early studies on its biochemical functions, the control of its expression, and its use in practical applications. Microorganisms 9(8):1637. https://doi.org/10.3390/microorganisms9081637
Xu Y, Shan L, Zhou Y, Xie Z, Ball AS, Cao W, Liu H (2019) Development of a Cre-loxP-based genetic system in Aspergillus niger ATCC 1015 and its application to construction of efficient organic acid-producing cell factories. Appl Microbiol Biotechnol 103(19):8105–8114. https://doi.org/10.1007/s00253-019-10054-3
Yu F, Zhao X, Wang Z, Liu L, Yi L, Zhou J, Li J, Chen J, Du G (2021a) Recent advances in the physicochemical properties and biotechnological application of Vitreoscilla hemoglobin. Microorganisms 9(7):1455. https://doi.org/10.3390/microorganisms9071455
Yu X, Zhang Z, Li J, Su Y, Gao M, Jin T, Chen G (2021b) Co-immobilization of multi-enzyme on reversibly soluble polymers in cascade catalysis for the one-pot conversion of gluconic acid from corn straw. Bioresour Technol 321:124509. https://doi.org/10.1016/j.biortech.2020.124509
Zhang H, Zhang J, Bao J (2016) High titer gluconic acid fermentation by Aspergillus niger from dry dilute acid pretreated corn stover without detoxification. Bioresour Technol 203:211–219. https://doi.org/10.1016/j.biortech.2015.12.042
Zhang X, Xu C, Liu Y, Wang J, Zhao Y, Deng Y (2020) Enhancement of glucaric acid production in Saccharomyces cerevisiae by expressing Vitreoscilla hemoglobin. Biotechnol Lett 42(11):2169–2178. https://doi.org/10.1007/s10529-020-02966-2
Funding
This study was funded by the National Key Research and Development Program of China (2021YFC2100700).
Author information
Authors and Affiliations
Contributions
Conceptualization: JL, HL; methodology: JL, QZ, XL, SZ; literature search: JL; validation: JL, QZ, XL, RZ, XH, SZ; data curation: JL, QZ, XL, RZ, XH, SZ; writing—original draft preparation: JL, HL; writing—review and editing: JL, HL; visualization: JL; supervision: JL, HL; project administration: JL, HL; funding acquisition: HL. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
The article does not contain any studies with human participants or animals where ethical approval was required.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(PDF 586 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, J., Zhang, Q., Liang, X. et al. Improving glucose oxidase catalysis in Aspergillus niger via Vitreoscilla hemoglobin fusion protein. Appl Microbiol Biotechnol 108, 48 (2024). https://doi.org/10.1007/s00253-023-12931-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00253-023-12931-4